
Abstract
Background
Moxifloxacin, a fluoroquinolone antibiotic, is used for the treatment of respiratory tract, pelvic inflammatory disease, skin, and intra-abdominal infections. Its safety profile is considered favorable in most reviews but has been challenged with respect to rare but potentially fatal toxicities (e.g. hepatic, cardiac, or skin reactions).
Objective
To analyze and compare the safety profile of moxifloxacin versus comparators in the entire clinical database of the manufacturer.
Setting
Data on the valid-for-safety population from phase II–IV actively controlled studies (performed between 1996 and 2010) were analyzed. Studies were either double blind (n = 22 369) or open label (n = 7635) and included patients with indications that have been approved in at least one country [acute bacterial sinusitis, acute exacerbation of chronic bronchitis, community-acquired pneumonia, uncomplicated pelvic inflammatory disease, complicated and uncomplicated skin and skin structure infections, and complicated intra-abdominal infections] (n = 27 824) and patients with other indications (n = 2180), using the recommended daily dose (400 mg) and route of administration (oral, intravenous/oral, intravenous only). The analysis included patients at risk (age ≥65 years, diabetes mellitus, renal impairment, hepatic impairment, cardiac disorders, or body mass index <18 kg/m2). Patients with known contraindications were excluded from enrollment by study protocol design, but any patient having entered a study, even if inappropriately, was included in the analysis.
Main Outcome Measure
Crude incidences and relative risk estimates (Mantel-Haenszel analysis) of patients with any adverse event (AE), adverse drug reaction (ADR), serious AE (SAE), serious ADR (SADR), treatment discontinuation due to an AE or ADR, and fatal outcomes related to an AE or ADR.
Results
Overall incidence rates of AEs were globally similar in the moxifloxacin and comparator groups. By filtering the data for differences in disfavor of moxifloxacin (i) at ≥2.5% for events with an incidence ≥2.5% or at ≥2-fold for events with an incidence <2.5% in one or both groups and (ii) affecting ≥10 patients in either group, we observed slightly more (i) AEs in double-blind intravenous-only and open-label oral studies, (ii) SAEs in double-blind intravenous-only studies, (iii) ADRs and SADRs in open-label oral studies, (iv) SADRs in open-label intravenous/oral studies, and (v) premature discontinuation due to AEs in open-label intravenous-only studies. The actual numbers of SADRs (in all studies) were small, with clinically relevant differences noted only in intravenous/oral studies and mainly driven by ‘gastrointestinal disorders’ (15 versus 7 patients) and ‘changes observed during investigations’ (23 versus 7 patients [asymptomatic QT prolongation: 11 versus 4 patients in double-blind studies]). Analysis by comparator (including another fluoroquinolone) did not reveal medically relevant differences, even in patients at risk. Incidence rates of hepatic disorders, tendon disorders, clinical surrogates of QT prolongation, serious cutaneous reactions, and Clostridium difficile-associated diarrhea were similar with moxifloxacin and comparators.
Conclusion
The safety of moxifloxacin is essentially comparable to that of standard therapies for patients receiving the currently registered dosage and for whom contraindications and precautions of use (as in the product label) are taken into account.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Moxifloxacin is approved for oral and intravenous administration in 123 and 108 countries, respectively, as a once-daily 400 mg antibiotic for the treatment of respiratory tract infections (community-acquired pneumonia [CAP], acute exacerbations of chronic bronchitis [AECB], and acute bacterial sinusitis [ABS]) and, depending on the country, pelvic inflammatory disease [PID], complicated and uncomplicated skin and skin structure infections [cSSSIs/uSSSIs], and complicated intra-abdominal infections [cIAIs]. An estimated 140 million prescriptions have been issued for moxifloxacin worldwide, and the drug is included as an effective alternative in guidelines and/or recommendations for each of these indications.[1–10]
The clinical efficacy of moxifloxacin has been unambiguously demonstrated,[11–30] and its safety profile has been analyzed periodically on the basis of pre-marketing studies,[21,31–35] including populations with risk factors,[36,37] such as the elderly[38,39] and those with hepatic or renal insufficiency.[37,40] These data did not show significantly higher toxicity of moxifloxacin compared with commonly used antibiotics if the contraindications and precautions of use mentioned in the Summary of Product Characteristics[41–43] are taken into account. Post-marketing studies[44–53] have confirmed that moxifloxacin is generally well tolerated in medical practice, without new or unanticipated serious adverse events (SAEs) beyond those already established from controlled clinical studies.
The safety profile of moxifloxacin has nevertheless been questioned for two main reasons. First, a number of initially promising fluoroquinolones have been withdrawn (e.g. temafloxacin, trovafloxaxin, sparfloxacin, and gatifloxacin[54–58]) or not approved in Europe (e.g. garenoxacin and gemifloxacin), partly because of toxicity concerns,[59,60] creating suspicion about the whole class. Second, the safety profile of fluoroquinolones has been challenged by the regulatory authorities, triggering (i) for all approved drugs in the US, the inclusion of a ‘black box warning’ for tendinitis;[41] and (ii) for moxifloxacin, the issue in European countries of ‘Dear Healthcare Provider’ letters[61] warning about rare but serious side effects related to hepatotoxicity and severe skin reactions, together with a statement by the European Medicines Agency that “due to safety concerns (hepatic, cardiac [in women and elderly patients], and intestinal problems), moxifloxacin should only be used when other antibiotics cannot be used or have stopped working”,[62] with corresponding label changes throughout the European Union.[42,43]
The current paper presents an in-depth analysis of the safety profile of moxifloxacin, based on the manufacturer’s clinical trial database comprising all actively controlled phase II–IV clinical trials. The objective of the analysis was to examine and compare the safety profile of moxifloxacin with those of the comparators that were all selected as reference therapies for the treatment of corresponding indications at the time the studies were designed.
Methods
Studies
The analysis comprised all double-blind and open-label actively controlled clinical trials included in the clinical trial database of moxifloxacin 400 mg once daily and performed by the manufacturer as part of the phase II–IV programs that were initiated and completed between 1996 and 2010, with the exception of one exploratory phase II study conducted in cirrhotic patients, most of them with Child–Pugh class C cirrhosis. All studies used the oral formulation (400 mg tablets), the 400 mg/250 mL solution for infusion formulation, or a sequence of intravenous and oral formulations. Forty-nine oral studies enrolled patients diagnosed with streptococcal pharyngitis (n = 1), ABS (n = 10), AECB (n = 17), CAP (n = 12), uSSSIs (n = 4), uncomplicated PID (uPID; n = 3), or uncomplicated (n = 3) or complicated (n = 1) urinary tract infection (UTI). Some patients could be enrolled in the same study looking at two different indications – for example, ABS and AECB, or AECB and CAP. Fifteen intravenous/oral studies enrolled patients with CAP (n = 7), cSSSIs (n = 3), cIAIs (n = 2), nosocomial pneumonia (n = 2), or lung abscess or aspiration pneumonia (n = 1). Four intravenous-only studies enrolled patients with CAP (n = 2), cIAIs (n = 2), or AECB (n = 1; this study also enrolled patients with CAP).
Patients
The studies were conducted in Europe, the Americas, the Middle East, Africa, and the Asia/Pacific region. Safety-valid patients were defined as those randomized within an actively controlled clinical trial, having received at least one dose of the study drug and having had at least one observation after initial drug intake. The following subgroups of patients with pre-existing risk factors were evaluated: elderly (age ≥65 years); diabetes mellitus (blood glucose level >200 mg/dL at baseline or at least one medical history finding coded to a preferred term [PT] with a primary path in the high-level term [HLT] diabetes [including subtypes]); renal impairment (serum creatinine ≥1.5 mg/dL for women and ≥1.8 mg/dL for men, or calculated creatinine clearance ≤89 mL/min and ≤59 mL/min for patients aged <65 and ≥65 years, respectively); hepatic impairment (alanine aminotransferase [ALT] or aspartate aminotransferase [AST] >3 × the upper limit of normal [ULN]; or alkaline phosphatase [ALP] >2 × ULN; or total bilirubin >2 × ULN and ALT or ALP >1 × ULN); cardiac disorders (at least one medical history finding coded to a cardiac PT in the Bayer MedDRA Query [BMQ] history of cardiac disease); and low body mass index (BMI) [<18 kg/m2]. Patients with known contraindications, according to what was known or included in the labeling at the time of enrollment, were excluded from entering the study as per the study protocol design. Conversely, no patient entering a study and receiving one or more doses of moxifloxacin or a comparator was excluded from the analysis, even if found later to be among those who should have been prevented from enrollment.
Analyses
All patients valid for the safety analysis from trials with oral, intravenous, or sequential intravenous/oral moxifloxacin and active comparators that were available in the most recent database (data lock point: March 31, 2010) were included in the analysis. The analysis examined all treatment-emergent events (that is, any event occurring after the first dose of medication until the end of follow-up [typically 10–27 days following the last dose]). The planned treatment duration as per the protocols varied from 5 to 21 days according to the indication and/or disease severity, except in one study (treatment duration determined by the investigator).
An overall analysis of safety data was carried out to estimate differences in incidence rates of treatment-emergent adverse events (AEs), adverse drug reactions (ADRs), SAEs, serious ADRs (SADRs), premature discontinuations due to AEs, premature discontinuations due to ADRs, AEs with fatal outcome, and ADRs with fatal outcome. The Medical Dictionary for Regulatory Activities (MedDRA; http://www.meddramsso.com/ [version 13.0]) was used for coding the events. The assessment of causality and seriousness of AEs was made by the study investigators. The incidence rates for events are presented overall, by system organ class (SOC), or by PT within SOC. The analysis was extended by looking specifically for rare events known to be associated with the use of fluoroquinolones, as defined by Standard MedDRA Queries (SMQs)[63] and customized BMQs developed by medical and coding experts (see table SDC-I in the Supplemental Digital Content [SDC]; available online at http://links.adisonline.com/DRZ/A6). Descriptive statistical methods were used to analyze the demographic and safety data.[64] Incidence rates were calculated as crude rates. To compare the risk of a specific AE for moxifloxacin relative to a comparator, relative risk estimates (with corresponding 95% confidence intervals) were calculated by a Mantel–Haenszel analysis stratified by study,[65] utilizing a constant continuity correction term of 0.1 in case of zero cells. Because of the large number of comparisons (several outcome variables, various study pools, and a large number of subgroups), no detailed assessment or exploration of heterogeneity of relative risks across studies is provided. The analyses presented are exploratory in nature; confirmatory statistics were not carried out.
For the present reporting, filters were applied to highlight incidence rates and numerical differences between groups. These are explicitly stated in the titles and/or captions of each table or figure. Although somewhat arbitrary, these filters were always set at a low value and were conservative to avoid missing potentially important signals. Highlighted differences were interpreted on the basis of the actual number of patients involved in the comparison. Unless stated otherwise, data are presented overall for the double-blind and the open-label studies, but separate reporting is available in the SDC.
Results
Population and Comparator Antibiotics
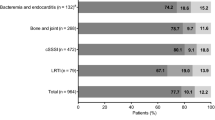
Table I shows the number of patients valid for the safety analysis who received moxifloxacin (n = 14 981) or comparator treatment (n = 15 023) by the oral, intravenous, or intravenous/oral routes, stratified by study design (double blind or open label). Approximately 75% of patients were enrolled in the double-blind studies. The percentage of patients with intravenous and intravenous/oral (sequential) treatments (29%) is substantially higher than that currently seen in clinical practice but reflects the design of studies and the severity of the studied indications. The choice of comparator(s) and dosage is consistent with standard therapies for the respective indications at the time each study was conducted.

Distribution of patients valid for the safety analysis, stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only) and by comparator
Demographics
Table II shows the demographics of the population analyzed (total = 30 004: see table SDC-II for stratification between double-blind and open-label studies). There was no meaningful difference between the patients receiving moxifloxacin and those receiving a comparator with respect to age, sex, BMI, race, indications, and pre-existing risk factors (renal or hepatic impairment, diabetes mellitus, cardiac disorders, or low BMI). Overall, the distribution of patients among the different indications mirrors the current prescribing patterns and clinical usage.[19,29] The majority of patients receiving oral moxifloxacin were treated for respiratory tract infections,[66] whereas patients receiving intravenous or intravenous/oral therapy (i) were older; (ii) were predominantly treated for CAP, cIAI and cSSSI; and (iii) had a higher incidence of pre-existing risk factors (related to the severity of their infection and their age).

Demographic parameters of the patients valid for the safety analysis, stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only). See table SDC-II or further stratification according to study design (double blind versus open label)
Overall Safety Data
Table III shows the summary of the safety data for all patients, subdivided between double-blind studies and open-label studies, respectively. As for any drug, a gradual decrease in the incidence of events was seen when looking from all AEs down to ADRs and further to SADRs. To help identify the highest incidence rates and imbalances between the treatment groups affecting a specific event, the data were filtered, and situations are highlighted where (i) there was a 2-fold difference between treatment arms for events with an incidence <2.5% in either of the treatment groups or a ≥2.5% difference between treatments for events with an incidence ≥2.5% in both groups and (ii) the number of patients experiencing an event was ≥10 in either treatment group. With these filters, the differences between moxifloxacin and comparators were related to (i) AEs and SAEs in the intravenous double-blind studies; and (ii) AEs, ADRs, and SADRs in the oral studies, SADRs in the intravenous/oral studies, and premature discontinuation due to AE in the intravenous open-label studies. Concerning SADRs reported in open-label oral and intravenous/oral studies, the numbers of patients with such events were small in each treatment group (moxifloxacin 12 [0.7%] versus comparator 5 [0.2%] in the oral studies; moxifloxacin 42 [2.7%] versus comparator 19 [1.2%] in the intravenous/oral studies). In the intravenous/oral studies, the difference in incidence rates (1.5%) was driven by gastrointestinal disorders (mostly diarrhea: 8 cases [0.5%] for moxifloxacin versus 1 case [<0.1%] for comparator) and results of investigations (10 cases [0.6%] for moxifloxacin versus 1 case [<0.1%] for comparator), including asymptomatic prolongation of the QT interval.

Summary of safety data for patients valid for the safety analysis, treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only) and by study design. An asterisk (*) indicates differences observed between treatment groups in disfavor of moxifloxacin that were ≥2.5% for events with an incidence ≥2.5% in both groups or ≥2-fold for events with an incidence <2.5% in one or both groups and for which the number of patients experiencing an event was ≥10 in either group
Adverse Events (AEs)
Rates of treatment-emergent AEs (classified by MedDRA SOC and PTs) based on study design are presented in table SDC-III. Reported AEs with ≥5% incidence for patients in the double-blind studies included wound infections (moxifloxacin 11.7% versus comparator 7.4% [intravenous; corresponding mainly to patients treated for cIAIs and cSSSI]); diarrhea (moxifloxacin 6.2% versus comparator 4.9% [oral], moxifloxacin 8.1% versus comparator 7.9% [intravenous/oral], moxifloxacin 6.3% versus comparator 4.4% [intravenous]); nausea (moxifloxacin 7.9% versus comparator 6.2% [oral], moxifloxacin 7.3% versus comparator 6.3% [intravenous/oral], moxifloxacin 5.4% versus comparator 3.5% [intravenous]); headache (moxifloxacin 5.6% versus comparator 5.9% [intravenous/oral]); constipation (moxifloxacin 7.7% versus comparator 6.1% [intravenous/oral]); hypokalemia (moxifloxacin 5.1% versus comparator 5.0% [intravenous/oral]); and insomnia (moxifloxacin 7.2% versus comparator 7.2% [intravenous/oral]). Reported AEs with ≥5% incidence for patients enrolled in open-label studies included diarrhea (moxifloxacin 3.6% versus comparator 7.4% [oral], moxifloxacin 6.1% versus comparator 6.5% [intravenous/oral]) and nausea (moxifloxacin 5.1% versus comparator 2.4% [oral]).
Again limiting the description to situations where (i) there was a 2-fold difference between treatment arms for events with an incidence <2.5% in either of the treatment groups or a ≥2.5% difference between treatments for events with an incidence ≥2.5% in both groups, and (ii) the number of patients experiencing an event was ≥10 in either treatment group, the following differences were noted in disfavor of moxifloxacin in the double-blind studies: (i) for patients treated with oral therapy (moxifloxacin 8822 versus comparator 8643): hyperhidrosis (36 [0.4%] versus 16 [0.2%]), tremor (35 [0.4%] versus 15 [0.2%]), atrial fibrillation (16 [0.2%] versus 3 [<0.1%]), and pleural effusion (12 [0.1%] versus 5 [<0.1%]); (ii) for patients treated with intravenous/oral therapy (moxifloxacin 1889 versus comparator 1856): incision site pain (21 [1.1%] versus 10 [0.5%]), erythema (19 [1.0%] versus 6 [0.3%]), hypophosphatemia (16 [0.8%] versus 3 [0.2%]), depression (15 [0.8%] versus 4 [0.2%]), increase in white blood cell (WBC) count (11 [0.6%] versus 5 [0.3%]), and increase in lactate dehydrogenase (LDH; 10 [0.5%] versus 4 [0.2%]); and (iii) in patients treated by the intravenous route (moxifloxacin 588 versus comparator 571): insomnia (11 [1.9%] versus 3 [0.5%]) and abdominal pain (10 [1.7%] versus 1 [0.2%]). Conversely, and with the same double filter, the following AEs were more frequently reported in the comparator group: (i) in oral studies: dysgeusia (moxifloxacin 74 [0.8%] versus comparator 179 [2.1%]), increase in gammaglutamyl transferase (GGT; moxifloxacin 20 [0.2%] versus comparator 41 [0.5%]), muscle spasms (moxifloxacin 12 [0.1%] versus comparator 25 [0.3%]), and myocardial infarction (moxifloxacin 2 [<0.1%] versus comparator 12 [0.1%]); and (ii) in intravenous/oral studies: cough (moxifloxacin 7 [0.4%] versus comparator 15 [0.8%]), myocardial infarction (moxifloxacin 5 [0.3%] versus comparator 10 [0.5%]), musculoskeletal pain (moxifloxacin 3 [0.2%] versus comparator 10 [0.5%]), and leukocytosis (moxifloxacin 2 [0.1%] versus comparator 10 [0.5%]).
In the open-label studies, the most common AEs in disfavor of moxifloxacin were nausea (in oral studies: moxifloxacin 91 [5.1%] versus comparator 50 [2.4%]) and dizziness (in oral studies: moxifloxacin 45 [2.5%] versus comparator 9 [0.4%]; in intravenous/oral studies: moxifloxacin 26 [1.7%] versus comparator 13 [0.8%]), and the most common AE in disfavor of the comparator was diarrhea (in oral studies: moxifloxacin 65 [3.6%] versus comparator 152 [7.4%]).
Adverse Drug Reactions (ADRs)
ADRs occurring in at least 0.5% of patients in either treatment group are shown in table IV. In the oral population enrolled in double-blind studies, the most common ADRs were nausea (moxifloxacin 602 [6.8%] versus comparator 457 [5.3%]), diarrhea (moxifloxacin 432 [4.9%] versus comparator 334 [3.9%]), dizziness (moxifloxacin 247 [2.8%] versus comparator 198 [2.3%]), headache (moxifloxacin 165 [1.9%] versus comparator 177 [2.0%]), and vomiting (moxifloxacin 162 [1.8%] versus comparator 150 [1.7%]). Only dysgeusia (moxifloxacin 66 [0.7%] versus comparator 171 [2.0%]) and increased GGT (moxifloxacin 11 [0.1%] versus comparator 30 [0.3%]) met the criteria set by the double filter used in table III. In the double-blind intravenous/oral population, diarrhea was the most common ADR (moxifloxacin 96 [5.1%] versus comparator 95 [5.1%]). Differences affected fewer than 10 patients in each treatment group, except for vomiting (moxifloxacin 13 [0.7%] versus comparator 26 [1.4%]). In the double-blind intravenous population, increased lipase (moxifloxacin 14 [2.4%] versus comparator 18 [3.2%]) and increased GGT (moxifloxacin 13 [2.2%] versus comparator 18 [3.2%]) were the most common ADRs, and only nausea showed a difference in disfavor of moxifloxacin versus comparator (12 [2.0%] versus 3 [0.5%], respectively) according to the double filter. In the open-label oral studies, nausea (moxifloxacin 77 [4.3%] versus comparator 44 [2.2%]) and diarrhea (moxifloxacin 54 [3.0%] versus comparator 141 [6.9%]) were again the most common ADRs across therapy arms, followed by dizziness (moxifloxacin 30 [1.7%] versus comparator 4 [0.2%]), upper abdominal pain (moxifloxacin 23 [1.3%] versus comparator 20 [1.0%]), and vomiting (moxifloxacin 20 [1.1%] versus comparator 14 [0.7%]), all experienced by >1% of patients in the moxifloxacin arm. Application of the double filter to the open-label oral population showed that diarrhea was more frequent with comparators (moxifloxacin 54 [3.0%] versus comparator 141 [6.9%]), whereas dizziness (moxifloxacin 30 [1.7%] versus comparator 4 [0.2%]), rash (moxifloxacin 16 [0.9%] versus comparator 8 [0.4%]), dysgeusia (moxifloxacin 13 [0.7%] versus comparator 2 [<0.1%]), and somnolence (moxifloxacin 10 [0.6%] versus comparator 2 [<0.1%]) were more frequent with moxifloxacin. In the open-label intravenous/oral population, diarrhea was the most common ADR for both moxifloxacin and comparator (61 [4.0%] and 60 [3.8%], respectively). Differences in disfavor of moxifloxacin versus comparator that met the double filter criteria concerned QT prolongation (moxifloxacin 19 [1.2%] versus comparator 3 [0.2%]) and dizziness (moxifloxacin 10 [0.6%] versus comparator 2 [0.1%]). For patients treated with intravenous therapy in the open-label population, all ADRs occurred in <10 patients in both treatment groups at low incidence rates, i.e. nausea (moxifloxacin 5 [1.4%] versus comparator 2 [0.6%]), dizziness (moxifloxacin 6 [1.7%] versus comparator 6 [1.7%]), increased ALT (moxifloxacin 9 [2.6%] versus comparator 8 [2.3%]), and rash (moxifloxacin 8 [2.3%] versus comparator 3 [0.9%]).

Adverse drug reactions occurring in either treatment group in ≧0.5% of patients valid for the safety analysis, treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only) and by study design (double blind, open label). Numbers in bold italic text correspond to events with an incidence ≥5% in either treatment group. A single asterisk (*) indicates differences observed between groups that were ≥2.5% for events with an incidence ≥2.5% in both groups or ≥2-fold for events with an incidence <2.5% in one or both groups (calculations were made using the number of patients [no rounding]; in the event of a null value for one treatment, only situations where ≥2 cases were observed in the other treatment group are indicated); the symbol is placed to the right of the value observed for the drug in disfavor. A double asterisk (**) indicates differences observed between treatment groups according to the same rule and where the number of patients experiencing an event was ≥10 in either group; the symbols are placed to the right of the value observed for the drug in disfavor
Serious AEs and Serious ADRs
Treatment-emergent SAEs are presented by SOCs for combined double-blind and open-label studies in table V. In the oral population, the overall incidence of SAEs (4.0% versus 3.9% in moxifloxacin- and comparator-treated patients) and those within the SOCs were very similar in the treatment groups. More SAEs were reported in the intravenous/oral studies in both treatment groups (moxifloxacin 595 [17.3%] versus comparator 527 [15.4%]), as expected given the increased severity of the disease. The SOCs associated with the highest incidences of events in both treatment groups, were ‘infections and infestations’ (moxifloxacin 219 [6.4%] versus comparator 165 [4.8%]) and ‘respiratory, thoracic, and mediastinal disorders’ (moxifloxacin 129 [3.8%] versus comparator 143 [4.2%]). Serious ‘cardiac disorders’ in the population treated by the intravenous/oral routes were reported with a similar incidence in the two groups (moxifloxacin 84 [2.4%] versus comparator 89 [2.6%]). In the intravenous-only trials, the overall rates were 7.9% and 6.0% in moxifloxacin- and comparator-treated patients, respectively, with SAEs from the SOC ‘infections and infestations’ being predominant (moxifloxacin 38 [4.1%] versus comparator 23 [2.5%]).

Serious adverse events presented by system organ class in patients valid for the safety analysis, treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only). A single asterisk (*) indicates differences observed between groups that were ≥2.5% for events with an incidence ≥2.5% in both groups or ≥2-fold for events with an incidence <2.5% in one or both groups (calculations were made using the number of patients [no rounding]; in the event of a null value for one treatment, only situations where ≥2 cases were observed in the other treatment group are indicated); the symbol is placed to the right of the value observed for the drug in disfavor. A double asterisk (**) indicates differences observed between treatment groups according to the same rule and where the number of patients experiencing an event was ≥10 in either group; the symbols are placed to the right of the value observed for the drug in disfavor
Table VI shows the incidences of SADRs in the combined double-blind and open-label studies, stratified by administration route. These were low considering the number of patients treated (oral: moxifloxacin 0.6% versus comparator 0.5%; intravenous/oral: moxifloxacin 2.8% versus comparator 1.9%; intravenous: moxifloxacin 1.0% versus comparator 0.8%). In the oral population, the incidences of SADRs within each SOC were similar between the treatment groups, with no individual SADR occurring at an incidence >0.15% in either the moxifloxacin or the comparator groups. In the intravenous/oral population, the SOCs associated with the highest incidence of events in both treatment groups were ‘infections and infestations’ (moxifloxacin 24 [0.7%] versus comparator 23 [0.7%]), [investigations’ (moxifloxacin 23 [0.7%] versus comparator 7 [0.2%]), and ‘gastrointestinal disorders’ (moxifloxacin 15 [0.4%] versus comparator 7 [0.2%]). Differences in disfavor of moxifloxacin versus comparator, using a 2-fold cut-off and events affecting at least 10 patients, were seen only for the SOCs ‘gastrointestinal disorders’ and [investigations’. Of note, ‘cardiac disorders’ were less frequent for moxifloxacin than for comparators (moxifloxacin 5 [0.1%] versus comparator 11 [0.3%] patients). In the intravenous-only population, the numbers were all very small, limiting the meaning and accuracy of any comparison. In the moxifloxacin and comparator intravenous groups, only one and two patients, respectively, experienced a cardiac disorder.

Serious adverse drug reactions presented by system organ class in patients valid for the safety analysis, treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only). A single asterisk (*) indicates differences observed between groups that were ≥2.5% for events with an incidence ≥2.5% in both groups or ≥2-fold for events with an incidence <2.5% in one or both groups (calculations were made using the number of patients [no rounding]; in the event of a null value for one treatment, only situations where ≥2 cases were observed in the other treatment group are indicated); the symbol is placed to the right of the value observed for the drug in disfavor. A double asterisk (**) indicates differences observed between treatment groups according to the same rule and where the number of patients experiencing an event was ≥10 in either group; the symbols are placed to the right of the value observed for the drug in disfavor
The nature of SADRs occurring in more than two patients in the oral, intravenous/oral, and intravenous populations was examined by the double-blind versus open-label design of the studies (see table SDC-IV). This showed that the occurrences of corrected QT (QTc) interval prolongation, for the studies where ECG data were available, were few in both the double-blind studies (intravenous/oral: moxifloxacin 11 versus comparator 4) and the open-label studies (moxifloxacin 2 versus comparator 0). Diarrhea was the most frequent SADR in both the double-blind and the open-label studies, but with quite small numbers: (i) in double-blind studies: oral, moxifloxacin 3 (<0.1%) versus comparator 3 (<0.1%); intravenous/oral, moxifloxacin 2 (0.1%) versus comparator 3 (0.2%); and (ii) in open-label studies: intravenous/oral, moxifloxacin 5 (0.3%) versus comparator 0 (0%). All other SADRs were rarely reported and with a similar incidence in the two groups, except that in the intravenous/oral double-blind studies, there were more ‘cardiac disorders’ with the comparator (moxifloxacin 2 [0.1%] versus comparator 10 [0.5%]) and more [investigations’ related to electrocardiographic QTc prolongation with moxifloxacin (moxifloxacin 11 [0.6%] versus comparator 4 [0.2%]), and in the intravenous/oral open-label studies, there were more [investigations’ with moxifloxacin (moxifloxacin 10 [0.6%] versus comparator 1 [<0.1%]). In the intravenous-only double-blind studies, more events related to ‘infections and infestations’ were reported for comparators (moxifloxacin 1 [0.2%] versus comparator 3 [0.5%]). Clostridium difficile colitis was reported in only one patient in each group in the oral and intravenous-only double-blind studies; in the intravenous/oral studies, it was reported in none of the moxifloxacin-treated patients but in four comparator-treated patients.
Selected AEs
The official labeling of fluoroquinolones in most countries mentions a series of AEs commonly associated with administration of these drugs. These include gastrointestinal effects, central nervous system [CNS] effects (headache, dizziness, and convulsion), cardiac effects (associated with prolongation of the QTc interval), dysglycemia, tendon disorders, phototoxicity, hypersensitivity, skin disorders, and hepatic toxicity. We therefore looked specifically for these events. The corresponding incidence rates (ranked by SMQs/BMQs and most frequent PTs [if ≧0.5%]) are presented in table VII. They are commented upon hereunder along with C. difficile-associated events (not organized as SMQs/BMQs), which are not displayed in the table.

Incidence of selected treatment-emergent adverse events presented by Standard MedDRA Queries/Bayer MedDRA Queries and preferred terms in patients valid for the safety analysis, treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only). Data are limited to events with an incidence ≧0.5% in either group of patients. A single asterisk (*) indicates differences observed between groups that were ≥2.5% for events with an incidence ≥2.5% in both groups or ≥2-fold for events with an incidence <2.5% in one or both groups (calculations were made using the number of patients [no rounding]; in the event of a null value for one treatment, only situations where ≥2 cases were observed in the other treatment group are indicated); the symbol is placed to the right of the value observed for the drug in disfavor. A double asterisk (**) indicates differences observed between treatment groups according to the same rule and where the number of patients experiencing an event was ≥10 in either group; the symbols are placed to the right of the value observed for the drug in disfavor
Drug-Related Hepatic Disorders – Comprehensive Search (Standard MedDRA Query [SMQ])
The overall incidences of the SMQs (AEs) designated as drug-related hepatic disorders in oral, intravenous/oral, and intravenous-only studies were similar in the moxifloxacin and comparator treatment groups, though in the oral studies more cases of abnormal hepatic function were observed in the moxifloxacin-treated patients. Four cases of hepatic failure were experienced in total, of which two due to the study drug occurred in moxifloxacin-treated patients and one occurred in a comparator-treated patient: with moxifloxacin, patient ♯1 (treated by the intravenous/oral routes for CAP) had a medical history of hepatitis C, alcohol abuse, and intravenous drug abuse, and developed acute hepatic failure after 2 days of therapy in the context of multi-organ failure with fatal outcome; patient ♯2 (treated orally for CAP) had a medical history of chronic hepatitis and developed hepatic failure after 4 days of therapy, which resolved spontaneously without discontinuation of the study drug; with the comparator, the patient (treated orally with levofloxacin for uncomplicated UTI) had no relevant medical history findings and developed hepatic failure 1 day after the study drug was stopped, which resolved spontaneously.
Severe Cutaneous Adverse Reactions (SMQ)
These were very rare and were reported with similar incidences in the moxifloxacin and comparator groups, with most events being non-serious (including conjunctivitis and stomatitis cases). One case of Stevens–Johnson syndrome (an ADR) was reported in a moxifloxacin-treated patient enrolled in a PID study. Three patients (one and two in the moxifloxacin and comparator groups, respectively) had skin necrosis (AEs), but these were not considered drug related.
Convulsions (SMQ)
These were very rarely reported in either treatment group.
Psychiatric Disorders (SMQ)
Psychiatric disorders (most often agitation and depression) were more frequent in the intravenous/oral and the intravenous-only studies but with no real difference between moxifloxacin and comparator, with the exception of depression, which was slightly more frequent in the moxifloxacin group in the intravenous/oral studies.
AEs Considered as Relevant Clinical Outcome of Corrected QT Interval Prolongation (Bayer MedDRA Query [BMQ])
These were reported with a similar frequency between the treatment groups in the oral studies and in the intravenous/oral studies. In the intravenous-only studies, they were slightly more frequent in the moxifloxacin group, mostly driven by a higher incidence of cardiac arrests. Only one of the eight cases of cardiac arrest reported, however, was considered to be related to the study drug (cardiac arrest in one cirrhotic patient treated with intravenous moxifloxacin for cIAI, who developed severe intra-abdominal sepsis secondary to a large intestine perforation, complicated by septic shock). Ventricular arrhythmia, tachycardia, and fibrillation were rare events in either treatment group.
Anaphylactic Reactions (SMQ)
These were rarely reported, with circulatory collapse and shock being the most frequent AEs in the intravenous/oral studies (none being drug related in moxifloxacin-treated patients). Anaphylactic/anaphylactoid reactions were seen only in three comparator-treated patients (drug related in all cases).
Photosensitivity Reactions (BMQ)
These were rarely reported and occurred exclusively in oral studies.
Tendinopathies (BMQ)
These were equally reported in both moxifloxacin- and comparator-treated patients.
Dysglycemia (SMQ/BMQ)
Incidence rates were similar between the treatment groups, with hyperglycemia being more frequently reported than hypoglycemia.
Clostridium difficile-Associated Diarrhea (Preferred Terms)
Incidence rates of ‘clostridial infection’, ‘Clostridium colitis’, ‘Clostridium difficile colitis’, and ‘pseudomembranous colitis’ were <0.1% in the oral studies but were higher in the intravenous/oral studies, although similar in moxifloxacin- and comparator-treated patients (moxifloxacin 0.6%, comparator 0.4%). The incidence rate in the intravenous-only studies was 0.1% in each treatment group.
Analysis by Comparator Class
In order to more specifically assess the toxicity pattern of moxifloxacin independently from those of other fluoroquinolones, we conducted an analysis by classes of antibiotics for all groups with sufficient numbers of patients (oral: moxifloxacin versus a β-lactam, versus a macrolide, versus another fluoroquinolone, or versus a β-lactam with a macrolide; intravenous/oral: moxifloxacin versus a β-lactam, versus a β-lactam with or without a macrolide, or versus another fluoroquinolone; intravenous: moxifloxacin versus a β-lactam or versus another fluoroquinolone). These data are presented as table SDC-V. Concentrating on differences in disfavor of moxifloxacin, there was a near to 2-fold increased risk estimate in intravenous-only studies for (i) discontinuation due to AEs in comparison with β-lactams (moxifloxacin 11 [2.7%] versus β-lactam 6 [1.5%]); (ii) discontinuation due to AEs in comparison with another fluoroquinolone (moxifloxacin 21 [6.0%] versus other fluoroquinolone 11 [3.1%]); and (iii) discontinuation due to ADRs also in comparison with another fluoroquinolone (moxifloxacin 17 [4.9%] versus other fluoroquinolone 9 [2.6%]).
Analysis by Main Indication
Moxifloxacin is indicated for infections of different levels of severity. The data were, therefore, stratified by the main approved indications for which there were sufficient numbers of patients to draw meaningful conclusions – namely ABS, AECB, CAP, uPID, cSSSI, and cIAI. The results are presented graphically in figure 1 with substratification by administration route (oral, intravenous/oral, intravenous). A 2-fold excess in event frequencies for moxifloxacin versus comparator was only seen (i) for SADRs in cIAI patients treated by the intravenous/oral routes, and (ii) for discontinuation due to AEs or to ADRs in AECB patients treated by the intravenous route only. However, in each case, there were relatively small numbers of patients (moxifloxacin 21 [3.4%] versus comparator 9 [1.4%] in patients with cIAI; moxifloxacin 7 [7.3%] versus comparator 2 [2.0%] in patients with AECB).

Relative risk estimates (moxifloxacin versus comparator) for adverse events from pooled data stratified according to indications (the most pertinent or most frequent ones). The data are substratified according to the route of administration approved or commonly used for the corresponding indication: (a) oral route; (b) intravenous route followed by oral route [sequential]; (c) intravenous route. The number of patients enrolled in each cohort (moxifloxacin versus the comparator) is shown at the top of each graph. Calculations were made using the Mantel–Haenszel method stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented on a 0–3 linear scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator). Values ≤3 are displayed as squares. Circles placed at the edge of the scale indicate that the actual value is >3 (the numbers of patients who received moxifloxacin versus the comparator are shown to the left of the circle). White symbols indicate values with a lower limit of the calculated 95% confidence interval >1, indicating a nominally significantly higher risk for moxifloxacin relative to the comparator (the number of patients in each group is shown to the right of the symbol). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ABS = acute bacterial sinusitis; ADR = adverse drug reaction; AE = adverse event; AECB = acute exacerbation of chronic bronchitis; CAP = community-acquired pneumonia; cIAI = complicated intra-abdominal infection; cSSSI = complicated skin and skin structure infection; IV = intravenous; PO = oral; SADR = serious ADR; SAE = serious AE; uPID = uncomplicated pelvic inflammatory disease.
Patients with Co-Morbidities
Because the safety of drugs can be adversely influenced by the patient status and may also worsen it, data were also stratified according to the main pertinent co-morbidities and elimination pathway disorders observed in the population – namely age, diabetes mellitus, renal impairment, hepatic impairment, cardiac disorders, and abnormally low BMI. First, patients were stratified by study design (double blind and open label) and administration route (oral, intravenous/oral, intravenous), and the results are presented in table VIII. To better apprehend potentially meaningful differences, relative risk estimates (moxifloxacin versus comparator) were then calculated for each patient group stratified according to the administration route. The results are presented graphically in figures 2 and 3. On the basis of a threshold of a 2–fold increase in risk estimates, the only difference seen in patients receiving oral treatment was in those with underlying cardiac disorders (more AEs with fatal outcome for comparator) [figure 3b]; and the only differences seen in those receiving intravenous treatment were in those with (i) age ≥65 years (more ADRs with fatal outcome for comparator [figure 2a]); (ii) diabetes mellitus (more discontinuations due to ADRs for comparator [figure 2b]); (iii) hepatic impairment (more SADRs, discontinuation due to ADRs, and AEs with fatal outcome for moxifloxacin [figure 3a]); (iv) cardiac disorders (more discontinuations due to AEs for moxifloxacin and more ADRs with fatal outcome for comparator [figure 3b]); and (v) BMI <18 kg/m2 (more discontinuations due to AEs or ADRs, and more AEs with fatal outcome for moxifloxacin [figure 3c]). However, numbers in the intravenous-only studies were small in all cases (1–7 patients). Lastly, the relative risk estimates (moxifloxacin versus comparator) were calculated after substratifying each group according to the comparator used, concentrating for each comparator on patients treated by the most frequent route of administration (if versus a β-lactam: oral, intravenous/oral and intravenous; if versus a macrolide alone: oral; if versus a β-lactam alone or a beta-lactam combined with a macrolide: intravenous/oral; if versus fluoroquinolone: intravenous only). The results are shown graphically in figures 4–6. Concentrating again on differences in disfavor of moxifloxacin, a >2-fold difference in disfavor of moxifloxacin in at least one safety variable was observed for patients with (i) age ≥65 years (intravenous/oral versus β-lactam alone or versus β-lactam ± macrolide [figure 5a or 5b]); (ii) diabetes mellitus (intravenous/oral versus β-lactam alone or versus β-lactam ± macrolide [figure 5a or 5b]); (iii) renal impairment (intravenous/oral versus β-lactam ± macrolide [figure 5b], and intravenous versus β-lactam [figure 6a]); (iv) hepatic impairment (oral versus β-lactam [figure 4a], and intravenous versus β-lactam or versus another fluoroquinolone [figure 6a or 6b]); (v) cardiac disorders (intravenous versus β-lactam [figure 6a]); and (vi) BMI <18 kg/m2 (oral versus β-lactam [figure 4a] and intravenous versus β-lactam or versus another fluoroquinolone [figure 6a or 6b]). However, the numbers of patients with events were very small in all cases (1–24).

Relative risk estimates (moxifloxacin versus the comparator) for adverse events from pooled data on (a) elderly patients, (b) patients with diabetes mellitus, and (c) patients with renal impairment. The data are stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only).The number of patients enrolled in each subgroup (moxifloxacin versus the comparator) is shown at the top of each graph, and the numbers of patients with each of the recorded events are shown to the left of the corresponding symbol. Calculations were made using the Mantel–Haenszel method (with the 95% confidence interval) stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented as black squares on a (0.1–10) logarithmic scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator), and the horizontal lines denote the confidence interval (limited to a maximum of 0.1 to 10 for reasons of legibility; lines that extend beyond these limits [or where the limits are masked by text] have an arrowhead symbol; when not visible, the lines is shorter than the corresponding symbol size). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ADR = adverse drug reaction; AE = adverse event; IV = intravenous; PO = oral; SADR = serious ADR; SAE = serious AE.

Relative risk estimates (moxifloxacin versus the comparator) for adverse events from pooled data on (a) patients with hepatic impairment, (b) patients with a cardiac disorder, and (c) patients with a body mass index <18 kg/m2. The data are stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only).The number of patients enrolled in each subgroup (moxifloxacin versus the comparator) is shown at the top of each graph, and the numbers of patients with each of the recorded events are shown to the left of the corresponding symbol. Calculations were made using the Mantel–Haenszel method (with the 95% confidence interval) stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented as black squares on a (0.1–10) logarithmic scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator), and the horizontal lines denote the confidence interval (limited to a maximum of 0.1 to 10 for reasons of legibility; lines that extend beyond these limits [or where the limits are masked by text] have an arrowhead symbol; when not visible, the lines is shorter than the corresponding symbol). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ADR = adverse drug reaction; AE = adverse event; BMI = body mass index; IV = intravenous; PO = oral; SADR = serious ADR; SAE = serious AE.

Relative risk estimates (moxifloxacin versus the comparator) for adverse events from pooled data on patients treated by the oral route with the most frequent or meaningful comparator antibiotic: (a) β-lactam or (b) a macrolide. The data are stratified according to risk factors (age ≥65 years, diabetes mellitus, renal impairment, hepatic impairment, cardiac disorders, body mass index <18 kg/m2). The number of patients enrolled in each subgroup (moxifloxacin versus the comparator) is shown at the top of each graph. Calculations were made using the Mantel–Haenszel method stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented on a 0–3 linear scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator). Values ≤3 are displayed by squares. Circles placed at the edge of the scale indicate that the actual value is >3 (the numbers of patients who received moxifloxacin versus the comparator are shown to the left of the circle). White symbols indicate values with a lower limit of the calculated 95% confidence interval >1, indicating a nominally significantly higher risk for moxifloxacin relative to the comparator (the numbers of patients in each group are shown to the right or left of the corresponding symbol). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ADR = adverse drug reaction; AE = adverse event; BMI = body mass index; SADR = serious ADR; SAE = serious AE.

Relative risk estimates (moxifloxacin versus the comparator) for adverse events from pooled data on patients treated by the intravenous route followed by the oral route (sequential) with the most frequent or meaningful comparator antibiotic(s): (a) β-lactam or (b) β-lactam ± macrolide (the design of the study did not allow differentiation between patients receiving only a β-lactam and those receiving the combination of a β-lactam and a macrolide). The data are stratified according to risk factors (age ≥65 years, diabetes mellitus, renal impairment, hepatic impairment, cardiac disorder, body mass index <18 kg/m2). The number of patients enrolled in each subgroup (moxifloxacin versus the comparator) is shown at the top of each graph. Calculations were made using the Mantel–Haenszel method stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented on a 0–3 linear scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator). Values ≤3 are displayed by squares. Circles placed at the edge of the scale indicate that the actual value is >3 (the numbers of patients who received moxifloxacin versus the comparator are shown to the left of the circle). White symbols indicate values with a lower limit of the calculated 95% confidence interval >1, indicating a nominally significantly higher risk for moxifloxacin relative to the comparator (the numbers of patients in each group are shown to the right or left of the corresponding symbol). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ADR = adverse drug reaction; AE = adverse event; BMI = body mass index; SADR = serious ADR; SAE = serious AE.

Relative risk estimates (moxifloxacin versus the comparator) for adverse events from pooled data on patients treated by the intravenous route with the most frequent or meaningful comparator antibiotic: (a) β-lactam or (b) another fluoroquinolone. The data are stratified according to risk factors (age ≥65 years, diabetes mellitus, renal impairment, hepatic impairment, cardiac disorder, body mass index <18 kg/m2). The number of patients enrolled in each subgroup (moxifloxacin versus the comparator) is shown at the top of each graph. Calculations were made using the Mantel–Haenszel method stratified by study, with a continuity correction of 0.1 in the event of a null value. The relative risk estimates are presented on a 0–3 linear scale (1 denotes no difference; values <1 and >1 denote a correspondingly lower and higher risk, respectively, associated with moxifloxacin treatment relative to the comparator). Values ≤3 are displayed by squares. Circles placed at the edge of the scale indicate that the actual value is >3 (the numbers of patients who received moxifloxacin versus the comparator are shown to the left of the circle). White symbols indicate values with a lower limit of the calculated 95% confidence interval >1, indicating a nominally significantly higher risk for moxifloxacin relative to the comparator (the numbers of patients in each group are shown to the right or left of the corresponding symbols). The light gray shaded area highlights the zone where the relative risk estimate (moxifloxacin/comparator) is between 0.5 and 2. ADR = adverse drug reaction; AE = adverse event; BMI = body mass index; SADR = serious ADR; SAE = serious AE.

Incidence of adverse events, adverse drug reactions, serious adverse events, serious adverse drug reactions, discontinuation due to adverse events, discontinuation due to adverse drug reactions, adverse events with fatal outcome, and adverse drug reactions with fatal outcome in patients with risk factors (age, diabetes mellitus, renal or hepatic impairment, cardiac disorder, low body mass index) treated with moxifloxacin or a comparator and stratified by route of administration (oral only; intravenous followed by oral [sequential]; intravenous only) and by study design
Discussion and Conclusion
By using the data on all valid-for-safety populations in the phase II–IV randomized actively controlled clinical trials, with stratification by study design (double blind or open label), route of administration (oral, intravenous with or without a subsequent switch to oral therapy), pre-existing risk factors, main indications, and types of comparator, the present paper may represent a new standard in the public reporting of adverse effects for a drug marketed over the past several years. Such data are usually communicated to regulatory authorities only (as part of registration applications, Periodic Safety Update Reports, and Risk Management Plans) and remain, therefore, largely unknown to the clinician. The benefit of using pooled randomized active-controlled clinical trial data, as has been done here, is that risks associated with the study drug can be directly compared with those of clinically valid comparators. This approach also allows estimation of the incidence of relatively rare effects with a fair degree of certainty. Since the data are from randomized studies, patients should be equally balanced with respect to known as well as unknown factors associated with the outcome variables, making comparisons between treatment groups as fair as possible.[64]
A first key observation is that moxifloxacin does not show a markedly different safety profile compared with comparator therapies. The filters used highlight situations where moxifloxacin caused more untoward effects than the comparator, but either the actual numbers of affected patients were close to those seen with the comparator or the differences were small. For ADRs, there were actually several situations where the comparator showed more untoward effects, especially in the double-blind studies. In the open-label studies, most moxifloxacin ADRs concerned nervous system disorders that are listed in the labeling, which may lead to over-reporting. Concentrating on SADRs, differences in the open-label studies mainly concerned gastrointestinal effects and the need for biological investigations. Here, also, the moxifloxacin labeling lists these effects; no difference in SADRs was seen between moxifloxacin and comparator when considering the double-blind studies. A higher incidence of AEs was also seen in patients receiving the intravenous formulation in double-blind studies. However, this was not seen for ADRs (except for nausea). The other global indicators of toxicity – ADRs, treatment discontinuation due to ADRs, or ADRs with fatal outcome – showed no clinically meaningful difference in frequency between moxifloxacin and comparator.
The second key observation is that the incidence of ADRs across the treatment groups was low. This may be explained by at least two factors – namely (i) patients with known contraindications were systematically excluded from participation in the studies; and (ii) all patients were closely monitored throughout the observation period, which may have prevented AEs developing into recognizable ADRs. While this could suggest that the patients analyzed do not correspond to those seen in routine clinical practice, excluding patients on the basis of contraindications and following them for occurrence of side effects should be the rule in actual prescribing situations. Excluding patients with risk factors that commonly occur alongside the primary pathology (e.g. CAP, cSSSI) but are not clear contraindications could confound results of large retrospective analyses such as that conducted in the current study. Yet, patients with risk factors were actually included in the studies, consistent with trials conducted during the whole phase II–IV development program. The impact of close monitoring of patients considered to be at high risk did not introduce bias to the reporting, since in none of these subgroups was early drug discontinuation reported more frequently (an increased frequency would, indeed, have prompted the investigators’ intervention to address the corresponding safety concern and to discontinue therapy). Thus, in the context of clinical trials involving about 15 000 patients treated with moxifloxacin, no clear differentiation could be made with respect to tolerance versus the comparators used, either as a group or individually. As all of the comparators were accepted standards of care at the time at which each study was designed, it is reasonable to consider that moxifloxacin has a safety profile that is comparable to that of the comparators.
The labeling of fluoroquinolones, and of moxifloxacin in particular, includes multiple side effects (e.g. tendon, cardiac, CNS, cutaneous, and hepatic effects, and C. difficile infections) that were not seen in substantial frequencies in the current analysis, despite careful investigation. When detected, the incidence of cardiac and hepatic AEs was slightly higher in patients receiving moxifloxacin treatment than in those receiving comparator treatment, but this related only to ‘hepatic function abnormal’ in oral and ‘cardiac arrest’ in intravenous studies, respectively. These events were no different in frequency when examining ADRs. The incidences of SADRs related to CNS, cutaneous, cardiac, or hepatic toxicity were similar in moxifloxacin- and comparator-treated patients. Although it has been suggested that patients with pre-existing risk factors or co-morbidities may be at particular risk of experiencing an AE, our data did not reveal any clinically relevant differences compared with the comparators in this context. This holds true not only for comparisons with other fluoroquinolones, but also for comparisons with other antibiotic classes.
All but one of the studies used in the present analysis had the evaluation of the clinical efficacy of moxifloxacin in the target indications as a primary goal, and the majority of the studies have been published in peer-reviewed journals (see references[26,27,29] for recent review papers). Most studies concluded that moxifloxacin was clinically as effective as the comparators or superior to them, which implies that moxifloxacin was not underdosed (all patients received the standard registered dose that has proven to be efficacious in all registered indications to date). This contrasts with some of the comparators (including those proposed as first-line therapies in applicable guidelines), for which higher dosages than those used in the studies pooled for the current analysis are now proposed. For β-lactams[67–69] and levofloxacin,[70] this reflects the progressive decrease in bacterial susceptibility over time and the corresponding attempts by clinicians to maintain sufficient treatment efficacy based on pharmacokinetic/dynamic principles and to avoid failures[71] and/or emergence of resistance.[72,73]
As with all meta-analyses, the present study and its conclusions have several limitations. Although we looked at specific risks, we did not reanalyze the original investigators’ statements or medical assessment of the corresponding cases, nor made any attempt at further adjudication of specific events. No exploration of heterogeneity of results across studies was done, because of the large number of comparisons. Lastly, although a large number of patients were included in the analysis, it may not be sufficient for detecting very rare side effects. These are usually captured from post-marketing spontaneous reports and larger non-interventional studies, but such reports are subject to other limitations relating to the quality of reporting, difficulties in ensuring unbiased data collection, and lack of detailed information on the patient characteristics. Moreover, while the population at risk is known for non-interventional studies, the actual number of exposed persons is difficult to determine for spontaneous reports. Thus, other approaches need to be followed to further define the safety profile of drugs when they are administered in a real-life setting. This has already been carried out for hepatotoxicity using a registry approach to compare telithromycin and several fluoroquinolones, including moxifloxacin[74] (that study did not reveal significant differences between moxifloxacin and the other fluoroquinolones marketed at that time in this context). It is important to stress that such studies should be applied to comparators as well, in order to correctly define their true safety profile. Centralization and cross-checking of product safety update reports and their publication by independent bodies would also be of significant interest. In the meantime, clinicians will need to rely on analyses such as those presented here for making informed choices on treatment options.
References
Woodhead M, Blasi F, Ewig S, et al. Guidelines for the management of adult lower respiratory tract infections: full version. Clin Microbiol Infect 2011; 17 Suppl. 6: E1–59.
Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis 2007; 44 Suppl. 2: S27–72.
Balter MS, La Forge J, Low DE, et al. Canadian guidelines for the management of acute exacerbations of chronic bronchitis. Can Respir J 2003; 10 Suppl. B: 3B–32B.
Solomkin JS, Mazuski JE, Bradley JS, et al. Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Clin Infect Dis 2010; 50 (2): 133–64.
Anon JB, Jacobs MR, Poole MD, et al. Antimicrobial treatment guidelines for acute bacterial rhinosinusitis. Otolaryngol Head Neck Surg 2004; 130 (1 Suppl.): 1–45.
Sociedad Española de Quimioterapia, Sociedad Española de Otorrinolaringología y Patología Cérvico-Facial. Diagnosis and antimicrobial treatment of sinusitis. Rev Esp Quimioter 2003; 16 (2): 239–51.
Clinical Effectiveness Group, British Association for Sexual Health and HIV. UK national guideline for the management of pelvic inflammatory disease 2011 (updated June 2011) [online]. Available from URL: http://www.bashh.org/documents/3572 [Accessed 2012 Jan 28].
Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41 (10): 1373–406.
Eron LJ, Lipsky BA, Low DE, et al. Managing skin and soft tissue infections: expert panel recommendations on key decision points. J Antimicrob Chemother 2003; 52 Suppl. 1: 13–17.
Pham PA, Bartlett JG. Moxifloxacin [online]. Available from http://www.hopkinsguides.com/hopkins/ub/view/Johns_Hopkins_ABX_Guide/540355/all/Moxifloxacin [Accessed 2012 Jan 28].
Balfour JA, Wiseman LR. Moxifloxacin. Drugs 1999; 57 (3): 363–73.
Krasemann C, Meyer J, Tillotson G. Evaluation of the clinical microbiology profile of moxifloxacin. Clin Infect Dis 2001; 32 Suppl. 1: S51–63.
Culley CM, Lacy MK, Klutman N, et al. Moxifloxacin: clinical efficacy and safety. Am J Health Syst Pharm 2001; 58 (5): 379–88.
Talan DA. Clinical perspectives on new antimicrobials: focus on fluoroquinolones. Clin Infect Dis 2001; 32 Suppl. 1: S64–71.
Zhanel GG, Ennis K, Vercaigne L, et al. A critical review of the fluoroquinolones: focus on respiratory infections. Drugs 2002; 62 (1): 13–59.
Blondeau JM. The role of fluoroquinolones in skin and skin structure infections. Am J Clin Dermatol 2002; 3 (1): 37–46.
Muijsers RB, Jarvis B. Moxifloxacin in uncomplicated skin and skin structure infections. Drugs 2002; 62 (6): 967–73.
Ball P, Stahlmann R, Kubin R, et al. Safety profile of oral and intravenous moxifloxacin: cumulative data from clinical trials and postmarketing studies. Clin Ther 2004; 26 (7): 940–50.
Keating GM, Scott LJ. Moxifloxacin: a review of its use in the management of bacterial infections. Drugs 2004; 64 (20): 2347–77.
Llor C, Naberan K, Cots JM, et al. Economic evaluation of the antibiotic treatment of exacerbations of chronic bronchitis and COPD in primary care. Int J Clin Pract 2004; 58 (10): 937–44.
Van Bambeke F, Michot JM, Van Eldere J, et al. Quinolones in 2005: an update. Clin Microbiol Infect 2005; 11 (4): 256–80.
Sethi S. Moxifloxacin for the treatment of acute exacerbations of chronic obstructive pulmonary disease. Clin Infect Dis 2005; 41 Suppl. 2: S177–85.
Grossman RF, Rotschafer JC, Tan JS. Antimicrobial treatment of lower respiratory tract infections in the hospital setting. Am J Med 2005; 118 Suppl. 7A: 29S–38S.
Ferrara AM. New fluoroquinolones in lower respiratory tract infections and emerging patterns of pneumococcal resistance. Infection 2005; 33 (3): 106–14.
Haggerty CL, Ness RB. Newest approaches to treatment of pelvic inflammatory disease: a review of recent randomized clinical trials. Clin Infect Dis 2007; 44 (7): 953–60.
Miravitlles M. Moxifloxacin in the management of exacerbations of chronic bronchitis and COPD. Int J Chron Obstruct Pulmon Dis 2007; 2 (3): 191–204.
Miravitlles M, Anzueto A. Moxifloxacin: a respiratory fluoroquinolone. Expert Opin Pharmacother 2008; 9 (10): 1755–72.
Simoens S, Decramer M. A pharmacoeconomic review of the management of respiratory tract infections with moxifloxacin. Expert Opin Pharmacother 2008; 9 (10): 1735–44.
Burkhardt O, Welte T. 10 years’ experience with the pneumococcal quinolone moxifloxacin. Expert Rev Anti Infect Ther 2009; 7 (6): 645–68.
Simoens S. Evidence for moxifloxacin in community-acquired pneumonia: the impact of pharmaco-economic considerations on guidelines. Curr Med Res Opin 2009; 25 (10): 2447–57.
Sprandel KA, Rodvold KA. Safety and tolerability of fluoroquinolones. Clin Cornerstone 2003; Suppl. 3: S29–36.
Iannini PB. Fluoroquinolone toxicity: a review of class- and agent-specific adverse effects. Drug Benefit Trends 2004; 16 Suppl. B: 34–41.
Andriole VT, Haverstock DC, Choudhri SH. Retrospective analysis of the safety profile of oral moxifloxacin in elderly patients enrolled in clinical trials. Drug Saf 2005; 28 (5): 443–52.
Choudri SH, Kuesmann K, Perroncel R. Cardiac safety of moxifloxacin in hospitalized patients with community-acquired pneumonia [abstract no. L-1079]. 46th Inter-science Conference on Antimicrobial Agents and Chemotherapy (ICAAC); 2006 Sep 27–30; San Francisco (CA).
Van Bambeke F, Tulkens PM. Safety profile of the respiratory fluoroquinolone moxifloxacin: comparison with other fluoroquinolones and other antibacterial classes. Drug Saf 2009; 32 (5): 359–78.
Iannini PB. Cardiotoxicity of macrolides, ketolides and fluoroquinolones that prolong the QTc interval. Expert Opin Drug Saf 2002; 1 (2): 121–8.
Iannini PB. The safety profile of moxifloxacin and other fluoroquinolones in special patient populations. Curr Med Res Opin 2007; 23 (6): 1403–13.
Stahlmann R, Lode H. Fluoroquinolones in the elderly: safety considerations. Drugs Aging 2003; 20 (4): 289–302.
Stahlmann R, Lode H. Safety considerations of fluoroquinolones in the elderly: an update. Drugs Aging 2010; 27 (3): 193–209.
Grange JD, Thabut D, Lucidarme D, et al. Randomized, comparative study of moxifloxacin versus amoxicillin-clavulanate in the treatment of bacterial infections in cirrhotic patients [abstract no. 1086]. Hepatology 2004; 40 Suppl. S4:631A.
Avelox®: US prescribing information [online]. Available from URL: http://www.univgraph.com/bayer/inserts/avelox.pdf [Accessed 2012 Jan 28].
Avelox® 400 mg/250 mL solution pour perfusion: résumé des caractéristiques du produit [online]. Available from URL: http://www.fagg-afmps.be/en/ [Accessed 2012 Jan 28].
Avelox® 400 mg comprimés: résumé des caractéristiques du produit [online]. Available from URL: http://www.faggafmps.be/en/ [Accessed 2012 Jan 28].
Landen H, Moller M, Tillotson GS, et al. Clinical experience in Germany of treating community-acquired respiratory infections with the new 8-methoxyfluoroquinolone, moxifloxacin. J Int Med Res 2001; 29 (2): 51–60.
Barth J, Landen H. Efficacy and tolerability of moxifloxacin in 2338 patients with acute exacerbation of chronic bronchitis. Clin Dug Invest 2003; 23 (1): 1–10.
Faich GA, Morganroth J, Whitehouse AB, et al. Clinical experience with moxifloxacin in patients with respiratory tract infections. Ann Pharmacother 2004; 38 (5): 749–54.
Elies W, Landen H, Stauch K. Efficacy and tolerability of moxifloxacin in patients with sinusitis treated in general practice: results of a post-marketing surveillance study. Clin Drug Investig 2004; 24 (8): 431–9.
Koch H, Landen H, Stauch K. Daily-practice treatment of acute exacerbations of chronic bronchitis with moxifloxacin in a large cohort in Germany. Clin Drug Investig 2004; 24 (8): 449–55.
Koch H, Landen H, Stauch K. Once-daily moxifloxacin therapy for community-acquired pneumonia in general practice: evidence from a post-marketing surveillance study of 1467 patients. Clin Drug Investig 2004; 24 (8): 441–8.
Barth J, Stauch K, Landen H. Efficacy and tolerability of sequential intravenous/oral moxifloxacin therapy in pneumonia: results of the first post-marketing surveillance study with intravenous moxifloxacin in hospital practice. Clin Drug Investig 2005; 25 (11): 691–700.
Schaberg T, Moller M, File T, et al. Real-life treatment of acute exacerbations of chronic bronchitis with moxifloxacin or macrolides: a comparative post-marketing surveillance study in general practice. Clin Drug Investig 2006; 26 (12): 733–44.
Liu LY, Landen H. Treatment of respiratory tract infections with moxifloxacin: results of postmarketing surveillance in China. Int J Clin Pract 2007; 61 (9): 1509–15.
Zhou B, Jiang X, Zhai L, et al. Moxifloxacin in the treatment of acute bacterial rhinosinusitis: results of a multicenter, non-interventional study. Acta Otolaryngol 2010; 130(9): 1058–64.
Norrby SR, Lietman PS. Safety and tolerability of fluoroquinolones. Drugs 1993; 45 Suppl. 3: 59–64.
Ball P, Tillotson G. Tolerability of fluoroquinolone antibiotics: past, present and future. Drug Saf 1995; 13 (6): 343–58.
Bertino Jr J, Fish D. The safety profile of the fluoroquinolones. Clin Ther 2000; 22 (7): 798–817.
Ball P. Adverse drug reactions: implications for the development of fluoroquinolones. J Antimicrob Chemother 2003; 51 Suppl. 1: 21–7.
Juurlink DN, Park-Wyllie LY, Kapral MK. The effect of publication on internet-based solicitation of personal-injury litigants. CMAJ 2007; 177 (11): 1369–70.
European Medicines Agency. Withdrawal assessment report for garenoxacin mesylate (Garenoxacin): EMEA/H/C/747 [online]. Available from http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500067888.pdf [Accessed 2011 Dec 13].
European Medicines Agency. Withdrawal assessment report for Factive. International nonproprietary name: gemifloxacin. Procedure no. EMEA/H/C/995 [online]. Available from URL: http://www.ema.europa.eu/docs/en_GB/document_library/Application_withdrawal_assessment_report/2010/01/WC500060988.pdf [Accessed 2011 Dec 13].
Bayer Schering Pharma-Bayer plc. Direct healthcare professional communication regarding moxifloxacin (Avelox®) and serious hepatic and bullous skin reactions [online]. Available from URL: http://www.mhra.gov.uk/home/groups/pl-p/documents/websiteresources/con014103.pdf [Accessed 2012 Jan 28].
European Medicines Agency. Moxifloxacin [online]. Available from URL: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/referrals/Moxifloxacin/human_referral_000114.jsp&mid=WC0b01ac0580024e9a [Accessed 2012 Jan 28].
Council for International Organizations of Medical Sciences Working Group. Introductory guide for Standardised MedDRA Queries (SMQs) Version 13.0. Chantilly (VA): MedDRA Maintenance and Support Services Organization, 2010.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH harmonized tripartite guideline. Statistical principles for clinical trials: E9 [online]. Available from URL: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E9/Step4/E9_Guideline.pdf [Accessed 2012 Jan 28].
Greenland S, Robins JM. Estimation of a common effect parameter from sparse follow-up data. Biometrics 1985; 41 (1): 55–68.
Miravitlles M. Moxifloxacin in respiratory tract infections. Expert Opin Pharmacother 2005; 6 (2): 283–93.
Craig WA. Overview of newer antimicrobial formulations for overcoming pneumococcal resistance. Am J Med 2004; 117 Suppl. 3A: 16S–22S.
File TM, Garau J, Jacobs MR, et al. Efficacy of a new pharmacokinetically enhanced formulation of amoxicillin/clavulanate (2000/125mg) in adults with community-acquired pneumonia caused by Streptococcus pneumoniae, including penicillin-resistant strains. Int J Antimicrob Agents 2005; 25 (2): 110–9.
Aspa J, Rajas O, de Castro FR. Pneumococcal antimicrobial resistance: therapeutic strategy and management in community-acquired pneumonia. Expert Opin Pharmacother 2008; 9 (2): 229–41.
Croom KF, Goa KL. Levofloxacin: a review of its use in the treatment of bacterial infections in the United States. Drugs 2003; 63 (24): 2769–802.
Klugman KP. Bacteriological evidence of antibiotic failure in pneumococcal lower respiratory tract infections. Eur Respir J Suppl 2002; 36: 3s–8s.
Odenholt I, Cars O. Pharmacodynamics of moxifloxacin and levofloxacin against Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae and Escherichia coli: simulation of human plasma concentrations after intravenous dosage in an in vitro kinetic model. J Antimicrob Chemother 2006; 58 (5): 960–5.
Lister PD. Pharmacodynamics of levofloxacin against characterized ciprofloxacin-resistant Streptococcus pneumoniae. Postgrad Med 2008; 120 (3 Suppl. 1): 46–52.
Brinker A. Telithromycin-associated hepatotoxicity [online]. Available from www.fda.gov/ohrms/dockets/AC/06/slides/2006-4266s1-01-07-FDA-Brinker.ppt [Accessed 2012 Jan 28].
Acknowledgments
Bayer Pharma AG provided all authors with free access to the moxifloxacin clinical database. Highfield Communication Consultancy Ltd (Oxford, UK) [funded by Bayer Pharma] provided editorial assistance in the preparation of this manuscript. The analysis was jointly designed and conducted and the results interpreted by all authors, who also prepared and approved the manuscript. The clinical relevance of all results has also been assessed by Paul M. Tulkens and Pierre Arvis. Paul M. Tulkens has received research grants and honoraria (related to published studies and presentations about moxifloxacin but not to this work) from Bayer Pharma, Sanofi-Aventis, Bristol-Myers/Squibb, Pfizer, and GlaxoSmithKline. Pierre Arvis and Frank Kruesmann are employees of Bayer Santé SAS and Bayer Pharma AG, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Tulkens, P.M., Arvis, P. & Kruesmann, F. Moxifloxacin Safety. Drugs R D 12, 71–100 (2012). https://doi.org/10.2165/11634300-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11634300-000000000-00000