
Abstract
Background: Safety is a primary concern with contrast agents used for MRI. If precautions could be taken before the repeated administration of gadolinium-based contrast media, then the awareness and management of adverse reactions would be more efficient.
Objectives: To assess the safety and efficacy of gadoterate meglumine (Gd-DOTA) [Magnescope® in Japan, Dotarem® in other countries], a gadolinium-based contrast agent, in patients undergoing imaging of the brain/spinal cord and/or trunk/limbs, and to identify factors associated with the onset of adverse reactions.
Methods: The study ran for 4 years and included 3444 cases. The study was conducted before it became known that gadolinium-based contrast agents could trigger the development of nephrogenic systemic fibrosis. Patients for whom the contrast agent was indicated and who underwent imaging of the brain/spinal cord and/or trunk/limbs by MRI were enrolled. There were 1300 inpatients who were followed up during hospitalization (for several days), and 2144 outpatients who were followed up for at least 2 hours on-site. After Gd-DOTA administration, 13 patient baseline characteristics were used to explore factors that might predict a greater likelihood of acute non-renal adverse reactions. The physician’s appraisal of the efficacy of Gd-DOTA was also assessed.
Results: A total of 40 adverse reactions were recorded in 32 patients, giving an overall incidence of adverse reactions of 0.93%. Gastrointestinal disorders were the most commonly reported adverse reactions (0.49%). Most adverse reactions reported were of mild intensity and no serious adverse reactions were reported. This study found that statistically significant risk factors for adverse reactions were general patient condition, liver disorder, kidney disorder, health complications, concomitant treatments, and Gd-DOTA dose (although the incidence of adverse reactions was not dose dependent). In the majority of cases (99.53%), the efficacy of Gd-DOTA was rated as ‘effective’ or ‘very effective’; only the presence of kidney disorder was associated with a significantly greater likelihood of Gd-DOTA inefficacy.
Conclusion: Overall, this postmarketing surveillance study did not reveal any untoward or unexpected findings concerning the safety or efficacy of Gd-DOTA. The low incidence of adverse reactions (<1%) and the absence of serious adverse reactions reported during the survey period showed that Gd-DOTA was very well tolerated. The use of Gd-DOTA as an MRI-enhancing contrast medium in the clinical practice setting appears to be safe and effective.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MRI is used for imaging of internal body tissues and organs, and provides excellent tissue contrast. However, improved contrast enhancement can be achieved by the use of contrast media, which has greatly enhanced the diagnostic accuracy of this imaging modality.
Gadolinium, which is widely used as a contrast agent, is intrinsically toxic, in part, by blocking calcium channels. Gadolinium must be chelated with an appropriate ligand to allow clinical use. These gadolinium complexes can be linear or macrocyclic.[1] Extracellular gadolinium-chelates are distributed throughout the extracellular space without crossing the normal blood-brain barrier, and are therefore efficient for detecting pathologic abnormalities, including brain metastases as well as detailed characterization of parenchymal lesions and lymph nodes. The clinical benefit associated with the use of gadolinium-chelates has been clearly established over the last 20 years.[2–6]
Most gadolinium-based agents are excreted by the kidneys, but elimination becomes problematic for individuals with reduced kidney function.[7] Gadolinium-containing contrast agents are rapidly cleared with a half-life of approximately 2 hours in patients with normal renal function, while in those with chronic renal failure this clearance may exceed 30–120 hours.[8]
A primary concern with all MRI contrast agents is safety. The safety profile of gadolinium-based contrast agents seems to be superior to that of iodinated contrast molecules used for x-ray based procedures.[9] Minor adverse effects associated with gadolinium-based agents include nausea, headache, and taste perversion.[9] A number of gadolinium agents are available and although they cannot be distinguished on the basis of their mild adverse effects, recent studies have highlighted chelate stability and safety issues.[9] Therefore, the stability of gadolinium-contrast agents seems to be an important factor in the pathogenesis of the serious complication of nephrogenic systemic fibrosis (NSF), and, in particular, clinical observations associated with the least stable of gadolinium-contrast agents. Importantly, it was demonstrated in 2006 that some gadolinium-based contrast agents may trigger the development of NSF in patients with impaired renal function.[8] Consequently, the European Medicines Agency (EMA) ruled that the use of gadodiamide, gadoversetamide, and gadopentate dimeglumine in patients with a glomerular filtration rate (GFR) <30 mL/min/1.73 m2 was contraindicated and that these agents should only be used with caution in patients with moderately reduced kidney function (30–60 mL/min/1.73 m2).
Among the gadolinium-based contrast media available in clinical practice, gadoterate meglumine (Gd-DOTA, Magnescope® in Japan, Dotarem® in other countries), a cyclic ionic gadolinium chelate, is approved for imaging cerebral and spinal lesions with abnormal blood-brain barrier or anomalous vascularity as well as for body imaging. Gd-DOTA has the highest thermodynamic stability (log10 KTHERM = 25.6), apparent stability (log10 Kcond = 19.3), kinetic stability, and decomplexation of all available gadolinium-chelate complexes.[1,9–12] The safety profile of Gd-DOTA has recently been established in a German post-marketing surveillance study involving more than 24 000 patients who underwent routine MRI examinations.[13] The German study aimed to assess the diagnostic value and safety of Gd-DOTA in clinical practice, but did not identify the factors that may increase the risk of adverse reactions.
The aim of this Japanese post-marketing surveillance study was to assess the safety and efficacy of Gd-DOTA (registered in Japan in September 2000) in patients undergoing MRI, and to identify factors associated with the onset of adverse reactions. This type of evaluation, mandatory in Japan, is designed to confirm the safety and efficacy of any new drug in several conditions in which the product is routinely used, in contrast with the pre-market, development phase.
Methods
Study Design and Patient Enrollment Conditions
This post-marketing surveillance study was conducted according to the protocol submitted by Eiken Chemical Co., Ltd (Tokyo, Japan) in accordance with the Pharmaceutical Affairs Law of Japan. The Pharmaceutical Affairs Act set up a drug re-examination system (i.e. a system that ensures marketers will report on safety and effectiveness of the contrast agents/drugs used in routine medical practice), which usually collects data 6 years after the launch of any product.
As a part of this Act, marketers were instructed by the Ministry of Health, Labor and Welfare to conduct a “drug use-results survey” during that period. A guideline for “drug use-results survey” implementation method defines that marketers shall collect a minimum of 3000 cases during the 3 years after product launch, and its purpose is to assure safety and effectiveness of routine medical treatment or administration of contrast agents.
Therefore, our post-marketing surveillance study was planned to run for at least 3 years following the Japanese launch in April 2001 of the Gd-DOTA vials and syringes (Guerbet, Roissy CdG Cedex, France) and was designed to enroll at least 3000 inpatients and/or outpatients. A continuous surveillance process was followed whereby patients who had been administered Gd-DOTA were registered consecutively until the cases reached the agreed numbers and the defined period of recruitment mentioned in the contract with each hospital site. Informed consent was considered unnecessary for this post-marketing surveillance study.
Patients for whom the contrast agent was indicated and who underwent imaging of the brain/spinal cord and/or trunk/limbs by MRI were enrolled.
Baseline Characteristics
Demographic characteristics (sex, age, weight) and clinical characteristics (patient status [inpatient/outpatient], indications for imaging, general condition, health complications, concomitant drugs), as well as Gd-DOTA dose, premedication, and medical history (liver disorder, kidney disorder, history of allergy/adverse reactions to drugs) were obtained. General condition and complication were determined by the physician’s appraisal at each site. No specific laboratory tests were required, but if patients had liver disorder and/or kidney disorder, the reports of the following tests conducted in routine medical practice were obtained: AST, ALT, alkaline phosphatase (ALP), lactate dehydrogenase (LDH), and γ-glutamyl transferase (γ-GT), total bilirubin for liver disorders, and serum creatinine and blood urea nitrogen (BUN) for kidney disorders.
Safety Evaluation
After administration of Gd-DOTA, adverse events for outpatients were recorded up to at least 2 hours on-site or until they left the MRI suite, and inpatients were followed up for several days (during their hospitalization). Every adverse event occurring during observation in routine medical practice was recorded in the case report form.
Patient baseline characteristics were used to explore factors that might predict a greater likelihood of acute non-renal adverse reactions. Late and very late adverse reactions were not systematically studied.
In addition, the main subsets of patients who were submitted to specific surveillance for adverse reactions were: (i) those who received Gd-DOTA >0.1 mL/kg in whom the kidney was the imaging target; (ii) those with decreased kidney function; (iii) those with decreased liver function; (iv) pediatric patients (<15 years); and (v) elderly patients (≥65 years).
Skin biopsy and inspection of the skin of the patients with reduced renal function 3 months after Gd-DOTA exposure were not planned in this study.
Efficacy Evaluation
The efficacy of Gd-DOTA as an image-enhancing agent was assessed by the physician’s appraisal according to the following four grades: (i) ‘very effective’ (i.e. diagnostic performance highly improved); (ii) ‘effective’ (i.e diagnostic performance improved); (iii) ‘ineffective’ (i.e. examination performed normally but diagnostic performance not improved or necessary information for diagnosis was not obtained); and (iv) ‘appraisal impossible’ (i.e. when no evaluable image was obtained as a result of the movement and/or lack of cooperation from the patient or when the MRI examination was not performed for whatever reason).
Statistical Evaluation
Descriptive statistics were used for patient baseline characteristics and the frequency of adverse reactions.
Paired t-test analyses were conducted on continuous data and chi-squared tests on categorical data. All comparisons were considered significant with a p-value of <0.05.
Results
Patient Characteristics and Datasets Analyzed
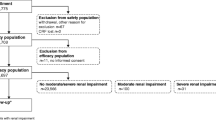
This post-marketing surveillance study was conducted over 4 years (from March 2001 to March 2005). A total of 3480 survey forms were returned from 127 participating institutions in Japan (mean number of cases per institution was 27.4; range 5–100) [figure 1]. A total of 36 cases were excluded from the analyses because of the inability to confirm continuous surveillance (n = 30), administration of Gd-DOTA outside the survey period (n = 4) or duplicate results (n = 2). Among these 36 cases, two patients experienced adverse reactions of mild intensity (nausea and injection-site inflammation following extravasation). As these two patients received Gd-DOTA outside the period defined in the contract with hospitals, they were excluded from the safety dataset. Consequently, a total of 3444 cases were included in the safety dataset. The characteristics of these patients are detailed in table I.

Process to determine the datasets analyzed.

Background summary of patients in the safety dataset (n = 3444)
For the efficacy population, 18 cases were excluded from the safety dataset because of the following reasons: images not evaluable (n = 9), artefact or equipment malfunction (n = 4), termination due to adverse reaction (n = 3), injection-site extravasation (n = 1), or loss of clinical record (n = 1). Accordingly, the efficacy population comprised 3426 cases (figure 1).
Safety
Adverse Reactions
A total of 40 adverse reactions were recorded in 32 patients. The incidence of adverse reactions was 0.93% (32 of 3444 patients) compared with 1.33% (11 of 829 patients) in trials conducted prior to approval (table II). There were no serious adverse reactions reported during the survey period. Most of the 40 adverse reactions reported were of mild intensity, except for four cases of moderate intensity (two patients with nausea and two with abnormal liver function).

Incidence of acute non-renal adverse reactions (including infection) to meglumine gadoterate (Gd-DOTA) according to the system organ class and preferred term
The most frequently reported adverse reactions in the current survey were gastrointestinal disorders (0.49%), abnormal laboratory test results (0.26%), and skin and subcutaneous tissue disorders (0.15%) [table II]. The incidences of these common adverse reactions found in trials conducted before Gd-DOTA approval were 0.48%, 0%, and 0.48%, respectively.
Factors Affecting the Onset of Adverse Reactions
Among the 13 patient baseline characteristics analyzed, sex, age, weight, status, indications, history of allergy/adverse drug reactions, and premedication were not associated with a significantly increased risk of adverse reactions. Conversely, statistically significant risk factors for experiencing adverse reactions were general condition, liver disorder, kidney disorder, complication, concomitant treatments, and Gd-DOTA dose (table III).

Patient background risk factors associated with statistically significant differences in adverse reaction rates
As shown in table III, patients with ‘poor’ general condition had a greater risk of experiencing an adverse reaction compared with those classified as ‘good’ or ‘fair’ (p = 0.0414).
Patients with liver and kidney disorders showed a significantly higher (p < 0.0001) risk of experiencing an adverse reaction than patients without liver and kidney disorders (see details in the ‘Patients with Specific Backgrounds’ section).
In addition, the incidence of adverse reactions was statistically significantly higher for patients with complications (p < 0.0003) than for those with no complications.
Concomitant therapy was used by 1160 patients. Twenty of these patients experienced an adverse reaction compared with only 12 of 2278 patients not using concomitant therapy. Simultaneous use of a concomitant drug was associated with a statistically significantly higher incidence of adverse reactions (p < 0.0005). The most frequently used concomitant drugs were peptic ulcer drugs (8.4%), vasodilators (6.2%), anti-hypertensive drugs (5.2%), antiepileptic drugs (4.6%), and adrenal hormone drugs (4.5%).
Finally, when analyzing the administration of the Gd-DOTA dose, the incidence of adverse reactions was 0.93% (18 of 1941) in patients receiving Gd-DOTA >0.1–0.2 mL/kg, and 0.84% (12 of 1437) in patients receiving Gd-DOTA >0.2 mL/.kg (table III). No dose-dependent increase in the incidence of adverse reactions was observed.
Patients with Specific Backgrounds
Patients Receiving Gd-DOTA >0.1 mL/kg in Whom the Kidney was the Imaging Target
MRI analysis of kidneys was performed in 60 patients and 52 of these patients received Gd-DOTA >0.1 mL/kg (mean Gd-DOTA dose 0.2176 mL/kg; range 0.1176–0.4255 mL/kg). No patients experienced adverse reactions and the results for the kidney function tests (serum creatinine levels) performed in 24 patients were not significantly different before and after Gd-DOTA administration.
Patients with Decreased Kidney Function
Kidney disorders were reported by 98 patients with mild to moderate renal impairment in most cases. The incidence of adverse reactions was significantly higher in patients with kidney disorders than in those with normal kidney function (table III; p < 0.0001). A total of ten adverse reactions (two cases of kidney disorders, two cases of elevated serum creatinine, two cases of nausea, and one case each of elevated blood urea, abnormal kidney function tests, abnormal liver function tests, and sneezing) occurred in eight patients with kidney disorders. Kidney function tests (conducted during routine medical practice) revealed no significant difference in serum creatinine and BUN before and after Gd-DOTA administration (table IV).

Variation in kidney and liver function test values before and after meglumine gadoterate (Gd-DOTA) administration in patients with decreased kidney or liver function
The incidence of adverse reactions was associated with the type of kidney disorder (kidney failure 25.00% [4 of 16 cases], diabetic nephropathy 20.00% [1 of 5], other 4.48% [3 of 67], nephritis 0% [0 of 1]; p = 0.0282) and the severity of kidney disorders (‘severe’ 30.00% [3 of 10], ‘moderate’ 13.33% [2 of 15], ‘mild’ 4.17% [3 of 72]; p = 0.0154).
Patients with Impaired Liver Function
A total of 328 patients had impaired liver function, which was considered to be mild to moderate in most cases. The incidence of adverse reactions was significantly higher in these patients than in those with normal liver function (table III; p < 0.0001). A total of 15 adverse reactions were experienced by 12 patients with liver disorders (four cases of nausea, two cases of abnormal liver function, two cases of abnormal liver function tests, one case each of elevated AST, elevated ALT, kidney disorder, abnormal kidney function tests, abnormal sensation, and two cases of drug eruptions).
Liver function tests (when performed) before and after Gd-DOTA administration revealed a statistically significant decrease in test values for LDH after administration (n = 178; 362.3 IU/L vs 341.1 IU/L, respectively; p = 0.0429), but variations in the levels of AST, ALT, ALP, γ-GT, and total bilirubin were not significant (table IV). No significant difference in the incidence of adverse reactions was observed according to underlying causal factor or the severity of the liver disorder.
Pediatric Patients (<15 years)
There were 41 pediatric patients (age range from 1 month to 14 years and 11 months) at the time of Gd-DOTA administration. The recommended dose of Gd-DOTA is 0.1 mmol/kg (0.2 mL/kg) for adults, children, and infants. The average dose of Gd-DOTA administered was 0.2049 mL/kg. Two children received a dose that was approximately 1.7-fold and 1.9-fold greater than the recommended dose (0.2 mL/kg) for brain/spinal cord imaging in both cases. No adverse reactions were reported for any of the pediatric patients.
Elderly Patients (≥65 years)
Patients aged ≥65 years accounted for 40.3% of patients of the survey (1388 of 3444; table I). The incidence of adverse reactions was not significantly different between those aged ≥65 years and those aged 16–64 years (0.79% vs 1.02%; p = 0.492).
Efficacy
According to the physician’s appraisal, Gd-DOTA was scored as ‘very effective’ or ‘effective’ in the majority of patients (1075 and 2335 cases, respectively, of 3426 cases in total). Gd-DOTA was considered to be ‘ineffective’ in only 16 cases. The overall efficacy rate was 99.53% (3410 of 3426 cases), which was similar to the results obtained in trials conducted prior to approval (efficacy rate 90.21% [553 of 613]).
Analysis of the factors affecting efficacy revealed that the presence of kidney disorders was associated with a statistically significantly greater likelihood of inefficacy (2.08% vs 0.44% for patients without kidney disorders; p = 0.0247). Other patient characteristics (including sex, age, weight, indications, liver disorder, Gd-DOTA dose) were not associated with any significant differences in efficacy (table V).

Summary of efficacy trial results classified according to risk factors
Discussion
This mandatory post-marketing surveillance survey was designed to assess the safety and efficacy of Gd-DOTA for brain/spinal cord and/or trunk/limb imaging in a wide range of patients, following its registration in Japan in September 2000. The results presented in this article show that Gd-DOTA, as used in clinical practice, was associated with a low rate of adverse reactions (<1%) and was effective as an image-enhancing agent in the majority of patients.
This survey was also designed to identify risk factors for the development of adverse reactions. The analysis showed that statistically significant risk factors for adverse reactions were general condition, liver disorders, kidney disorders, complications, concomitant treatments, and Gd-DOTA dose (although the incidence of adverse reactions was not dose dependent). However, these data should be interpreted cautiously in view of the small number of patients for whom historical and clinical data were available at screening in this study.
The incidence of adverse reactions following Gd-DOTA administration in the majority of published studies to date is less than 1%, ranging from 0.4%[13] to 0.97%.[14] The one notable exception is a reported incidence of 17.3%;[15] however, in this study, the authors noted that 71% of adverse reactions were reported 24 hours after the procedure, which, due to the rapid clearance rate reported for gadolinium-based contrast agents, raises the question as to whether these events were drug related. The most frequently reported adverse reactions in our post-marketing surveillance survey were gastrointestinal disorders (0.49% compared with 0.48% in previous trials conducted before Gd-DOTA approval), laboratory test abnormalities (0.26%), and skin and subcutaneous tissue disorders (0.15% compared with 0.48% in previous trials). Most of the reported adverse reactions were of mild intensity and there were no serious adverse reactions. The results of the current survey are in agreement with a large-scale post-marketing surveillance study conducted in Germany.[13] Herborn et al.[13] reported that minor adverse events occurred infrequently (0.4%; 94/24 308 patients) and included nausea (0.17%; 42/24 308 patients), vomiting (0.05%; 13/24 308 patients), feeling of warmth (0.020%; six patients), and taste alterations (0.02%; four patients). Interestingly, patients with liver disease experienced nausea after Gd-DOTA injection significantly more frequently than any other subgroup (p < 0.001) but for unknown reasons.
Anaphylactoid reactions induced by gadolinium-containing products are rare and although >15 million patients worldwide have been exposed to Gd-DOTA, there are few published case reports of anaphylactic shock following Gd-DOTA administration. The occurrence of anaphylactic shock in one patient during a large-scale post-marketing surveillance study of Gd-DOTA allowed Herborn and colleagues[13] to calculate the incidence of anaphylactic shock associated with Gd-DOTA as 0.004%. Indeed, to our knowledge, only four cases of anaphylactic shock have been published[13,16,17] and, in every case, the patient developed an immediate reaction to Gd-DOTA despite the absence of any history of allergy. No patients in the present study experienced an anaphylactoid reaction following Gd-DOTA administration, although 4.44% of patients had a history of allergy or adverse drug reactions.
Several recent publications[7,18–21] have suggested an association between administration of gadolinium-based contrast agents and NSF, and there appears to be a difference between the various contrast agents in their trigger of NSF.[22]
Cyclic gadolinium-based contrast agents (such as Gd-DOTA and others) are more stable and less likely to release free gadolinium3+,[23] and to our knowledge, no validated case of NSF has been solely attributed to Gd-DOTA. Based on the results obtained in the current survey and our study design, which did not allow accurate assessment of NSF, it is difficult to draw conclusions about NSF risk for this study. Tsushima et al.[24] suggested that the small number of reported cases of NSF in Japan compared with the US and Europe can be attributed to the uncommon use of high-dose (>0.2 mmol/kg) administration of gadolinium-based contrast agents.
The 99.53% global efficacy rate reported in the present study is in line with the study conducted by Herborn et al.[13] in which the efficacy of Gd-DOTA was rated as ‘excellent’ or ‘very good’ in the majority of cases (97.5%). Interestingly, the present survey demonstrated a significantly greater likelihood of inefficacy associated with kidney disorders, whereas Herborn et al.[13] found that image quality was significantly poorer in patients with a body mass index >25 kg/m2 (p < 0.001). Unfortunately, detailed reasons for the inefficacy associated with kidney disorder were not declared during our study.
This study is not free of limitations. Logistic regression analysis for evaluating the confounding factors on the onset of adverse reactions was not performed. This post-marketing surveillance study was limited by a lack of clear definitions of general condition and complications that would ensure homogeneity between sites. As contrast-induced nephropathy (CIN) has become a significant source of hospital morbidity, CIN incidence and risk factors have not been evaluated in this survey. Another limitation of this post-marketing surveillance study was the evaluation of only acute non-renal adverse reactions and not delayed ones. Thus, it seems likely that some adverse reactions may have been missed and underreported in this patient cohort. Finally, the use of questionnaires for data collection in such a study limits the quality of the results compared with controlled clinical trials.
Conclusion
Overall, this post-marketing surveillance study did not reveal any untoward or unexpected findings concerning the safety or efficacy of Gd-DOTA. The low incidence of adverse reactions (<1%) and the absence of serious adverse reactions reported during the survey period showed that Gd-DOTA was very well tolerated. In conclusion, the use of Gd-DOTA as an MRI-enhancing contrast medium in the clinical practice setting appears to be safe and effective.
References
Idee JM, Port M, Raynal I, et al. Clinical and biological consequences of transmetallation induced by contrast agents for magnetic resonance imaging: a review. Fundam Clin Pharmacol 2006; 20 (6): 563–76
Ruehm SG, Goyen M, Barkhausen J, et al. Rapid magnetic resonance angiography for detection of atherosclerosis. Lancet 2001; 357 (9262): 1086–91
Claussen C, Laniado M, Schorner W, et al. Gadolinium-DTPA in MR imaging of glioblastomas and intracranial metastases. AJNR Am J Neuroradiol 1985; 6 (5): 669–74
Healy ME, Hesselink JR, Press GA, et al. Increased detection of intracranial metastases with intravenous Gd-DTPA. Radiology 1987; 165 (3): 619–24
Meaney JF, Weg JG, Chenevert TL, et al. Diagnosis of pulmonary embolism with magnetic resonance angiography. N Engl J Med 1997; 336 (20): 1422–7
Semelka RC, Helmberger TK. Contrast agents for MR imaging of the liver. Radiology 2001; 218 (1): 27–38
Thomsen HS, Marckmann P, Logager VB. Nephrogenic systemic fibrosis (NSF): a late adverse reaction to some of the gadolinium based contrast agents. Cancer Imaging 2007; 7: 130–7
Grobner T. Gadolinium: a specific trigger for the development of nephrogenic fibrosing dermopathy and nephroge-nic systemic fibrosis? Nephrol Dial Transplant 2006; 21 (4): 1104–8
Kirchin MA, Runge VM. Contrast agents for magnetic resonance imaging: safety update. Top Magn Reson Imaging 2003; 14 (5): 426–35
Laurent S, Elst LV, Muller RN. Comparative study of the physicochemical properties of six clinical low molecular weight gadolinium contrast agents. Contrast Media Mol Imaging 2006; 1 (3): 128–37
Port M, Idee JM, Medina C, et al. Stability of gadolinium chelates and their biological consequences: new data and some comments. Br J Radiol 2008; 81 (963): 258–9
Allard M, Doucet D, Kien P, et al. Experimental study of DOTA-gadolinium: pharmacokinetics and pharmacologic properties. Invest Radiol 1988; 23 (1): S271–4
Herborn CU, Honold E, Wolf M, et al. Clinical safety and diagnostic value of the gadolinium chelate gadoterate meglumine (Gd-DOTA). Invest Radiol 2007; 42 (1): 58–62
Oudkerk M, Sijens PE, Van Beek EJ, et al. Safety and efficacy of dotarem (Gd-DOTA) versus magnevist (Gd-DTPA) in magnetic resonance imaging of the central nervous system. Invest Radiol 1995; 30 (2): 75–8
Brugieres P, Gaston A, Degryse HR, et al. Randomised double blind trial of the safety and efficacy of two gadolinium complexes (Gd-DTPA and Gd-DOTA). Neuroradiology 1994; 36 (1): 27–30
Beaudouin E, Kanny G, Blanloeil Y, et al. Anaphylactic shock induced by gadoterate meglumine (DOTAREM). Eur Ann Allergy Clin Immunol 2003; 35 (10): 382–5
Hasdenteufel F, Luyasu S, Renaudin JM, et al. Anaphylactic shock after first exposure to gadoterate meglumine: two case reports documented by positive allergy assessment. J Allergy Clin Immunol 2008; 121 (2): 527–8
DeHoratius DM, Cowper SE. Nephrogenic systemic fibrosis: an emerging threat among renal patients. Semin Dial 2006; 19 (3): 191–4
Cowper SE. Nephrogenic fibrosing dermopathy: the first 6 years. Curr Opin Rheumatol 2003; 15 (6): 785–90
Scheinfeld N. Nephrogenic fibrosing dermopathy: a comprehensive review for the dermatologist. Am J Clin Dermatol 2006; 7 (4): 237–47
Cowper S. Nephrogenic fibrosing dermopathy [online]. Available from URL: http://www.icnfdr.org [Accessed 2010 Aug 27]
Penfield JG, Reilly RF. Nephrogenic systemic fibrosis risk: is there a difference between gadolinium-based contrast agents? Semin Dial 2008; 21 (2): 129–34
Kimura J, Ishiguchi T, Matsuda J, et al. Human comparative study of zinc and copper excretion via urine after administration of magnetic resonance imaging contrast agents. Radiat Med 2005; 23 (5): 322–6
Tsushima Y, Takahashi-Taketomi A, Endo K. Nephrogenic systemic fibrosis in Japan: advisability of keeping the administered dose as low as possible. Radiology 2008; 247 (3): 915–6
Acknowledgements
The authors would like to thank all the 127 institutions that participated in this Magnescope post-marketing surveillance study. This post-marketing surveillance study was supported by the Guerbet group. The authors have no conflicts of interest that are directly relevant to the contents of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ishiguchi, T., Takahashi, S. Safety of Gadoterate Meglumine (Gd-DOTA) as a Contrast Agent for Magnetic Resonance Imaging. Drugs R D 10, 133–145 (2010). https://doi.org/10.2165/11539140-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11539140-000000000-00000