
Abstract
Background: Combination therapy with a long-acting bronchodilator and an inhaled corticosteroid (ICS) is recommended in patients with chronic obstructive pulmonary disease (COPD) who have frequent exacerbations. The efficacy and tolerability of the combination of budesonide/formoterol have been demonstrated in patients with COPD when administered via the dry powder inhaler (DPI) in a 1-year study and when administered via the hydrofluoroalkane (HFA) pressurized metered-dose inhaler (pMDI) in a 6-month study.
Objective: This study assessed the long-term efficacy and tolerability of budesonide/formoterol HFA pMDI in patients with moderate to very severe COPD.
Methods: This was a 12-month, randomized, double-blind, double-dummy, parallel-group, active- and placebo-controlled, multicentre study (NCT00206167) of 1964 patients aged ≥40 years with moderate to very severe COPD conducted from 2005 to 2007 at 237 sites in the US, Europe and Mexico. After 2 weeks of treatment based on previous therapy (ICSs, short-acting bronchodilators allowed), patients received one of the following treatments twice daily: budesonide/formoterol pMDI 160/4.5 μg × two inhalations (320/9 μg); budesonide/formoterol pMDI 80/4.5 μg × two inhalations (160/9 μg); formoterol DPI 4.5 μg × two inhalations (9 μg); or placebo.
Main outcome measures: The co-primary efficacy variables were pre-dose forced expiratory volume in 1 second (FEV1) and 1-hour post-dose FEV1.
Results: Budesonide/formoterol 320/9 μg demonstrated greater improvements in pre-dose FEV1 versus formoterol (p = 0.008), and both budesonide/formoterol doses demonstrated greater improvements in 1-hour post-dose FEV1 versus placebo (p < 0.001). The rate of COPD exacerbations was lower in both budesonide/formoterol groups compared with formoterol and placebo (p ≤ 0.004). Both budesonide/formoterol doses were more effective than placebo (p ≤ 0.006) for controlling dyspnoea and improving health status (St George’s Respiratory Questionnaire). All treatments were generally well tolerated. The incidence of pneumonia was not different for active (3.4–4.0%) and placebo (5.0%) groups.
Conclusions: Budesonide/formoterol pMDI (320/9 μg and 160/9 μg) improved pulmonary function and reduced symptoms and exacerbations over 1 year in patients with moderate to very severe COPD. Only budesonide/formoterol pMDI 320/9 μg demonstrated greater efficacy for both co-primary variables compared with formoterol DPI 9 μg. Both budesonide/formoterol pMDI dosages were well tolerated relative to formoterol and placebo.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
In patients with chronic obstructive pulmonary disease (COPD) and frequent exacerbations, the combination of a long-acting bronchodilator and an inhaled corticosteroid (ICS) is recommended.[1] Budesonide and formoterol delivered via a single dry powder inhaler (DPI; Symbicort® Turbuhaler®; AstraZeneca, Lund, Sweden) have been shown to improve pulmonary function[2,3] and reduce COPD exacerbation rates.[2,3] Fixed-combination budesonide/formoterol is available in the US in a hydrofluoroalkane pressurized metered-dose inhaler (pMDI; Symbicort® Inhalation Aerosol; AstraZeneca LP, Wilmington, DE, USA). While most patients with COPD can benefit from either a pMDIor DPI, the pMDI formulation allows patients whomay have inadequate peak inspiratory flow for optimal delivery from a DPI to use the pMDI effectively. A recent 6-month study demonstrated the efficacy and tolerability of budesonide/formoterol pMDI in patients with moderate to very severe COPD.[4] Treatment with budesonide/formoterol pMDI resulted in significant improvements in pulmonary function, dyspnoea and health-related quality of life; however, the duration of the study was limiting with respect to the analysis of low-frequency events, such as COPD exacerbations, and precluded an assessment of safety outcomes with long-term exposure.[4] In this study, the long-term efficacy and tolerability of two dosage strengths of budesonide/formoterol pMDI were assessed over 12 months in patients with moderate to very severe COPD.
Patients and Methods
Patients
The inclusion criteria were designed to select a population with moderate to very severe COPD with previous exacerbations (i.e. appropriate candidates for combination ICS/long-acting β2-adrenoceptor agonist [LABA] therapy): age ≥40 years, diagnosis of symptomatic COPD for >2 years, ≥10 pack-year smoking history, prebronchodilator forced expiratory volume in 1 second (FEV1) of ≤50% of predicted normal and prebronchodilator FEV1/forced vital capacity (FVC) of <70%. Patients were to have a Modified Medical Research Council dyspnoea scale score of ≥2 and a history of at least one COPD exacerbation requiring oral corticosteroids or antibacterials within 1–12 months before the first study visit. Additional enrolment criteria were the same as those in a similar 6-month study by Tashkin et al.[4]
Study Design
This was a randomized, double-blind, double-dummy, parallel-group, active- and placebo-controlled, multicentre trial (ClinicalTrials.gov identifier: NCT00206167) conducted from 2005 to 2007 at 237 sites in the US, Europe and Mexico. The study protocol was approved by a local institutional review board and ethics committee, and written informed consent was obtained from patients. The study was designed to conform with the Declaration of Helsinki, and was consistent with the International Conference on Harmonisation and Good Clinical Practice and applicable regulatory requirements.
After meeting eligibility criteria, patients entered a 2-week run-in period, during which they received ICS monotherapy if previously stable on ICS (alone or in combination) and ipratropium bromide at a fixed dose if previously receiving anticholinergics. Albuterol (salbutamol) was permitted for rescue use throughout the study. At randomization, all previous ICSs were discontinued and patients were randomized to one of four treatments (figure 1). Patients who were treated with ipratropium bromide during the run-in period were allowed to continue on it throughout the study period. Other allowed and disallowed concomitant medications were the same as those reported in the study by Tashkin et al.[4]

Study design. To maintain blinding, patients received both a pressurized metered-dose inhaler (pMDI) and a dry powder inhaler (DPI) containing either active treatment or double-dummy placebo (PL) as appropriate. This study used formoterol (FM) DPI (Oxis® Turbuhaler®, AstraZeneca, Lund, Sweden) as the FM comparator because FM is not available as a hydrofluoroalkane pMDI in the US. A previous study in asthma patients reported equivalent FM-related bronchodilatory effects when FM was administered in combination with budesonide (BUD) via pMDI or alone via DPI.[5] Patients were asked to return to the clinic for follow-up visits 3–8 at the end of months 1, 2, 4, 6, 9 and 12, and received a telephone call 4 weeks after the last clinic visit. bid = twice daily; R = randomization.
Efficacy Evaluations
The co-primary efficacy variables, measured at all clinic visits, were pre-dose FEV1 and 1-hour post-dose FEV1. Additional pulmonary function variables included pre-dose and 1-hour post-dose FVC measured at all clinic visits, and morning and evening peak expiratory flow (PEF) recorded daily in patient diaries. In a subset of patients (n = 491 of 1964; 25%) who agreed to undergo spirometry testing, 12-hour serial spirometry was performed and pre-dose and 1-hour post-dose inspiratory capacity (IC) were collected at randomization and months 6 and 12. FEV1 was measured pre-dose and at 5, 15, 30, 60, 120, 180, 240, 360, 480, 600 and 720 minutes post-dose. Baseline-adjusted average 12-hour FEV1 was calculated as the area between the 12-hour post-dose FEV1-over-time curve and the baseline pre-dose FEV1, divided by observation time. Spirometry was performed according to American Thoracic Society guidelines.[6] Crapo-predicted normals for FEV1 were used.[7]
Secondary efficacy endpoints included COPD exacerbations, dyspnoea (separate and composite scores) and health status, all assessed as previously described.[4] A COPD exacerbation was defined as worsening of COPD requiring an oral corticosteroid or hospitalization.[4] Dyspnoea was assessed daily before the evening dose of study medication using the Breathlessness Diary, a validated single-item component of the Breathlessness Cough and Sputum Scale (BCSS).[8] Health status was assessed at months 1, 2, 6 and 12 using the St George’s Respiratory Questionnaire (SGRQ).[9,10] Sleep score, percentage of awakening-free nights (sleep score of 0) and study rescue medication use were assessed as previously described by Tashkin et al.[4]
Safety Evaluations
Safety was assessed by adverse event (AE) reporting. Pneumonia events were reported by physicians based on the Medical Dictionary for Regulatory Activities (version 10.0) pneumonia-related preferred terms (pneumonia, bronchopneumonia, lobar pneumonia or pneumonia staphylococcal). Vital signs and 12-lead ECGs were evaluated as previously described by Tashkin et al.[4] Subsets of patients were assessed for 24-hour urinary cortisol (n = 179), 24-hour Holter monitoring (n = 520), bone mineral density (BMD) at the hip and spine regions (n = 326), and ophthalmological assessments (n = 461), including intraocular pressure and progression of lenticular opacities. In all patients, blood and urine samples were collected, and comprehensive physical examinations were carried out at the time of screening and at the end of month 12. Blood and urine samples were analysed by a central laboratory (Quest Diagnostics Clinical Laboratories, Van Nuys, CA, USA). Vital signs, including heart rate and blood pressure, were measured at all study visits.
Samples to assess 24-hour urinary cortisol levels were collected in a subset of patients at or before randomization and within 1 week before the 6- and 12-month visits, and analysed by Quest Diagnostics. 24-Hour Holter monitoring was performed at screening and months 1 and 4, BMD assessments (two sets of dual energy x-ray absorptiometry scans in the hip and spine regions) were performed at screening and the end of month 12, and ophthalmology assessments (intraocular pressure and lenticular opacities) were performed at screening and the end of months 6 and 12.
Statistical Analyses
The efficacy analysis set (i.e. intent-to-treat population) included all randomized patients who received at least one dose of randomized study medication and contributed sufficient data for at least one co-primary or secondary efficacy endpoint to be calculated during the randomized treatment period. The safety analysis population included all randomized patients who received at least one dose of randomized study medication and from whom any post-randomization data were available. For the subsets of patients who underwent serial spirometry, 24-hour urinary cortisol, BMD, 24-hour Holter monitoring and ophthalmological analyses, analysis sets were defined as those patients who received at least one dose of randomized study medication and had baseline and on-treatment values for the variable being assessed. Patients who discontinued prematurely completed final visit (month 12) assessments at the time of discontinuation, followed by the 4-week follow-up. A sample size of approximately 400 patients per treatment group was calculated to allow 90% power to detect a reduction in the number of COPD exacerbations of approximately 30%, adjusting for overdispersion of 2.3. This sample size ensured >95% power to detect a difference of 0.10L in FEV1, given an estimated standard deviation of 0.3 L. All tests were two-sided, with p ≤ 0.05 considered significant.
The prespecified primary comparators were formoterol DPI for pre-dose FEV1 to demonstrate the contribution of budesonide and placebo for 1-hour post-dose FEV1. To address issues of multiplicity relating to multiple-dose comparisons and multiple variables of interest, a fixed-sequence testing procedure was adopted. The primary comparisons were prespecified as budesonide/formoterol pMDI 320/9 μg compared with (i) placebo for pre-dose FEV1 and 1-hour post-dose FEV1; and (ii) formoterol DPI 9 μg for pre-dose FEV1. If significant differences were obtained, comparisons continued with budesonide/formoterol pMDI 320/9 μg compared with placebo for (i) dyspnoea; (ii) SGRQ total score; and (iii) number of exacerbations. If significant differences were obtained for each of these key secondary variables, the lower dose of budesonide/formoterol pMDI was tested on the co-primary variables, as previously described, and, if significant differences were obtained, testing continued with number of exacerbations, dyspnoea and SGRQ total score compared with placebo. For all secondary efficacy variables, the primary comparison was budesonide/formoterol pMDI versus placebo.
Primary and secondary variables were assessed as the change from baseline to the average over the randomized treatment period except for average 12-hour FEV1, mean FEV1 at 12 hours and SGRQ at end of treatment. Changes from baseline in the co-primary efficacy variables were analysed via analysis of co-variance (ANCOVA), adjusting for treatment, country and baseline value. The following three key secondary efficacy variables were identified: (i) dyspnoea; (ii) SGRQ total score; and (iii) exacerbations. Secondary variables were analysed similar to the co-primary efficacy variables; however, exacerbation rate (number/patient-treatment year) was analysed using a Poisson regression model, adjusted for country and differential randomization time, and time to first COPD exacerbation was described via Kaplan-Meier plot and analysed using the log-rank test. The number and percentage of patients experiencing clinically meaningful changes (based on a prespecified minimal important difference) in dyspnoea were analysed using the Cochran-Mantel-Haenszel test, adjusting for country.
Geometric mean 24-hour urinary cortisol levels at end of treatment were compared between treatment groups using a multiplicative ANCOVA model. Mean changes from baseline to the average during the randomized treatment period in 12-lead ECG, and Holter variables and ophthalmology variables were analysed using a model similar to that used for the co-primary variables. BMD variables (natural logarithm of the analysis timepoint minus the natural logarithm of the respective baseline value for hip and spine) were analysed using an ANCOVA model adjusting for country, treatment and natural logarithm of the baseline value. Other safety data were summarized using descriptive statistics. No formal hypothesis testing of the safety data was performed.
Results
Patients
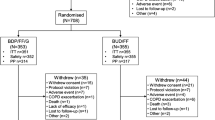
Of 1964 randomized patients, 1355 completed the study (figure 2). Discontinuation was greater with placebo versus budesonide/formoterol and formoterol driven by withdrawal of consent. Time to discontinuation was significantly (p ≤ 0.004) prolonged in both budesonide/formoterol groups versus placebo. AE was the most common reason for study discontinuation, with a similar incidence across treatments (12.1–13.5%). Approximately 60% of patients had documented use of an ICS, either alone or in combination before entering the study. The percentage of patients who discontinued from the study in the formoterol and placebo groups was slightly higher in patients previously treated with ICS (34.9% and 38.0%, respectively) compared with those not previously receiving ICS before the study (26.8% and 33.9%, respectively). In contrast, patients in the budesonide/formoterol 320/9-μg group previously receiving ICS had a lower discontinuation rate compared with those not previously receiving ICS (23.1% vs 34.1%, respectively). In the budesonide/formoterol 160/9-μg group, the percentage of patients who discontinued the study was similar for those who were previously receiving an ICS (29.5%) and those who were not (28.2%).

Patient disposition. AE = adverse event; bid = twice daily; BUD = budesonide; DPI = dry powder inhaler; FM = formoterol; med = medication; PL = placebo; pMDI = pressurized metered-dose inhaler.
Most demographic and disease characteristics at baseline were similar across treatments (table I). However, in the serial spirometry subset (n = 491), mean percentage reversibility was greater in the budesonide/formoterol 320/9 μg (19.9%), budesonide/formoterol 160/9 μg (20.7%) and placebo (19.5%) groups versus formoterol (16.9%). COPD severity[1] was moderate in 17.8%, severe in 60.4% and very severe in 21.5% of patients. Common co-morbid conditions included hypertension (41.6%), lipid profile abnormalities (22.0%), cardiac disease (17.7%), diabetes mellitus (11.0%), osteoporosis (10.5%), cataracts (5.2%), atrial fibrillation/arrhythmia (4.3%) and congestive cardiac failure (2.9%).

Patient demographic and baseline clinical characteristics of randomized patients
Efficacy Evaluations
Pulmonary Function
Co-Primary Assessments
Improvements in pre-dose FEV1 were significantly greater for budesonide/formoterol 320/9 μg compared with formoterol (primary comparison; p = 0.008) and for both budesonide/formoterol dosages compared with placebo (p < 0.001) [figure 3a]. Although improvements in 1-hour post-dose FEV1 were significantly greater for both budesonide/formoterol dosages compared with placebo (primary comparison; p < 0.001), budesonide/formoterol 320/9 μg also resulted in significantly greater improvements compared with formoterol (p = 0.023) [figure 3b]. Improvements from baseline were apparent at the first assessment (pre-dose FEV1 at end of month 1; 1-hour post-dose FEV1 at day of randomization) and overall maintained over the 12-month treatment period for both budesonide/formoterol dosages (figure 3a and b).

Co-primary efficacy endpoints. Least squares mean change from baseline by study visit over the randomized treatment period in (a) pre-dose forced expiratory volume in 1 second (FEV1) and (b) 1-hour post-dose FEV1. BUD = budesonide; DPI = dry powder inhaler; FM = formoterol; pMDI = pressurized metered-dose inhaler. * p < 0.001 vs placebo; † p ≤ 0.023 vs FM.
Secondary Assessments
Morning and evening PEF improved significantly for all active treatments compared with placebo (p ≤ 0.012) and for both budesonide/formoterol dosages compared with formoterol (p ≤ 0.017) [table II]. Results for pre-dose and 1-hour post-dose FVC are presented in the online supplement (see figures S1a and b in the supplementary material [‘ArticlePlus’] at http://links.adisonline.com/DGZ/A5).

Mean (SD) changes from baseline in additional pulmonary function assessments
In the serial spirometry subset, a ≥15% improvement in FEV1 was observed at 5 minutes after dose administration (first assessment) with all active treatments on the day of randomization (budesonide/formoterol 320/9 μg, 17.7%; budesonide/formoterol 160/9 μg, 20.3%; formoterol 9 μg, 16.5%) and at end of treatment (budesonide/formoterol 320/9 μg, 21.8%; budesonide/formoterol 160/9 μg, 22.4%; formoterol 9 μg, 15.0%) [figures 4a, b and c]. There was no evidence of a diminished effect at end of treatment over the 12-hour period in the budesonide/formoterol groups. Mean FEV1 at 12 hours and baseline-adjusted average 12-hour FEV1 were significantly improved with both budesonide/formoterol dosages compared with placebo on the day of randomization and at end of treatment (p ≤ 0.002) [table II]. Although both budesonide/formoterol dosages demonstrated significant improvements in mean FEV1 at 12 hours and in baseline-adjusted average 12-hour FEV1 compared with formoterol on the day of randomization (p ≤ 0.029), only budesonide/formoterol 320/9 μg demonstrated this effect at the end of treatment (p ≤ 0.004) [table II].

Mean percentage change from baseline in forced expiratory volume in 1 second (FEV1) over 12 hours at randomization and end of treatment (EOT) for (a) budesonide (BUD)/formoterol (FM) 320/9 μg twice daily (bid) vs placebo, (b) BUD/FM 160/9 μg bid vs placebo and (c) FM 9 μg bid vs placebo. DOR = day of randomization; DPI = dry powder inhaler; pMDI = pressurized metered-dose inhaler.
No significant differences in pre-dose IC were observed among the treatment groups (figure 5a). Improvements from baseline to the average over the randomized treatment period in 1-hour post-dose IC were significantly greater with both budesonide/formoterol dosages compared with placebo (p < 0.001) and formoterol (p ≤ 0.018) [figure 5b]. Both dosages of budesonide/formoterol resulted in a 350 mL (≈25%) improvement from baseline to the average over the randomized treatment period in 1-hour post-dose IC, and formoterol resulted in a 210 mL (17%) improvement in 1-hour post-dose IC.

Least squares mean change from baseline by study visit over the randomized treatment period in (a) pre-dose inspiratory capacity (IC) and (b) 1-hour post-dose IC. BUD = budesonide; DPI = dry powder inhaler; FM = formoterol; pMDI = pressurized metered-dose inhaler. * p < 0.001 vs placebo; † p = 0.01 vs FM; ‡ p < 0.05 vs FM; § p < 0.01 vs placebo.
Chronic Obstructive Pulmonary Disease (COPD) Exacerbations
Time to first COPD exacerbation was significantly prolonged with both budesonide/formoterol dosages compared with placebo (p ≤ 0.004) and with budesonide/formoterol 320/9 μg compared with formoterol (p = 0.026) [figure 6]. In addition, significant reductions in the overall number of exacerbations per patient-treatment year were observed with budesonide/formoterol 320/9 μg and 160/9 μg versus placebo (37% and 41%, respectively; p < 0.001) and formoterol (25% and 29%, respectively; p ≤ 0.004) [see figure S2 in the supplementary material]. These reductions were driven by exacerbations treated with oral corticosteroids, the rate of which was reduced with budesonide/formoterol 320/9 μg and 160/9 μg versus placebo (37.5% and 42.9%, respectively) and formoterol (24.1% and 30.6%, respectively) [p ≤ 0.006]. In addition, the percentage of patients in the budesonide/formoterol 320/9 μg, budesonide/formoterol 160/9 μg, formoterol and placebo groups who experienced an exacerbation was greater in those receiving ICS therapy either alone or in combination before the study (33.7%, 38.2%, 39.9% and 40.1%, respectively) compared with those not previously receiving ICS (25.8%, 24.9%, 29.4% and 32.8%, respectively).

Kaplan-Meier probability curve for the time to first chronic obstructive pulmonary disease exacerbation during randomized treatment. BUD = budesonide; DPI = dry powder inhaler; FM = formoterol; pMDI = pressurized metered-dose inhaler. * p ≤ 0.004 vs placebo; † p = 0.026 vs FM.
Health Status and COPD Symptoms
Improvements in SGRQ total score were significantly greater for both budesonide/formoterol dosages compared with placebo (p ≤ 0.006) and for budesonide/formoterol 160/9 μg compared with formoterol (p = 0.006; table III). The improvements from baseline in SGRQ total score in the active treatment groups were not clinically meaningful versus placebo (i.e. decrease of ≥4 points). Significantly greater improvements in COPD symptom variables were observed for both budesonide/formoterol dosages versus placebo for all variables (BCSS, dyspnoea score, cough score, sputum score, sleep score, percentage of awakening-free nights and total daily rescue medication use) [p ≤ 0.003]. Compared with formoterol, mean improvements were significantly greater with budesonide/formoterol 320/9 μg (p ≤ 0.038) for all COPD symptom variables except sputum score and percentage of awakening-free nights, and with budesonide/formoterol 160/9 μg (p ≤ 0.047) for all COPD symptom variables except BCSS, dyspnoea and sputum score (table IV).

Mean changes (SD) in SGRQ total and domain scores from baselinea to end of treatmentb

Mean (SD) changes in chronic obstructive pulmonary disease (COPD) symptom variables from baselinea to the average over the randomized treatment period
Improvements (i.e. reductions) in dyspnoea scores were significantly greater for all active treatments versus placebo (p ≤ 0.003) and for budesonide/formoterol 320/9 μg versus formoterol (p = 0.032; table IV). All active treatment arms demonstrated ≥0.2 points (minimal important difference) change from baseline, but only budesonide/formoterol 320/9 μg had an increase of ≥0.2 points over placebo. A significantly (p < 0.001) greater percentage of patients in both budesonide/formoterol groups and in the formoterol group experienced clinically meaningful improvements in dyspnoea versus placebo (see figure S3 in the supplementary material).
Safety Evaluations
Mean treatment exposure was lowest for placebo (270 days) compared with budesonide/formoterol 320/9 μg (305 days), budesonide/formoterol 160/9 μg (299 days) and formoterol (289 days). The most commonly reported AE (irrespective of causality) was COPD, which had a lower incidence with budesonide/formoterol 320/9 μg versus placebo and a slightly higher incidence with budesonide/formoterol 160/9 μg versus placebo (table V). AEs considered by the investigator to be related to study medication were generally similar among treatment groups with the most commonly reported being oral candidiasis, COPD and dysphonia (see table SI in the supplementary material).

Overall adverse events (AEs) [irrespective of relationship to study medication] reported by ≥3% of patients
The total incidence of pneumonia-related AEs (pneumonia, bronchopneumonia, lobar pneumonia and pneumonia staphylococcal) was similar for budesonide/formoterol 320/9 μg and 160/9 μg (4.0% and 3.4%, respectively) compared with formoterol (3.4%) and placebo (5.0%) [see table SII in the supplementary material]. For potential lung infections other than pneumonia, the incidence was slightly higher in the active treatment groups versus placebo, which was driven largely by bronchitis (see table SII in the supplementary material). AEs typically or potentially associated with local and systemic effects of inhaled corticosteroids (local effects: aphonia, dysphonia, oral candidiasis and candidiasis; systemic effects: weight gain, adrenal suppression, ocular effects, skin effects, psychiatric disorder, diabetes control, thirst, taste effects and bone effects) were more frequent with budesonide/formoterol 320/9 μg (10.3% and 4.0%, respectively) and budesonide/formoterol 160/9 μg (5.7% and 4.5%, respectively) than formoterol (0.6% and 2.6%, respectively) and placebo (2.5% and 2.7%, respectively). The overall incidence of LABA class effects (i.e. tremor, palpitation, tachycardia, potassium changes, glucose changes, headache, agitation, anxiety, sleep effects and muscle cramp) was low, but higher among active treatment groups (budesonide/formoterol 320/9 μg [9.5%], budesonide/formoterol 160/9 μg [8.9%] and formoterol [6.5%]) versus placebo (4.8%). The incidence of cardiac-related AEs was higher for all active treatments (10.5–11.3%) versus placebo (6.9%). The most common cardiac-related AEs were hypertension and angina pectoris (budesonide/formoterol 320/9 μg [2.4% and 1.2%, respectively], budesonide/formoterol 160/9 μg [1.6% and 0.8%, respectively], formoterol [2.6% and 1.8%, respectively] and placebo [2.1% and 1.0%, respectively]). Although the number of atrial fibrillation AEs was low, all cases occurred within the active treatment groups.
Discontinuations due to AEs (DAEs) occurred in 238 patients, with a similar incidence observed across treatment groups (range 11.3–12.5%) [see table SIII in the supplementary material]. The most common DAE was COPD, which was highest in the formoterol group (7.3%) and lowest in the budesonide/formoterol 320/9 μg group (4.0%); the incidence was similar in the budesonide/formoterol 160/9 μg (6.1%) and placebo (6.0%) groups (see table SIII in the supplementary material). The overall incidence of non-fatal serious adverse events (SAEs) was highest with formoterol (17.8%) compared with the other treatment groups and higher with budesonide/formoterol 320/9 μg (15.6%) and budesonide/formoterol 160/9 μg (13.6%) versus placebo (12.1%) [see table SIV in the supplementary material]. The most common non-fatal SAEs (occurring in five or more patients [all treatment groups combined]) were COPD (6.8%), pneumonia (1.5%), atrial fibrillation (0.5%), angina pectoris (0.3%), bronchitis (0.3%) and coronary artery disease (0.3%). These non-fatal SAEs occurred at a similar incidence in all treatment groups, except for COPD, which was slightly more common in the active treatment groups (budesonide/formoterol 320/9 μg [7.1%], budesonide/formoterol 160/9 μg [6.7%] and formoterol [7.9%]) versus placebo (5.6%). Fifteen patients died during the randomized treatment period with no imbalance across treatments: three in the budesonide/formoterol 320/9 μg group; six in the budesonide/formoterol 160/9 μg group; two in the formoterol group; and four in the placebo group. Fifteen patients died after the randomized treatment period (≥2 days after stopping study treatment) with no imbalance across treatments: five in the budesonide/formoterol 320/9 μg group; two in the budesonide/formoterol 160/9 μg group; four in the formoterol group; and four in the placebo group. None of the deaths were considered drug related by the investigator.
Clinically significant changes in vital signs, 24-hour urinary cortisol, and 12-lead ECGs, QT interval and Holter assessments were rare. The geometric mean values of 24-hour urinary cortisol at 6 months and end of treatment were lower in both budesonide/formoterol dosage groups compared with formoterol and placebo (see table SV in the supplementary material). Significant differences in 24-hour urinary cortisol were observed between the budesonide/formoterol 320/9 μg and placebo groups (p = 0.035) at month 6 and between the budesonide/formoterol 320/9 μg and formoterol groups (p = 0.044) at end of treatment.
Corrected QT (QTc) intervals ≥450 msec were experienced by similar percentages of patients across all treatment groups, whereas few patients experienced QTc intervals ≥500 msec or a QTc change ≥60 msec (see table SVI in the supplementary material). Data from ECG and Holter recordings showed a similar incidence of new onset atrial fibrillation in the active and placebo groups.
BMD was stable over the 1-year study period in all treatment groups, with small but statistically significant differences in changes from baseline observed for budesonide/formoterol 320/9 μg compared with all other treatments for total lumbar spine BMD (p ≤ 0.037) and for budesonide/formoterol 320/9 μg versus formoterol for total hip BMD (p = 0.012) [see table SVII in the supplementary material]. The differences between the treatment groups in changes from baseline in BMD were of unclear clinical relevance, as the geometric least squares mean ratios for these treatment group comparisons were close to 1 (range 0.98–0.99).
Minor increases in lenticular opacities and intraocular pressure were noted across all treatment groups in the ophthalmological analysis subset; the magnitude of these changes was similar among the treatment groups (see table SVIII in the supplementary material). Small but significant differences were observed for the change from baseline in posterior subcapsular score between the budesonide/formoterol groups (p = 0.022). Clinically significant changes in ophthalmological assessment were infrequent.
Discussion
In this study, both budesonide/formoterol dosage strengths resulted in significant improvements from baseline over the randomized treatment period in pulmonary function, health status and COPD symptoms, as measured by dyspnoea, cough and sputum production, and a clinically significant reduction in COPD exacerbation rate compared with placebo. In addition, both budesonide/formoterol pMDI dosages were well tolerated relative to formoterol alone and placebo.
The results of this 12-month study confirm those from the earlier related 6-month study by Tashkin et al.[4] and are consistent with those from studies of budesonide/formoterol DPI.[2,3] Taken together, the results demonstrate that this ICS/LABA combination provides benefits beyond formoterol alone in the treatment of COPD. The contribution of budesonide to improvements from baseline in pulmonary function and COPD symptoms was demonstrated by the greater efficacy of budesonide/formoterol 320/9 μg compared with formoterol alone for the co-primary variables of pre-dose FEV1 and 1-hour post-dose FEV1, and for dyspnoea and total BCSS scores, supporting use of the higher-dose combination for COPD symptoms. Both budesonide/formoterol dosage strengths demonstrated greater improvements from baseline compared with formoterol alone in morning and evening PEF, 12-hour FEV1, symptom scores and rescue medication use.
In addition, the 25–29% reduction in exacerbation rate observed with both budesonide/formoterol dosage strengths beyond the substantial reduction achieved with formoterol alone further demonstrates the important contribution of budesonide to the combination product. The magnitude of the reduction in exacerbation rates reported in this study is similar to that reported previously for fixed-dose regimens of budesonide/formoterol 320/9 μg twice daily[2–4] and fluticasone propionate/salmeterol[11–13] relative to LABA alone. A key difference between this 12-month study and the 6-month study of similar design reported by Tashkin et al.[4] is that this study was powered to show a difference in exacerbations, while the 6-month study was not. This study demonstrated a significant reduction in exacerbation rates in patients treated with budesonide/formoterol 320/9 μg and 160/9 μg compared with those receiving formoterol or placebo.
The fact that a greater percentage of patients in the formoterol and placebo groups who previously received ICS therapy, either alone or in combination, experienced exacerbations compared with those who did not previously receive ICS therapy may result in a skewing of the data; however, a similar response was observed in combination budesonide/formoterol patients with a greater percentage of patients who previously received ICS experiencing exacerbations compared with patients who were not previously treated with ICS therapy. Similarly, a greater percentage of patients who previously received ICS therapy discontinued in the formoterol and placebo groups than those who did not previously receive ICS therapy; however, this trend was reversed in the combination budesonide/formoterol groups.
The long-term nature of this study allowed for a more thorough assessment of tolerability compared with the 6-month study.[4] No differences in pneumonia incidence were observed among treatment groups in this study or in the 6-month study, despite a shorter exposure time in the placebo group.[4] These results contrast with previous COPD studies that showed an increased incidence of pneumonia with fluticasone propionate/salmeterol treatment.[11–14] However, in this study and the 6-month study,[4] the incidence of potential lung infections other than pneumonia was slightly higher in all active treatments compared with placebo; these differences were largely driven by an increased incidence of bronchitis. The incidence of SAEs, including COPD SAEs, was slightly higher in both budesonide/formoterol groups compared with placebo. This finding may be attributed to the earlier discontinuation in the placebo group, which resulted in a shorter study drug exposure (35 and 29 fewer days vs budesonide/formoterol 320/9 μg and 160/9 μg, respectively). Although the overall incidence of atrial fibrillation, a known LABA class effect, was small (1.1%), an imbalance was noted between the formoterol-containing arms and placebo. However, no differences in new-onset atrial fibrillation were apparent among groups based on 24-hour Holter monitoring or ECG data collected at repeated visits during the randomized treatment period. Overall, the safety profile of budesonide/formoterol pMDI in this 12-month study was similar to that reported in the 6-month study by Tashkin et al.,[4] suggesting no increase in safety concerns with a longer duration of treatment.
Conclusions
Both dosage strengths of budesonide/formoterol pMDI (320/9 μg and 160/9 μg) were effective and well tolerated over 1 year of treatment in this population of patients with moderate to very severe COPD. In addition, budesonide/formoterol pMDI 320/9 μg demonstrated greater efficacy compared with formoterol for pre-dose FEV1, suggesting that the higher dosage strength containing twice the daily dose of budesonide is appropriate in this patient population.
References
Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: updated 2007 [online]. Available from URL: http://www.goldcopd.com. [Accessed 2008 Feb 12]
Calverley PM, Boonsawat W, Cseke Z, et al. Maintenance therapy with budesonide and formoterol in chronic obstructive pulmonary disease. Eur Respir J 2003 Dec; 22(6): 912–9
Szafranski W, Cukier A, Ramirez A, et al. Efficacy and safety of budesonide/formoterol in the management of chronic obstructive pulmonary disease. Eur Respir J 2003 Jan; 21(1): 74–81
Tashkin DP, Rennard SI, Martin P, et al. Efficacy and safety of budesonide and formoterol in one pressurized metered-dose inhaler in patients with moderate to very severe chronic obstructive pulmonary disease. Drugs 2008; 68(14): 1975–2000
Miller CJ, Senn S, Mezzanotte WS. Bronchodilation of formoterol administered with budesonide: device and formulation effects. Contemp Clin Trials 2008 Mar; 29(2): 114–24
American Thoracic Society. Standardization of spirometry, 1994 update. Am J Respir Crit Care Med 1995 Sep; 152(3): 1107–36
Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis 1981 Jun; 123(6): 659–64
Leidy NK, Schmier JK, Jones MKC, et al. Evaluating symptoms in chronic obstructive pulmonary disease: validation of the Breathlessness, Cough and Sputum Scale. Respir Med 2003 Jan; 97 Suppl. A: S59–70
Jones PW, Quirk FH, Baveystock CM, et al. A self-complete measure of health status for chronic airflow limitation. The St George’s Respiratory Questionnaire. Am Rev Respir Dis 1992 Jun; 145(6): 1321–7
Jones PW. St George’s Respiratory Questionnaire: MCID. COPD 2005 Mar; 2(1): 75–9
Kardos P, Wencker M, Glaab T, et al. Impact of salmeterol/fluticasone propionate versus salmeterol on exacerbations in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2007 Jan 15; 175(2): 144–9
Ferguson GT, Anzueto A, Fei R, et al. Effect of fluticasone propionate/salmeterol (250/50 μg) or salmeterol (50 μg) on COPD exacerbations. Resp Med 2008 Aug; 102(8): 1099–108
Wedzicha JA, Calverley PMA, Seemungal TA, et al. The prevention of chronic obstructive pulmonary disease exacerbations by salmeterol/fluticasone propionate or tiotropium bromide. Am J Respir Crit Care Med 2008 Jan 1; 177(1): 19–26
Calverley PMA, Anderson JA, Celli B, et al. Salmeterol and fluticasone propionate and survival in chronic obstructive pulmonary disease. N Engl J Med 2007 Feb 22; 356(8): 775-89
Acknowledgements
The authors acknowledge Lisa M. Klumpp, PhD, Cynthia Gobbel, PhD, and Lisa Feder, PhD, from Scientific Connexions (Newtown, PA, USA) for writing assistance funded by AstraZeneca LP, the study sponsor, which designed the study and participated in the management, analysis and interpretation of the data and in the review and approval of the article. The study was supported by AstraZeneca LP, Wilmington, DE, USA.
Dr Tashkin has served as consultant for AstraZeneca, Boehringer-Ingelheim, Dey Laboratories and Schering-Plough; received honoraria from AstraZeneca, Boehringer-Ingelheim and Dey Laboratories; and received or will receive grants from Almirall, AstraZeneca, Boehringer-Ingelheim, Dey Laboratories, GlaxoSmithKline, NIH, Novartis, Pfizer, Sepracor and Forest Laboratories. Dr Rennard has served as a consultant or on the advisory board for Adams, Almirall, Altana, APT, Aradigm, AstraZeneca, Bend, Biolipox, Centocor, Critical Therapeutics, Dey, GlaxoSmithKline, ICOS, Johnson & Johnson, Novartis, Nycomed, Ono Pharma, Perengenix, Pfizer, Pharmaxis, Roche, Sankyo, Sanofi, Schering-Plough and Talecris; has served as a speaker for AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Otsuka and Pfizer; and has received grants from Almirall, Altana, Astellas, AstraZeneca, Biomark, Centocor, GlaxoSmithKline, Mpex, Nabi, Novartis and Pfizer. Ms McElhattan and Dr Goldman are employees of AstraZeneca LP. Dr Ramachandran and Dr Martin hold stock in AstraZeneca LP and are employees of AstraZeneca LP. Dr Silkoff is a former employee of AstraZeneca LP.
Investigator List:
We thank the following investigators for their participation in the study: Bulgaria: Vasil Dimitrov, Sofia; Ognian Georgiev, Sofia; Yavor Ivanov, Pleven; Grozdanka Ivanova, Plovdiv; Krassimira Kissiova, Varna; Rossen Marinov, Sofia; Hristo Metev, Russe; Dencho Osmanliev, Sofia; Dimitar Popov, Sofia; Denmark: John Arnved, Copenhagen; Vibeke Backer, Copenhagen; Asger Dirksen, Hellerup; Flemming Egede, Svendborg; Henriette Enevoldsen, Copenhagen; Niels Frølich, Viborg; Michael Hansen, Frederikssund; Michael Jensen, Skive; Kim Mølenberg, Aalborg; Carl Nielsen, Frederikshavn; Povl Revsbech, Silkeborg; Dan Rost, Christiansfeld; Torben Sørensen, Vaerløse; Germany: Helen Arievich, Berlin; Klaus Colberg, Bad Segeberg; Regina Deckelmann, Leipzig; Jörg Eller, Berlin; Wolfram Feussner, Kassel; Karin Förster, Berlin; Adrian Gillisssen, Leipzig; Ulf Harnest, Munich; Andrea Koch, Cologne; Marc Oliver Kornmann, Mainz; Thea Krecker, Bad Salzungen; Ludger Lindemann, Gelsenkirchen; Anneliese Linnhoff, Berlin; Helgo Magnussen, Grosshansdorf; Petra Mikloweit, Geesthacht; Wolfgang Mitlehner, Berlin; Stephan Molitor, Hanover; Herbert Querfurt, Bochum; Gisela Schirrmeister, Postdam; Sören Schmidtmann, Berlin; Wwinfried Schröder-Babo, Gelnhausen; Wolfgang Schürmann, Marburg; Peter-Michael Stutz, Bonn; Hermann Trauth, Marburg; Bernd-Georg Trümper, Erfurt; Anette Weihrich, Neuruppin; Greece: Konstantionos Gourgoulianus, Larisa; Marianna Kakoura, Thessaloniki; Adonis Rasidakis, Athens; Nicolaos Siafakas, Hraklion-Crete; Lazaros Sichletidis, Thessaloniki; Michael Toumbis, Athens; Hungary: Anna Bartha, Siófok; Márta Bisits, Tatabánya; István Boros, Deszk; György Böszörményi Nagy, Budapest; Mária Csányi, Gyöngyös; Zsuzsanna Cseke, Szombathely; Ilona Dalmadi, Keszthely; Radeczky Éva, Százhalombatta; Eva Farkas, Gödöllö; Katalin Gömöri, Balassagyarmat; Zsuzsa Györi, Érd; László Hajdu, Kiskunhalas; Mária Hegedüs, Salgótarján; Mária Jedlinszki, Szarvas; Teréz Kecskés, Pécs; Gábor Kelen, Füzesabony; Tamás Major, Mosdós; Ida Nagy, Jászberény; Miklós Naményi, Veszprém; Emil Prugberger, Sárvár; Katalin Puha, Györ; Katalin Radich, Budapest; Katalin Rajkay, Cegléd; Mária Schreiner, Pécs; Zsuzsanna Szalai, Mosonmagyaróvár; Gyöngyvér Szalóky, Gyula; Sándor Tehenes, Zalaegerszeg; Iceland: Magni Jónsson, Reykjavík; Andrés Sigvaldason, Reykajvík; Mexico: Eliseo Espinoza, Mexico City; Norma Martínez, Mexico City; Raul Ortiz Peregrina, Guadalajara; Alicia Ramirez Rivera, Monterrey; Enrique Rojas, Mexico City; Romania: Ioana Agache, Brasov; Nicoleta Bisca, Bucharest; Ioan Dorin Petrui, Deva; Traian Mihaescu, Iasi; Angelica Savu, Bucharest; Octavian Verescu, Bucharest; USA: Connie Abarikwu, Phoenix, AZ; Salim Abou-Jaoude, Youngstown, OH; John Adams, Houston, TX; Lawrence Alwine, Downingtown, PA; C. Lynn V Anderson, Bay Pines, FL; Charles Andrews, San Antonio, TX; Raiqua Arastu, San Antonio, TX; William Bailey, Birmingham, AL; Jose Bardelas, High Point, NC; Kendrick Bashor, Mogadore, OK; George Bensch, Stockton, CA; David Bernstein, Cincinnati, OH; Eugene Bleecker, Winston-Salem, NC; Vinko Bogdanic, Waterloo, IA; Joseph Boscia, Gaffney, SC; Sidney Braman, Providence, RI; Wesley Bray, Marietta, GA; Shari Anne Brazinsky, San Diego, CA; Dwayne Brown, Lafayette, LA; Jaine Brownell, Omaha, NE; Timothy Bruya, Spokane, WA; John Burk, Fort Worth, TX; Anthony Cagino, Albany, NY; Brian Cain, Mogadore, OK; Edward Campbell, Provo, UT; Sammy Campbell, Tucson, AZ; Francisco Candal, Slidell, LA; Brian W. Carlin, Pittsburgh, PA; Wissam Chatila, Philadelphia, PA; Jim Christensen, Las Vegas, NV; Dariusz Chrostowski, Ithaca, NY; John Condemi, Rochester, NY; Clinton Corder, Oklahoma City, OK; Arthur Degraff, Hartford, CT; Edward Diamond, Elk Grove Village, IL; Joel Diamond, Pittsburgh, PA; Mark Dransfield, Birmingham, AL; Gustav A. Dubois, Birmingham, AL; Leonard Dunn, Clearwater, FL; Gordon Eck, Elverson, PA; Darlene Elias, La Jolla, CA; Steven Elliott, Evansville, IN; David Erb, Union, SC; Barry Feinstein, Shreveport, LA; Gregory Feldman, Spartanburg, SC; Roan L. Flenniken, Shreveport, LA; Charles Fogarty, Spartanburg, SC; Toby Free, McCook, NE; Eli Freilich, Clearwater, FL; Fred Garfinkel, Tulsa, OK; Sandra Gawchik, Upland, PA; Harry Geisburg, Anderson, SC; William George, Cadillac, MI; Ronald Gilman, East Providence, RI; Joan Gluck, Miami, FL; Daniel Goldberger, Portage, MI; Gary Greenwald, Rancho Mirage, CA; Gary Gross, Dallas, TX; Roger Guthrie, Pismo Beach, CA; James Haden, Fort Worth, TX; Jeffrey Hammersley, Toledo, OH; James Hansbrough, Bowling Green, KY; Duane Harris, Salt Lake City, UT; James Harris, South Bend, IN; David Hassman, Berlin, NJ; Delmer Henninger, Murrieta, CA; Ira Horowitz, Cherry Hill, NJ; Gary Hunt, Blue Ridge, GA; Sean Hurley, Spokane, WA; Thomas Hyers, St. Louis, MO; Thomas Kaelin, Charleston, SC; Richard Kahn, Biddleford, ME; Harold Kaiser, Minneapolis, MN; Jill Karpel, New Hyde Park, NY; F. Adam Kawley, Houston, TX; Edward Kerwin, Minneapolis, MN; W. Ken Kleinsteuber, Louisville, KY; Andras Koser, Greenville, SC; Ritsu Kuno, Richmond, VA; Ware Kuschner, Palo Alto, CA; Mark Kutner, Miami, FL; David Laman, Pittsburgh, PA; Lauro Lapuz, Henderson, NV; Michael Lawrence, Taunton, MA; Dennis Ledford, Tampa, FL; Theodore Lee, Atlanta, GA; Lyndon Mansfield, El Paso, TX; Dennis McCluskey, Mogadore, OK; Arturo Meade, Fort Smith, AR; James Meli, Henderson, NV; Edward Millermaier, Portage, MI; Martin Mollen, Phoenix, AZ; Melvin Morganroth, Portland, OR; Timothy Moriarty, Panama City, FL; James Moy, Chicago, IL; Anjuli Nayak, Normal, IL; Brooke Nevins, Bronxville, NY; Michael Noonan, Portland, OR; Amir Ali Noorani, New Port Richey, FL; Eric Overland, Medford, OR; Parimal Parikh, Kenner, LA; Amit Patel, Riverside, CA; Rafael Perez, Decatur, GA; Stephen Pollard, Louisville, KY; Rana Rab-Hasan, Decatur, GA; Albert Razzetti, DeLand, FL; Frank Reda, Pittsburgh, PA; Emory Robinette, Abingdon, VA; Christine Rohr, Saginaw, MI; Bradley Sakran, O’Fallon, IL; Paul J. Scheinberg, Atlanta, GA; E. Joe Schelbar, Tulsa, OK; Robert Schilz, Cleveland, OH; Nathan Segall, Stockbridge, GA; Pedro Sepulveda, San Antonio, TX; Paul Shapero, Bangor, ME; Amir Sharafkhaneh, Houston, TX; Mandy Shaw, Omaha, NE; Tanna Shaw, Oklahoma City, OK; Mike Shedd, Scottsbluff, NE; T. Shull Lemire, Missoula, MT; Thomas Siler, St. Charles, MO; Wayne Sinclair, Missoula, MT; Deren M. Sinkowitz, Torrance, CA; Emil Skobeloff, Swarthmore, PA; Andrew Slaski, Tucson, AZ; Christopher Smith, Ithaca, NY; Gregory Smith, Sacramento, CA; Philip Snell, Greer, SC; John Southard, Council Bluffs, IA; Michael Spandorfer, Charleston, SC; Sheldon Spector, Los Angeles, CA; Mark Stang, Edina, MN; Ronald Stegemoller, Avon, IN; Clement Strumil, Las Vegas, NV; Patrick Sturm, Beaver, PA; Raul Tamayo, Longwood, FL; George Tannoury, Pahrump, NV; Gary Tarshis, Colorado Springs, CO; Stuart Thorson, Medford, OR; Melvin Tonkon, Santa Ana, CA; Diego Torres, Ormond Beach, FL; Miguel Trevino, Largo, FL; John Updegrove, Corsicana, TX; Robert Venuti, Turnersville, NJ; Thomas Wade, Indianapolis, IN; John Weiler, Iowa City, IA; Jan Westerman, Jasper, AL; Douglas Young, Carmichael, CA.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Rennard, S.I., Tashkin, D.P., McElhattan, J. et al. Efficacy and Tolerability of Budesonide/Formoterol in One Hydrofluoroalkane Pressurized Metered-Dose Inhaler in Patients with Chronic Obstructive Pulmonary Disease. Drugs 69, 549–565 (2009). https://doi.org/10.2165/00003495-200969050-00004
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003495-200969050-00004