
Abstract
Activation of inflammatory pathways is known to accompany development of obesity-induced nonalcoholic fatty liver disease (NAFLD), insulin resistance and type 2 diabetes. In addition to caspase-1, the neutrophil serine proteases proteinase 3, neutrophil elastase and cathepsin G are able to process the inactive proinflammatory mediators interleukin (IL)-1β and IL-18 to their bioactive forms, thereby regulating inflammatory responses. In this study, we investigated whether proteinase 3 is involved in obesity-induced development of insulin resistance and NAFLD. We investigated the development of NAFLD and insulin resistance in mice deficient for neutrophil elastase/proteinase 3 and neutrophil elastase/cathepsin G and in wild-type mice treated with the neutrophil serine proteinase inhibitor human α-1 antitrypsin. Expression profiling of metabolically relevant tissues obtained from insulin-resistant mice showed that expression of proteinase 3 was specifically upregulated in the liver, whereas neutrophil elastase, cathepsin G and caspase-1 were not. Neutrophil elastase/proteinase 3-deficient mice showed strongly reduced levels of lipids in the liver after being fed a high-fat diet. Moreover, these mice were resistant to high-fat-diet-induced weight gain, inflammation and insulin resistance. Injection of proteinase 3 exacerbated insulin resistance in caspase-1−/− mice, indicating that proteinase 3 acts independently of caspase-1. Treatment with α-1 antitrypsin during the last 10 d of a 16-wk high-fat diet reduced hepatic lipid content and decreased fasting glucose levels. We conclude that proteinase 3 is involved in NAFLD and insulin resistance and that inhibition of proteinase 3 may have therapeutic potential.
Similar content being viewed by others


Introduction
Nonalcoholic fatty liver disease (NAFLD) refers to a disease spectrum ranging from simple steatosis with no inflammation through hepatic necroinflammation (nonalcoholic steatohepatitis) to cirrhosis (1). It is strongly associated with obesity, insulin resistance and type 2 diabetes (T2D) and has become the most frequent liver disease worldwide (1,2).
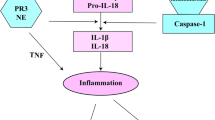
Over the last decade, it has become clear that inflammation has an important role in obesity and NAFLD (2). However, the detailed underlying mechanisms of obesity-induced inflammation are not fully understood. Important modulatory effects on both immunity and metabolism have been attributed to tumor necrosis factor (TNF), interleukin (IL)-1β and IL-18 (3). Many cytokines of the IL-1 family are secreted as inactive precursors and require processing by proteases. One protease able to process both pro-IL-1β and pro-IL-18 to their bioactive forms is caspase-1, a cysteine protease regulated by a protein complex called the inflammasome. Indeed, recent studies showed that mice deficient for caspase-1 or individual components of the inflammasome have improved insulin sensitivity and decreased liver steatosis in diet-induced obesity models (4,5). However, production of active IL-1β and IL-18 is not exclusively dependent on caspase-1. Other proteases highly important for processing of cytokine precursors are the neutrophil serine proteases. These proteases, namely neutrophil elastase, cathepsin G and proteinase 3, are released by neutrophils and are able to cleave/process membrane-bound TNF, mature TNF, pro-IL-1β and pro-IL-18, thereby regulating inflammatory responses (Figure 1). It is also known that a large proportion of the released serine proteases bind to the plasma membrane with their catalytic activity preserved (6,7). Recent studies showed that mice deficient for neutrophil elastase are resistant to high-fat-diet-induced body-weight gain, insulin resistance (8,9) and fatty liver (9). Relatively little is known about proteinase 3 in relation to insulin resistance and NAFLD. One study reported that a single injection of commercially available proteinase 3 induced insulin resistance in healthy mice (10). However, endogenous proteinase 3 has not been investigated for its capability to induce insulin resistance and NAFLD in diet-induced obese animal models.

The role of neutrophil serine proteases in inflammation. Following activation, neutrophils will release their serine proteases in the extracellular environment. A portion of these released serine proteases will bind to the membrane. Once released, serine proteases (both membrane-bound and in the extracellular environment) are able to process (activate and/or inactivate) cytokines, thereby regulating inflammation. Human α-1 antitrypsin has potent anti-inflammatory properties. It complexes with proteinase 3, neutrophil elastase and cathepsin G, thereby preventing their enzymatic activities. It also inhibits caspase-1 and enhances the production of the anti-inflammatory mediators IL-10 and IL-1-Ra. All these actions result in a strong anti-inflammatory effect. PR3, proteinase 3; NE, neutrophil elastase; CG, cathepsin G; Casp-1, caspase-1; IL-1Ra, interleukin 1 receptor antagonist.
Proteinase 3 is strongly inhibited by the human protease inhibitor α-1 antitrypsin, a multifunctional protein with potent anti-inflammatory properties. It is primarily synthesized in the liver but is also expressed in other cell types including neutrophils, monocytes and macrophages. Besides its capacity to inhibit proteinase 3, α-1 antitrypsin also blocks neutrophil elastase and cathepsin G (Figure 1) (11), as well as caspase-1 and caspase-3 (12,13). In addition, α-1 antitrypsin enhances the production of anti-inflammatory mediators such as IL-10 and IL1-Ra (14). In light of the recent evidence that both the caspase-1 and the neutrophil serine proteases are important for inducing insulin resistance and NAFLD, α-1-antitrypsin treatment might also be beneficial for the treatment of these conditions. In this study, we engaged a number of in vivo approaches to pursue the hypothesis that endogenous proteinase 3 is important in the induction of NAFLD and insulin resistance in mouse models of diet-induced obesity and to assess whether treatment with α-1 antitrypsin protects against obesity-induced development of NAFLD and insulin resistance.
Materials and Methods
Animals
Casp-1−/−, ela−/−/ctsg−/− (neutrophil elastase/cathepsin G double knock-out mice; NE−/−/CG−/−) and ela−/−/prtn3−/− (neutrophil elastase/proteinase 3 double knock-out mice; NE−/−/PR3−/−) were back-crossed to C57BL/6J background for at least 10 generations and have been described previously (15,16). Age-matched wild-type C57BL/6J mice were used as controls and obtained from Charles River or Jackson Laboratories. Mice were housed in a pathogen-free facility and maintained at 23°C with water and food ad libitum and a 12 h:12 h light:dark cycle (lights on 6 am to 6 pm). All mice were 10- to 12-wk-old males at the start of diet intervention. Animal studies were approved by the Animal Welfare Committee of the Radboudumc, Nijmegen, The Netherlands and the Washington University School of Medicine, St. Louis, Missouri, USA.
Diet Intervention and Treatment Regime
Male mice received a low-fat diet or a high-fat diet for 16 wks, providing 10% or 45% energy percent in the form of triglycerides (D12450B or D12451; Research Diets). Regarding the experiments in which mice were treated with α-1 antitrypsin or proteinase 3, a subgroup of mice receiving a high-fat diet was divided in bodyweight-matched treatment groups during the last 2 wks of dietary intervention. These mice were injected five times (every other day for 10 d) intraperitoneally (IP) with either α-1 antitrypsin (Zemaira; 2.0 mg per injection) or proteinase 3 (0.25 mg/kg; Athens Research & Technology Inc.) in vehicle (phosphate-buffered saline (PBS)) or vehicle alone. Also the low-fat-diet control group was treated with vehicle (five times every other day) for the last 10 d of dietary intervention. Insulin sensitivity was assessed by insulin tolerance tests and oral glucose tolerance tests. Mice were fasted for 4 h and overnight, respectively, before IP injection with insulin (0.75 U/kg bodyweight) or oral glucose administration (2 g/kg D glucose; Life Technologies). Blood samples were taken by tail-cut at baseline and after 15, 30, 45, 60, 90 and 120 min of insulin or glucose administration. Blood glucose concentrations were measured with a blood glucose meter (Accu-Chek Avia; Roche Diagnostics). At the end of the experiments, blood was collected in heparin-coated tubes and centrifuged to collect plasma. Liver and adipose tissue were weighed and collected for further analysis.
Histology/Immunohistochemistry
Hematoxylin and eosin (HE) staining of sections and detection of F4/80-positive macrophages were done using standard protocols. Quantification of lipid droplets in HE liver sections was performed by using the ImageJ software (http://rsbweb.nih.gov). Macrophages and crown-like structures were counted in four images per sample. Sections were scored in a blinded fashion.
Liver Triglycerides
For measuring liver triglycerides, l0% liver homogenates were prepared in buffer containing 250 nmol/L sucrose, 1 mmol/L EDTA and 10 mmol/L Tris-HCL at pH 7.5. Triglycerides were determined enzymatically following manufacturers’ protocols (Liquicolor; Human GmbH).
RNA Isolation and Quantitative PCR
Total RNA was isolated using TRIzol reagent (Life Technologies Europe BV) and quantity was controlled using a NanoDrop spectrophotometer (NanoDrop Technologies). Total RNA (1 µg) was reverse transcribed (iScript; Bio-Rad Laboratories) and quantitative polymerase chain reaction (qPCR) was performed by SYBR Green-based quantification (Applied Biosystems). Primers were developed using Primer3 (Supplementary Table S1; http://primer3.ut.ee/). Primer pairs were exon-spanning. The genes Gapdh and B2m were chosen as endogenous controls. Differences in expression were calculated by the 2ΔΔCt method (17).
Statistical Analysis
Data are represented as mean ± SEM unless stated otherwise. Data were analyzed using, as appropriate, the Student t test or analysis of variance followed by the Tukey post hoc test (GraphPad Prism 5.0). When necessary, results were log-transformed to normalize the data. A p value <0.05 was considered significant.
All supplementary materials are available online at www.molmed.org .
Results
Proteinase 3 Expression Is Increased in Livers from High-Fat-Diet-Induced Insulin-Resistant Mice
To explore the role of proteinase 3 in diet-induced NAFLD and insulin resistance, we first examined proteinase 3 messenger RNA (mRNA) expression in liver tissues obtained from independent studies investigating NAFLD and insulin resistance in mice. Relative expression of proteinase 3 (Prtn3) in the liver was increased after 4 wks of high-fat-diet feeding (2.7-fold induction; p = 0.0096). Noticeably, caspase-1, neutrophil elastase (Elane) and cathepsin G (Ctsg) gene expression levels were not affected (Figure 2A). In addition, whole-genome expression profiling results showed that proteinase 3 expression was induced more than 80-fold (corrected p = 3.78E-08) in livers from arthritic mice that showed inflammatory-induced insulin resistance. Caspase-1, neutrophil elastase and cathepsin G expression were not affected when compared with controls (18) (microarray data available; www.ebi.ac.uk/arrayexpress; accession number E-MTAB-1765). These results were validated by qPCR analysis (Figure 2B). The observation that proteinase 3 mRNA expression, but not caspase-1, neutrophil elastase and cathepsin G mRNA expression, is upregulated in livers from animals with diet-and inflammation-induced NAFLD and insulin resistance suggests an unique role of this protease in the liver.

Relative mRNA expression of proteinase 3, caspase-1, neutrophil elastase and cathepsin G in liver. (A) Relative mRNA expression of proteinase 3, caspase-1, neutrophil elastase and cathepsin G after 4 wks of diet intervention. (B) Relative mRNA expression of proteinase 3, caspase-1, neutrophil elastase and cathepsin G in nonarthritic and arthritic mice. PR3, proteinase 3; NE, neutrophil elastase; CG, cathepsin G; LFD, low-fat diet; HFD, high-fat diet; **significant difference p ≤ 0.01; NS, nonsignificant.
NE−/−/CG−/− and NE−/−/PR3−/− Mice Have Reduced Levels of Lipids in the Liver as well as Improved Insulin Sensitivity and Glucose Tolerance, Independently of Bodyweight
To further investigate the role of neutrophil serine proteases, especially proteinase 3, in diet-induced NAFLD and insulin resistance, we performed a 16-wk high-fat-diet intervention study in which wild-type mice and animals deficient for ela/ctsg (NE−/−/CG−/−) and ela/prtn3 (NE−/−/PR3−/−) were compared (16).
Before the start of the diet intervention, no differences in bodyweight were observed between the three genotypes. High-fat-diet feeding induced weight gain in all groups, although weight gain after high-fat diet was significantly less in both knock-out mice as compared with the wild-type animals (Figure 3A). No differences in weight gain were observed between the two double knockout genotypes. No differences in food intake were observed between the groups.

Improved NAFLD status and metabolic profiles in NE−/−/CG−/− and NE−/−/PR3−/− mice after a high-fat diet. (A) Bodyweight development of wild-type (n = 10), NE−/−/CG−/− (n = 15) and NE−/−/PR3−/− (n = 15) mice during diet intervention. (B) Insulin tolerance test and ITT AUC values, oral glucose tolerance test and oGTT AUC values of wild-type, NE−/−/CG−/− and NE−/−/PR3−/−. (C) Plasma insulin levels. (D) HE staining of liver sections. (E) Percentage of lipid droplets in HE-stained liver sections, liver weights and liver triglycerides levels. (F) Relative mRNA expression of Cd36, Fabp1, Dgat1, Fas, Ppar α and Cpt1a in liver. HFD, high-fat diet; WT, wild-type; ITT, insulin tolerance test; oGTT, oral glucose tolerance test; TG, triglycerides; *significant difference p ≤ 0.05; “significant difference p ≤ 0.01; ***significant difference p ≤ 0.001.
To assess insulin sensitivity, insulin tolerance tests and oral glucose tolerance tests were performed. Both NE−/−/CG−/− and NE−/−/PR3−/− mice were more insulin sensitive after high-fat diet when compared with wild-type mice. Insulin tolerance test area under the curve (AUC) values were significantly lower in NE−/−/CG−/− (p < 0.0001) and NE−/−/PR3−/− mice (p < 0.0001) when compared with the wild-type controls. Also the oral glucose tolerance tests AUC values were decreased in NE−/−/CG−/− (p = 0.02) and NE−/−/PR3−/− mice (p < 0.0001) when compared with those of wild-type mice, indicating improved glucose tolerance. Blood glucose levels were lower in NE−/−/PR3−/− mice after 120 min during the insulin tolerance test and after 60 min during the oral glucose tolerance test when compared with those of NE−/−/CG−/− mice (p = 0.0034 and p = 0.0374, respectively). However, differences in AUC values between NE−/−/CG−/− and NE−/−/PR3−/− mice were not significant for both the insulin tolerance test and the oral glucose tolerance test (Figure 3B). No differences in plasma insulin levels were observed between the three groups (Figure 3C).
NAFLD was assessed by quantifying lipid droplets in HE-stained liver sections. Large differences in the quantity of lipid droplets in the liver were observed between the three genotypes after high-fat diet. Both NE−/−/CG−/− and NE−/−/PR3−/− mice had a significantly reduced liver weight and reduced lipid droplets in the liver when compared with wild-type mice (p = 0.0035 and p < 0.0001, respectively). In accordance, liver triglyceride levels were significantly decreased in both NE−/−/CG−/− (p = 0.0001) and NE−/−/PR3−/− (p < 0.0001) mice when compared with those of wild-type mice (Figures 3D, E). Strikingly, and despite similar bodyweight gain upon high-fat-diet feeding, NE−/−/PR3−/− mice displayed a significant reduction in liver weight and lipid droplet accumulation in the liver when compared with NE−/−/CG−/− mice (p < 0.0001).
To identify molecular pathways that may be responsible for the observed reduction in lipid accumulation in the liver, relative expression of several genes involved in lipid metabolism was measured (Figure 3F). Expression of Cd36 (Cd36 molecule), involved in fatty acid uptake and transport, was significantly decreased in NE−/−/PR3−/− (p = 0.0073), but not in NE−/−/CG−/− mice, when compared with wild-type mice. Fabp1 (fatty acid-binding protein 1), a gene involved in intracellular fatty acid transport, was downregulated in NE−/−/CG−/− mice (p = 0.0297), but not in NE−/−/PR3−/− mice. In addition, two genes involved in lipogenesis (diacylglycerol O-acyltransferase 1 [Dgat1] and fatty acid synthase [Fas]) were significantly downregulated in NE−/−/PR3−/− (p < 0.0001 and p = 0.0002, respectively), but not in NE−/−/CG−/− mice. Also the genes Pparα (peroxisome proliferator-activated receptor α) and Cpt1a (carnitine palmitoyltransferase 1A, liver), both involved in fatty acid oxidation, were downregulated in NE−/−/PR3−/−, but not in NE−/−/CG−/− mice compared with wild-type animals (p = 0.0065 and p = 0.0037, respectively). Overall, gene expression data suggest that processes involved in fatty acid uptake and lipogenesis are less active in livers from NE−/−/PR3−/− mice when compared with NE−/−/CG−/− mice and wild-type mice. Altogether, our interim conclusion is that these results point toward an important role for neutrophil serine proteases in the development of high-fat-diet-induced obesity, insulin resistance, glucose intolerance and NAFLD. Moreover, the absence of proteinase 3 specifically causes an additional reduction in lipid accumulation in the liver as compared with cathepsin G.
NE−/−/CG−/− and NE−/−/PR3−/− Mice Displayed Reduced Inflammation in Response to High-Fat-Diet Feeding
Given the role of neutrophil serine proteases in the activation of inflammatory processes, we next investigated inflammation in white adipose tissue and liver of NE−/−/CG−/−, NE−/−/PR3−/− and wild-type mice. Immunohistochemical localization of F4/80-positive macrophages in white adipose tissue of high-fat-diet-fed mice was analyzed and showed a strong reduction in the number of macrophages in both NE−/−/CG−/− (p < 0.0001) and NE−/−/PR3−/− mice (p < 0.0001) when compared with wild-type mice (Figures 4A, B). No differences in the weight of white adipose tissue were observed between the groups. No significant difference in the number of macrophages was observed between the two knock-out mice groups. In addition, the number of crown-like structures that infiltrated the white adipose tissue was significantly reduced in both high-fat-diet-fed NE−/−/CG−/− (p = 0.0002) and NE−/−/PR3−/− mice (p < 0.0001) when compared with wild-type mice (Figure 4C). No differences in the number of crown-like structures were observed between NE−/−/CG−/− and NE−/−/PR3−/− mice.

NE−/−/CG−/− and NET−/−/PR3−/− mice have reduced inflammation after a high-fat diet. (A) Representative images of immunostaining of the macrophage marker F4/80 in white adipose tissue of wild-type, NE−/−/CG−/−and NE−/−/PR3−/− mice. (B) Average number of macrophages per field. (C) Average number of CLS per field. Secretion of (D) IL-6 and (E) KC by white adipose tissue explants. (F) Relative mRNA expression of IL-1β, Cd68, IL-6, Mcp-1 and IL-1Ra in liver. WT, wild-type; HFD, high-fat diet; CLS, crown-like structures; *significant difference p ≤ 0.05; **significant difference p ≤ 0.01 ***significant difference p ≤ 0.001; NS, nonsignificant.
Subsequently, we assessed the inflammatory status of white adipose tissue ex vivo. After isolation, white adipose tissue from all three genotypes was cultured in medium for 24 h and the release of IL-6 and KC (CXCL1) was measured. Excretion of IL-6, an interleukin induced by IL-1β, was significantly reduced in white adipose tissue explants of NE−/−/CG−/− mice (p = 0.069). IL-6 excretion was also reduced in white adipose tissue explants of NE−/−/PR3−/− mice when compared with those of wild-type mice but differences were not statistically significant. No differences in IL-6 levels were observed between the two knock-out groups (Figure 4D). Also no differences in KC (CXCL1) levels were observed between the three experimental groups (Figure 4E).
To investigate whether the reduction in lipid accumulation in livers from NE−/−/CG−/− and NE−/−/PR3−/− mice correlated with the inflammatory status in the liver, we examined the relative expression of several inflammatory genes (Figure 4F). Interestingly, while mRNA expression of IL-1β and Cd68 molecule (Cd68), a marker for monocyte and macrophage infiltration, were significantly downregulated in NE−/−/PR3−/− mice when compared with wild-type mice (p = 0.0325 and p < 0.0001, respectively), no reduction was observed in livers of NE−/−/CG−/− mice versus wild-type mice. A similar trend was observed for IL6 and monocyte chemotactic protein 1 (Mcp-1). The anti-inflammatory gene interleukin-1 receptor antagonist (IL-1Ra) was down-regulated in NE−/−/PR3−/− mice when compared with wild-type mice (p = 0.0085). To conclude this section, these results show that NE−/−/CG−/− and NE−/−/PR3−/− mice display reduced inflammation in adipose tissue as compared with wild-type mice. Interestingly, only NE−/−/PR3−/− mice, but not NE−/−/CG−/− mice, showed reduced hepatic inflammation, suggesting that proteinase 3 specifically plays an important role in the activation of inflammatory pathways in the liver.
Proteinase 3 Induces In Vivo Insulin Resistance
To investigate whether the induction of insulin resistance and NAFLD by proteinase 3 acts through a mechanism independently of caspase-1, we treated caspase-1−/− mice with proteinase 3 (0.25 mg/kg in vehicle) or vehicle (PBS) during the last 10 d of a 16-wk high-fat diet. No differences in bodyweight were observed between vehicle- and proteinase 3-treated animals (Figure 5A). Casp-1−/− mice injected with proteinase 3 had significantly higher fasting glucose levels at baseline (p = 0.0021) (Figure 5B) and were more insulin resistant, as assessed by the insulin tolerance test, when compared with casp-1−/− mice injected with vehicle (p = 0.021) (Figure 5C). Plasma insulin levels tended to be higher in proteinase 3-treated mice but differences were not statistically significant (Figure 5D). The quantity of lipid droplets in the liver, liver weights and liver triglycerides were not different between caspase-1−/− mice treated with vehicle or proteinase 3 (Figures 5E, F). In accordance, proteinase 3 treatment did not affect regulation of the hepatic lipid metabolism genes Cd36, Fabp1, Dgat1, Fas, Pparα and Cpt1a (Supplementary Figure S1). To evaluate whether proteinase 3 treatment affects the inflammatory status of white adipose tissue and liver, macrophage infiltration and ex vivo secretion of proinflammatory cytokines were determined, but no differences between vehicle- and proteinase 3-treated animals were observed. In addition, no differences in expression were observed for the inflammatory genes IL-1β, Cd68, IL6, Mcp-1 and IL-1Ra in either white adipose tissue or liver (data not shown). These results indicate that the induction of insulin resistance by PR3 acts independently of caspase-1. Moreover, short-term treatment with proteinase 3 enhances basal glucose levels and induces insulin resistance independent of changes in hepatic lipid metabolism or activation of genes involved in inflammation. Notably, bodyweight was not different between proteinase 3- and vehicle-treated animals, indicating that the observed insulin resistance is not caused by differences in bodyweight.

Injected proteinase 3 induces insulin resistance in caspase-1-deficient mice during the last 10 d of a high-fat diet. (A) Body-weight of caspase-1−/− mice treated with vehicle (n = 10) or proteinase 3 (n = 10) during last 10 d of a high-fat diet. (B) Fasting glucose levels at baseline. (C) Glucose levels during insulin tolerance test and ITT AUC values. (D) Plasma insulin levels. (E) HE staining of liver sections. (F) Percentage of lipid droplets in HE-stained liver sections, liver weights and liver triglycerides. ITT, insulin tolerance test; PR3, proteinase 3; TG, triglycerides; *significant difference p ≤ 0.05; **significant difference p ≤ 0.01.
Treatment with Human α-1 Antitrypsin Decreases Fasting Glucose Levels and Reduced Lipid Droplet Accumulation in the Liver
We next investigated whether inhibition of neutrophil serine proteases would reduce NAFLD and improve insulin resistance in vivo in an established animal model of diet-induced obesity. Wild-type C57BL/6J mice were divided in bodyweight-matched treatment groups and treated (IP injection every other day) either with α-1 antitrypsin (Zemaira 2.0 mg/mouse) or vehicle (PBS) during the last 10 d of a 16-wk high-fat-diet-feeding period. As a control, low-fat-diet-fed mice were also treated with vehicle.
After treatment, no differences in bodyweight were observed between vehicle- and α-1-antitrypsin-treated mice (Figure 6A). However, fasting glucose levels decreased substantially after α-1-antitrypsin treatment and were comparable with the fasting glucose levels of mice receiving the low-fat diet (p = 0.001) (Figure 6B). Despite the decreased fasting glucose levels in the α-1-antitrypsin-treated group, the insulin tolerance test AUC values and insulin levels did not differ between the two high-fat diet groups (Supplementary Figures S2A, B). NAFLD was assessed by quantifying lipid droplets in HE-stained liver sections. Lipid droplet accumulation in the liver was significantly lower in mice treated with α-1 antitrypsin when compared with mice treated with vehicle (p = 0.003). Liver weights and triglyceride levels were not different between groups after α-1-antitrypsin treatment (Figures 6C, D).

Treatment with α-1 antitrypsin improves NAFLD status and reduces fasting glucose during the last 10 d of a high-fat diet. (A) Bodyweight of mice after a low-fat diet and treated with vehicle and after a high-fat diet treated with either vehicle or α-1 antitrypsin. (B) Fasting glucose levels. (C) HE staining of liver sections. (D) Percentage of lipid droplets in HE-stained liver sections, liver weights and liver triglycerides. LFD, low-fat diet; HFD, high-fat diet; hAAT, human α-1 antitrypsin; TG, triglycerides; **significant difference p ≤ 0.01; ***significant difference p ≤ 0.001; NS, nonsignificant.
To identify the underlying pathways that may be responsible for the observed reduction in lipid accumulation in the liver after α-1-antitrypsin treatment, the relative expression of the genes Cd36, Fabp1 (fatty acid transport and uptake), Dgat1, Fas (lipogenesis), Ppara and Cpt1a (fatty acid oxidation) was evaluated in liver tissue. Although these genes were not significantly regulated after α-1-antitrypsin treatment, a trend toward increased expression was observed in the α-1-antitrypsin-treated group for Dgat1, Fas, Pparα and Cpt1a (Supplementary Figure S3). Overall, these results show that inhibition of neutrophil serine proteases by α-1 antitrypsin improves fasting glucose and reduces lipid droplet accumulation in the liver of mice fed a high-fat diet.
Treatment with α-1 Antitrypsin Reduces Macrophage Influx into the Adipose Tissue
It is a well-known fact that α-1 anti-trypsin has strong anti-inflammatory properties (14). We therefore investigated whether α-1 antitrypsin is capable of reducing low-grade inflammation as it is seen in obesity. The number of F4/80-positive macrophages that infiltrated the white adipose tissue was substantially reduced in high-fat-diet-fed mice treated with α-1 antitrypsin when compared with high-fat-diet-fed mice treated with vehicle (p = 0.0405) (Figures 7A, B). In line with the reduction in macrophage infiltration, there was a clear trend toward a reduction in the number of crown-like structures in adipose tissue of α-1-antitrypsin-treated mice (Figure 7C). Ex vivo culturing of white adipose tissue revealed no effect of α-1-antitrypsin treatment on the secretion of IL-6 and KC (Figures 7D, E).

Treatment with human α-1 antitrypsin reduces macrophage infiltration into the adipose tissue during the last 10 d of a high-fat diet. (A) Representative images of immunostaining of the macrophage marker F4/80 in white adipose tissue of low-fat-diet-fed mice treated with vehicle, high-fat-diet-fed mice treated with vehicle and high-fat-diet-fed mice treated with human α-1 antitrypsin. (B) Average number of macrophages per field. (C) Average number of crown-like structures (CLS) per field. Secretion of (D) IL6 and (E) KC by white adipose tissue explants. LFD, low-fat diet; HFD, high-fat diet; hAAT, human α-1 antitrypsin; CLS, crown-like structures; *P ≤ 0.05; NS, nonsignificant.
Subsequently, we investigated whether this reduced inflammatory status in the adipose tissue was also present in the liver after α-1-antitrypsin treatment. The relative expression of the genes IL1β, Cd68, IL6, Mcp-1 and IL1Ra was evaluated, but no differences between vehicle- and α-1-antitrypsin-treated mice were observed (data not shown). In addition, no differences in relative expression between the groups were observed for the genes proteinase 3 (Prtn3), neutrophil elastase (Elane) and cathepsin G (Ctsg) (data not shown). Overall, these results show that treatment with α-1 antitrypsin reduces adipose tissue macrophage content.
Discussion
Our results demonstrate that neutrophil serine proteases have a prominent role in the development of NAFLD and insulin resistance. We also show that treatment with the neutrophil serine proteases inhibitor α-1 antitrypsin strongly reduces lipid droplets in the liver, improves fasting glucose levels and decreases adipose tissue inflammation in mice with high-fat-diet-induced NAFLD and insulin resistance. Together, these findings provide evidence that proteinase 3 is a novel mechanism involved in the development of NAFLD and insulin resistance and that inhibition of proteinase 3, combined with inhibition of other neutrophil serine proteases, may have therapeutic potential.
Our results, indicating that neutrophil serine proteases are involved in the development of insulin resistance and NAFLD, are supported by others studies as well. Recently, it was reported that mice deficient for neutrophil elastase were resistant to high-fat-diet-induced inflammation, insulin resistance (8,9) and NAFLD (9). Concerning proteinase 3, relatively little is known and studies investigating the role of endogenous proteinase 3 in models for obesity, insulin resistance and NAFLD were lacking. Our results provide evidence that, in addition to neutrophil elastase, endogenous proteinase 3 is also important for high-fat-diet-induced development of obesity, insulin resistance and NAFLD. Both NE−/−/CG−/− and NE−/−/PR3−/− mice were more insulin sensitive when compared with wild-type mice. Regarding NAFLD, liver lipid content and liver weights were reduced in NE−/−/CG−/− mice, and this effect was even stronger in NE−/−/PR3−/− mice. Given that the absence of proteinase 3 specifically causes an additional reduction in lipid content in the liver as compared with cathepsin G, our results point out to a prominent role for proteinase 3 in the development of NAFLD. This additional effect of proteinase 3 is also seen at the level of transcription. Relative expression of genes involved in fatty acid uptake and transport (Cd36), lipogenesis (Dgat1, Fas) and fatty acid oxidation (Pparα, Cpt1a) was downregulated in livers from NE−/−/PR3−/− mice when compared with those of wild-type and NE−/−/CG−/− mice. These results suggest that lipid metabolism pathways in the liver are, at least partly, regulated by proteinase 3. We hypothesized that neutrophil serine proteases induce metabolic disturbances by altering and/or activating inflammatory processes. Indeed, our results showed that mice deficient for NE/CG and NE/PR3 had, in addition to the improved metabolic profile, a reduced adipose tissue macrophage infiltration after high-fat-diet feeding when compared with wild-type mice. Also here, the results suggest a prominent role for proteinase 3 in the liver as the relative expression of the inflammatory genes IL1b, Cd68 and IL1ra was down-regulated in livers from NE−/−/PR3−/− mice when compared with those of wild-type and NE−/−/CG−/− mice. Also IL6 and Mcp1 showed a strong tendency toward down-regulation in livers from NE−/−/PR3−/− mice. These results strengthen the hypothesis that neutrophil serine proteases induce metabolic dysfunction by the activation and/or prolongation of inflammatory processes.
The influence of weight differences between experimental groups is, as a confounding factor, a problem for studying metabolic disorders. Although bodyweight was different between wildtype and the two knock-out groups, no difference in bodyweight was observed between NE−/−/CG−/− and NE−/−/PR3−/− mice. Also no differences in bodyweight were observed between α-1-antitrypsin-and vehicle-treated mice. These results indicate that the improved metabolic profiles in NE−/−/PR3−/− mice and α-1 antitrypsin-treated mice were not caused by differences in bodyweight.
Given the prominent role of IL-1β in inflammation-induced T2D and NAFLD and the role of neutrophil serine proteases in processing pro-IL-1β into its active form, it is a logical next step to point out IL-1β as the prime suspect for the observed metabolic disturbances. However, that is not necessarily the case. Besides cytokine processing, neutrophil serine proteases are also known to regulate other molecular mechanisms involved in inflammation. Neutrophil serine proteases are able to proteolytically modify chemokines such as, for instance, CXC-chemokine 8 (CXCL8) (19). They also play a role in receptor activation (20) and have been shown to cleave cell-surface adhesion molecules, thereby potentially participating in neutrophil migration (21). These examples indicate that, in addition to the processing of pro-IL-1β, neutrophil serine proteases also regulate many other inflammatory pathways that might be involved in the induction of metabolic complications.
In recent years, it has become evident that NAFLD and T2D are complex disorders in which multiple mechanisms are involved. In search for new therapeutics, the effectiveness of targeting a single protein may therefore be limited and a more comprehensive approach is needed that integrates multiple mechanisms of action. In this perspective, α-1 antitrypsin is of particular interest as it targets multiple inflammatory proteins known to be involved in obesity-induced development of T2D and NAFLD. Given the recent evidence that caspase-1 (5) and both neutrophil elastase (8,9) and proteinase 3 (this study) are involved in the development of T2D and NAFLD, it is very well possible that α-1 antitrypsin therapy would be beneficial for the treatment of these conditions. Mansuy-Aubert et al. showed that transgenic mice over-expressing α-1 antitrypsin are protected against high-fat-diet-induced insulin resistance and NAFLD (9). Our results showed that α-1-antitrypsin treatment reduced liver lipid content and improved fasting glucose. In addition, α-1-antitrypsin treatment reduced obesity-induced inflammation. These results support our hypothesis that α-1-antitrypsin treatment is capable of improving metabolic status by reducing inflammation.
One limitation of this study is that a solo proteinase 3 knock-out mouse model is not available. As a result, it is difficult to exactly discriminate between effects induced by proteinase 3 or neutrophil elastase. Another limitation is that we were able to test only one dose of α-1 antitrypsin. We suggest that in future experiments multiple doses of α-1 antitrypsin need to be tested, thereby providing the opportunity to study its effects in a dose-response manner.
Conclusion
We identified proteinase 3 as a new factor involved in NAFLD and insulin resistance. Inhibition of proteinase 3 and other neutrophil serine proteases by α-1 antitrypsin reduced lipid content in the liver and decreased fasting glucose levels and therefore may have therapeutic potential.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Schuppan D, Schattenberg JM. (2013) Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic approaches. J. Gastroenterol. Hepatol. 28 Suppl 1:68–76.
Donath MY, Shoelson SE. (2011) Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 11:98–107.
Netea MG, et al. (2006) Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 12:650–6.
Stienstra R, et al. (2011) Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 108:15324–9.
Dixon LJ, Flask CA, Papouchado BG, Feldstein AE, Nagy LE. (2013) Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS One. 8:e56100.
Armstrong L, Godinho SI, Uppington KM, Whittington HA, Millar AB. (2009) Tumour necrosis factor-alpha processing in interstitial lung disease: A potential role for exogenous proteinase-3. Clin. Exp. Immunol. 156:336–43.
Coeshott C, et al. (1999) Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. U. S. A. 96:6261–6.
Talukdar S, et al. (2012) Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 18:1407–12.
Mansuy-Aubert V, et al. (2013) Imbalance between neutrophil elastase and its inhibitor alpha1-antitrypsin in obesity alters insulin sensitivity, inflammation, and energy expenditure. Cell Metab. 17:534–48.
Bae S, et al. (2012) Neutrophil proteinase 3 induces diabetes in a mouse model of glucose tolerance. Endocr. Res. 37:35–45.
Macen JL, Upton C, Nation N, McFadden G. (1993) SERP1, a serine proteinase inhibitor encoded by myxoma virus, is a secreted glycoprotein that interferes with inflammation. Virology. 195:348–63.
Toldo S, et al. (2011) Alpha-1 antitrypsin inhibits caspase-1 and protects from acute myocardial ischemia-reperfusion injury. J. Mol. Cell Cardiol. 51:244–51.
Greene CM, et al. (2010) Anti-apoptotic effects of Z alpha1-antitrypsin in human bronchial epithelial cells. Eur. Respir. J. 35:1155–63.
Grimstein C, et al. (2011) Alpha-1 antitrypsin protein and gene therapies decrease autoimmunity and delay arthritis development in mouse model. J. Transl. Med. 9:21.
Zaki MH, et al. (2010) The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 32:379–91.
Schreiber A, et al. (2012) Neutrophil serine proteases promote IL-1beta generation and injury in necrotizing crescentic glomerulonephritis. J. Am. Soc. Nephrol. 23:470–82.
Livak KJ, Schmittgen TD. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–8.
Ellero-Simatos S, Fleuren WW, Bauerschmidt S, Dokter WH, Toonen EJ. (2014) Identification of gene signatures for prednisolone-induced metabolic dysfunction in collagen-induced arthritic mice. Pharmacogenomics. 15:629–41.
Padrines M, Wolf M, Walz A, Baggiolini M. (1994) Interleukin-8 processing by neutrophil elastase, cathepsin G and proteinase-3. FEBS Lett. 352:231–5.
Pham CT. (2006) Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 6:541–50.
Levesque JP, Takamatsu Y, Nilsson SK, Haylock DN, Simmons PJ. (2001) Vascular cell adhesion molecule-1 (CD106) is cleaved by neutrophil proteases in the bone marrow following hematopoietic progenitor cell mobilization by granulocyte colony-stimulating factor. Blood. 98:1289–97.
Acknowledgments
The authors thank Mark Gresnigt for his help in the design of Figure 1. ET was supported by an EFSD/Novo Nordisk research grant provided by the European Foundation for the Study of Diabetes (EFSD). MGN was supported by an ERC Consolidator Grant (#310372). TC was supported by the German Research Foundation (CH279/5-1) and an ERC grant (ENDHOMRET). This work was also supported by a grant of the Else-Kröner-Fresenius-Stiftung (to ET, LJ and TC).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Toonen, E.J.M., Mirea, AM., Tack, C.J. et al. Activation of Proteinase 3 Contributes to Nonalcoholic Fatty Liver Disease and Insulin Resistance. Mol Med 22, 202–214 (2016). https://doi.org/10.2119/molmed.2016.00033
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2016.00033