
Abstract
Simvastatin has been shown to have antiinflammatory effects that are independent of its serum cholesterol lowering action, but the mechanisms by which these antiinflammatory effects are mediated have not been elucidated. To explore the mechanism involved, the effect of simvastatin on toll-like receptor (TLR) signaling in primary human monocytes was investigated. A short pretreatment with simvastatin dose-dependently inhibited the production of tumor necrosis factor (TNF)-α in response to TLR8 activation (but not TLR2, -4 or -5). Statins are known inhibitors of the cholesterol biosynthetic pathway, but, intriguingly, TLR8 inhibition could not be reversed by addition of mevalonate or geranylgeranyl pyrophosphate, downstream products of cholesterol biosynthesis. TLR8 signaling was examined in HEK 293 cells stably expressing TLR8, where simvastatin inhibited I kappa B kinase (IKK)α/β phosphorylation and subsequent nuclear factor (NF)-κB activation without affecting the pathway to activating protein-1 (AP-1). Because simvastatin has been reported to have antiinflammatory effects in RA patients and TLR8 signaling contributes to TNF production in human RA synovial tissue in culture, simvastatin was tested in these cultures. Simvastatin significantly inhibited the spontaneous release of TNF in this model, which was not reversed by mevalonate. Together, these results demonstrate a hitherto unrecognized mechanism of simvastatin inhibition of TLR8 signaling that may in part explain its beneficial antiinflammatory effects.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is a debilitating inflammatory disease that affects ∼1% of the population. It is characterized by autoimmune synovial inflammation with consequent destruction of cartilage and bone (1). The release of proinflammatory cytokines and matrix metalloproteinases by immune cells that infiltrate the synovial membrane is an important factor in the pathogenesis of RA, as demonstrated by the clinical effectiveness of anti-cytokine biologicals (antibodies/soluble receptors), in particular, those that target tumor necrosis factor (TNF)-α (2,3). A compounding problem associated with RA is an increased risk of cardiovascular mortality (4,5). Consequently, HMG-CoA inhibitors (statins) that inhibit the production of mevalonate, thereby preventing the biosynthesis of cholesterol, are often prescribed to RA patients. In addition to the lipid-lowering effects, which are beneficial in the treatment of cardiovascular disease, statins also have antiinflammatory actions, such as inhibition of nuclear factor (NF)-κB activity and consequent reduction in the production of proinflammatory cytokines (6,7). This antiinflammatory effect of statins has been reported to decrease clinical symptoms of disease in patients with RA (8–11) and have also been observed to have prophylactic and therapeutic efficacy in the collagen-induced arthritis mouse model (12). However, the precise mechanisms by which statins exert these effects have not yet been fully elucidated.
Statins inhibit the production of mevalonate, the committed step in cholesterol biosynthesis, thereby preventing the production of cholesterol and its precursors. They not only reduce cholesterol synthesis, but also reduce mevalonate-derived molecules, such as the isoprenoid intermediates, farnesyl pyrophosphate and geranylgeranyl pyrophosphate (GGPP). The function of these isoprenoid intermediates is to induce protein prenylation, that is, addition of a prenyl group to C-terminal cysteine residues of proteins. The functions of several intracellular signaling proteins such as Rac and Rho have been demonstrated to be controlled by prenylation (13). Thus, it is possible that statins could mediate inflammatory pathways by blocking this process.
The potential role of innate immunity in sustaining chronic inflammation in RA is now well recognized (14,15). In particular, toll-like receptors (TLRs), a family of pattern recognition receptors of which there are 10 in humans, have been implicated in the pathogenesis of RA (rev. in 16). Upon activation, TLRs initiate signaling cascades leading to the activation of transcription factors activating protein-1 (AP-1), nuclear factor (NF)-κB or interferon (IFN) regulatory factors leading to the production of cytokines. These signaling pathways are initiated by the adaptor proteins myeloid differentiation primary response 88 (MyD88) (17,18), MyD88 adaptor-like (Mal) protein (19), TIR domain-containing adaptor inducing IFN-β (TRIF) (20) and TRIF-related adaptor molecule (TRAM) (21). The main signaling pathway activated by most TLRs is the MyD88-dependent pathway that signals to the TRAF6 complex, which includes IRAK1, -2 and -4. This then activates TAK1 binding protein (TAB)-1 and -2 and transforming growth factor β-activated kinase (TAK)-1, at which point the signal splits to activate the I kappa B kinase (IKK) complex consisting of IKKα, IKKβ and nuclear factor-kappaB essential modulator (NEMO) leading to NF-κB activation or to the mitogen-activated protein kinases (MAPKs) that activate downstream AP-1 (22).
Interestingly, statins have previously been demonstrated to modulate TLR2 and -4 signaling and expression (23,24), making TLRs good candidates for the antiinflammatory mechanism of statins in RA. In an attempt to gain a further understanding of the mechanism by which statins may be benefit from an immunomodulatory perspective, the effect of simvastatin on TLR signaling in primary human monocytes and in human rheumatoid synovial membrane cell cultures that spontaneously produce inflammatory cytokines were investigated.
Materials and Methods
Reagents
Cell culture reagents used were penicillin-streptomycin, RPMI 1640 and Dulbecco’s modified Eagle’s medium (DMEM) and were products of Lonza. Fetal calf serum was a product of Sigma. The TLR ligands used were chloroform-extracted Escherichia coli, lipopolysaccharide (LPS) and resiquimod (R-848) and were products of InvivoGen. Flagellin (purified) and Pam3Cys-Ser(Lys)4·3HCl (Pam3) were products of Alexis (Enzo Life Sciences). Simvastatin and geranylgeranylpyrophosphate (GGPP) were products of Sigma.
Cell Culture
RA synovial membrane cells were isolated from patients undergoing joint replacement surgery as previously described (25,26). Immediately after isolation, cells were cultured at 1 × 105 cells/well in 96-well tissue culture plates (Falcon) in RPMI 1640 containing 5% (v/v) fetal bovine serum and 100 U/mL penicillin-streptomycin. All RA patients gave written informed consent, and the study was approved by the Riverside Research Ethics Committee (REC number 07/H0706/81). Primary human monocytes were isolated from single donor plateletphoresis residues obtained from the North London Blood Transfusion Centre (United Kingdom) by Ficoll-Hypaque centrifugation after peripheral blood mononuclear cell isolation by using lympholyte gradients (27). The use of peripheral blood mononuclear cells was approved by the Brighton and Sussex Medical School Research Governance and Ethics Committee. Cells were cultured in RPMI medium 1640 with 25 mmol/L HEPES and 2 mmol/L L-glutamine supplemented with 5% heat-inactivated fetal calf serum and 100 units/mL penicillin/streptomycin at 37°C. HEK-Blue hTLR8 cells (InvivoGen) were routinely cultured in DMEM (Sigma) supplemented with 10% fetal bovine serum and 100 U/mL penicillin-streptomycin. These cells stably express human TLR8 and an inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene driven by an NF-κB/Ap-1 promoter. Upon activation of TLR8, SEAP is secreted into the cell supernatant and is detected by using QUANTI-Blue (InvivoGen), a medium that turns purple/blue in the presence of SEAP.
Plasmids
The NF-κB reporter plasmid, pGNL6 (a gift from David Gould, Queen Mary, University of London), consists of six repeats of the NF-κB site upstream of a minimal cytomegalovirus promoter controlling expression of firefly luciferase (28). The prenylation reporter system was based on the lentiviral vector constructed by Chinault et al. (29), except that two separate plasmids were constructed for use in transient transfections rather than lentiviral delivery. The sequences coding for the Gal4BD-VP16-GFP-CDC42 tail was amplified from the lentiviral vector previously described (29) using the primers 5′-CTAGG CTAGC GATGA AGCTA CTGTC TTCTA TCGAA-3′ and 5′-CTAGG CGGCC GCTCA TAGCA GCACA CACCT GCGGC T-3′ and ligated into pcDNA6 (Life Technologies) by using NheI and NotI restriction enzymes. The sequences coding for five copies of Gal4 upstream of firefly luciferase were amplified from the lentiviral vector described above using primers 5′-CTAGG GTACC AGATC CAGTT TGGTT ACGAC GGAT-3′ and 5′-CTAGG CTAGC GGTGG CTTTA CCAAC AGTAC CGGA-3′ and ligated into pGL2-Basic (Promega) by using restriction sites for KpnI and NheI. All constructs were verified by sequencing. The Renilla luciferase plasmid (pRL-SV40; Promega) was used to normalize for transfection efficiency in all experiments.
Transient Transfection of HEK-Blue hTLR8 Cells
Transient transfections were performed by using 25-kDa linear polyethylenimine (PEI) (Polysciences Inc.). Stock solutions of PEI were prepared in water at a concentration of 1 mg/mL, and the pH was adjusted to 7.0. The transfection conditions were as previously described (30). Briefly, cells were plated at a density of 4 × 105 cells/well in 96-well plates and transfected with a total of 0.5 µg DNA per well. The transfection complex was formed at a DNA:PEI ratio of 1:3 in OPTI-MEM (Life Technologies), with a 30-min incubation at room temperature before addition to the cells. Culture media were replaced 24 h later with 100 µL fresh DMEM containing 5 µg/mL R-848 and cultured for a further 24 h before collecting supernatants or assaying luciferase activity in cell lysates.
Western Blotting
Primary human monocytes or HEK-Blue TLR8 cells were preincubated with simvastatin for 30 min before stimulation with 2 or 5 µg/mL R-848, respectively, for 30 min. Cells were lysed by the addition of 100 µL sodium dodecyl sulfate sample buffer. Extracts were separated on 12% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, and proteins were transferred to a nitrocellulose membrane. Membranes were blocked in 5% bovine serum albumin in Tris-buffered saline containing 0.1% Tween 20 (TBST) and probed with antibodies recognizing phosphorylated forms of p38, TAK-1 or IKKα/β, or anti-GAPDH as a loading control (all from Cell Signaling Technologies). Membranes were then incubated with anti-rabbit IgG-horseradish peroxidase conjugate (Sigma), followed by detection with enhanced chemiluminescence (ECL) reagents (GE Healthcare, Amersham, UK) and exposed to autoradiography by using Hyperfilm (GE Healthcare). Films were developed by using an AGFA Curix 60 developer (Agfa Healthcare).
Enzyme-linked Immunosorbent Assay
Sandwich enzyme-linked immunosorbent assays (ELISAs) were used to measure TNF and interferon gamma-induced protein 10 (IP-10) (BD) according to the manufacturer’s recommendations. Standards were a product of Peprotech. Optical density was read on a spectrophotometric ELISA plate reader (BioTek) and analyzed by using Gen5 software V2.6.
Assays
Firefly and Renilla luciferase activities were assayed in cell lysates by using the Steady-Glo Luciferase Assay System (Promega) and Renilla Luciferase Assay System (Promega), respectively, according to the manufacturer’s instructions.
Luminescence was measured by using a BioTek plate reader. SEAP activity was measured in cell culture supernatants by using a Quanti-Blue reagent (InvivoGen) according to the manufacturer’s instructions. Measurements were taken at 630 nm. Cell viability was assessed by MTT assay (Sigma) (31).
Statistical Methods
Mean, standard deviation (SD), standard error of the mean (SEM) and statistical significance were calculated by using GraphPad version 3 (GraphPad Software). For statistical analysis, a one-tailed t test of paired data was used with a 95% confidence interval or a Wilcoxon matched-pairs two-tailed signed rank test.
Results
Simvastatin Inhibits TLR8 Signaling in Primary Human Monocytes and Is Not Reversed by Treatment with GGPP
It has previously been suggested that the antiinflammatory effects of simvastatin could be mediated by inhibition of TLR signaling. Therefore, the release of TNF from primary human monocytes pre-incubated with 10 µg/mL simvastatin for 30 min before stimulation for 18 h with ligands specific for TLR2 (Pam3), TLR4 (LPS), TLR5 (flagellin) and TLR7/8 (R-848) was measured. Simvastatin had no effect on TNF secretion in response to stimulation of TLR2, -4 or -5 but did result in a decrease in TNF produced in response to R-848 (Figure 1A), with no effect on cell viability as shown by MTT assay (Figure 1B). This effect was assumed to be due to inhibition of TLR8, rather than TLR7, signaling, since it has previously been demonstrated that monocytes do not produce TNF in response to TLR7 ligands (32). To investigate the mechanism by which simvastatin inhibits R-848-induced TNF secretion, a downstream product of the cholesterol pathway (GGPP) was added to the cultures together with simvastatin in an attempt to reverse this effect. Treatment with simvastatin dose-dependently inhibited TNF secretion from primary human monocytes, but could not be reversed by addition of 10 µmol/L GGPP (Figure 1C). Similar results were found in experiments where mevalonate (also downstream of HMG-Co inhibition) rather than GGPP was added to the monocytes (Figure 1D).

Simvastatin inhibits TLR8, but not TLR2, -4 or -5, signaling and is not reversed by GGPP in primary human monocytes. (A) Cells were incubated with 10 µg/mL simvastatin for 30 min and then stimulated with 10 ng/mL Pam3, 100 ng/mL LPS, 2 µg/mL R-848 or 10 ng/mL flagellin for 16 h. Supernatants were analyzed for TNF. Data pooled from four separate donors are shown as a percentage of the appropriate maximal response (*p < 0.0001). (B) Cell viability was measured by MTT assay. Data are pooled from four separate donors as a percentage of the appropriate maximal response. (C) Primary human monocytes were cultured in the presence of 10 µg/mL simvastatin ± 10 µmol/L GGPP for 30 min before stimulation with 2 µg/mL R-848. Supernatants were analyzed for TNF production. Data are representative of five separate experiments using five different donors. (D) Primary human monocytes were cultured in the presence of 0 or 10 µg/mL simvastatin ± 1 µmol/L mevalonate for 30 min before stimulation with R-848. Supernatants were analyzed for TNF production. Data are representative of four separate experiments using four different donors.
Simvastatin-Mediated Inhibition of TLR8 Signaling in HEK-Blue TLR8 Cells Is Independent of the Cholesterol Pathway
To model these responses in a more genetically tractable system, we also tested the effect of simvastatin on TLR8 signaling in HEK-Blue TLR8 cells that stably express both TLR8 and an AP-1/NF-κB SEAP reporter. These cells constitutively express human TLR8 and also have the advantage that NF-κB/AP-1 activation can be measured via the Quanti-Blue assay. Treatment of HEK-Blue TLR8 cells with simvastatin dose-dependently inhibited activation of NF-κB in response to treatment with R-848, which could not be reversed by addition of 10 µmol/L GGPP (Figure 2A), with no effect on cell viability (Figure 2B).

Simvastatin inhibits TLR8 signaling in HEK Blue TLR8 cells, which is not reversed by treatment with GGPP. (A) HEK Blue TLR8 cells (which stably express TLR8 and a reporter gene expressing a SEAP under the control of a NF-κB/AP-1-inducible promoter) were cultured for 30 min in the presence of 10 µg/mL simvastatin ± 10 µmol/L GGPP, before stimulation with 5 µg/mL R-848 for 16 h. Supernatants were analyzed for SEAP activity by using the Quanti-Blue assay and cell viability (B) was measured by MTT assay. Data are representative of three independent experiments.
GGPP Can Reverse the Effects of Simvastatin on Prenylation, but Not the Effects of Simvastatin, on Inhibition of TLR8 Signaling
To confirm that the GGPP used in these experiments was functional and could reverse the effects of simvastatin treatment, a prenylation reporter system was constructed, based on one previously described (29). The system is based on transient transfection of two plasmids: one coding for a chimeric Gal4-VP16-GFP transcription factor with a prenylation motif (the cdc42 tail) and a second plasmid coding for 5 Gal4 DNA-binding sites upstream of the gene for firefly luciferase. Once prenylated, the recombinant cdc42 tail associates with cell membranes, thereby preventing its translocation into the nucleus via the intrinsic nuclear localization sequences within the Gal4 DNA-binding domain. However, in the absence (or inhibition) of prenylation, the unprenylated chimeric transactivator can accumulate in the nucleoplasm, where it can drive the expression of firefly luciferase (Figure 3A). The reporter system was tested by transfection of both plasmids into HEK-Blue-TLR8 cells, followed by treatment with 10 µg/mL simvastatin ± 10 µmol/L GGPP. As expected, treatment with simvastatin dramatically increased luciferase expression, and the addition of GGPP reversed this effect completely (Figure 3B). Since GGPP did not reverse the inhibition of TLR8 signaling in HEK Blue TLR8 cells, the effect of simvastatin ± GGPP on prenylation was tested in these cells. Simvastatin blocked prenylation at all concentrations tested, whereas 10 µmol/L GGPP reversed this effect of simvastatin (Figure 3C), but not the effects on NF-κB activation (Figure 3D).

Inhibition of TLR8 signaling by simvastatin is independent of the prenylation pathway. (A) Schematic showing the principle of the bioluminescence reporter for detection of inhibition of protein prenylation in cells. HEK Blue TLR8 cells were transfected with a plasmid coding for a Gal4-VP16-GFP transcription factor bearing a prenylation (geranylgeranylation) site from Cdc42 at its C-terminus (Gal4-VP16-GFP-Cdc42 tail). This drives transcription of firefly luciferase (F Luc) from a second transfected plasmid under the control of a synthetic promoter containing 5 Gal4 DNA-binding sites. When prenylated, the Gal4-VP16-GFPCdc42 tail associates with membranes, reducing its ability to drive firefly luciferase (F Luc) expression. (B) Inhibition of prenylation by 10 µg/mL simvastatin for 30 min prevents association of Gal4-VP16-GFP-Cdc42 tail with membranes, allowing the transactivator to accumulate in the nucleoplasm and augment reporter expression. The presence of 10 µmol/L GGPP fully reverses the effect of simvastatin. (C) Prenylation is inhibited by 10 µg/mL simvastatin and reversed by 10 µmol/L GGPP in HEK Blue TLR8 cells treated with 5 µg/mL R-848 for 16 h. (D) NF-κB/AP-1 activation is inhibited in HEK-Blue TLR8 cells by simvastatin, but is unaffected by 10 µmol/L GGPP.
Simvastatin-Mediated Inhibition of TLR8 Signaling Is Independent of AP-1 Activation
To investigate the possible mechanisms by which simvastatin inhibits TLR8 signaling, experiments were performed to determine if activation of both NF-κB and AP-1 transcription factors were inhibited by simvastatin, since the SEAP reporter would not discriminate between them. Stimulation of HEK-Blue TLR8 cells with R-848 induces the production of SEAP via activation of both of these transcription factors. Hence, the effect of simvastatin on activation of NF-κB alone, using a reporter plasmid coding for the firefly luciferase gene downstream of six NF-κB binding sites, was tested. Transient transfection of HEK-Blue TLR8 cells with the NF-κB reporter plasmid was followed by stimulation with R-848 ± simvastatin. Activation of NF-κB was dose-dependently inhibited by simvastatin (Figure 4A). The effect of simvastatin on the activation of AP-1 was explored by analysis of phosphorylation of p38 by Western blotting. Stimulation of HEK-Blue TLR8 cells with R-848 resulted in a marked increase in the level of intracellular phosphorylated p38, and this increase was not affected by simvastatin (Figure 4B). These results were confirmed in primary human monocytes (Figure 4C). Investigation of the NF-κB activation pathway in primary human monocytes revealed that treatment of monocytes with R-848 resulted in a marked increase in phosphorylated IKKα/β, which was inhibited by simvastatin and not reversed by GGPP (Figures 4C, D). To further elucidate the mechanism by which simvastatin inhibited phosphorylation of IKKα/β, activation of TGF-β-activated kinase-1 (TAK-1) was analyzed by Western blot in primary human monocytes, since this lies upstream of both p38 and IKKα/β. Treatment of human monocytes with R-848 resulted in phosphorylation of TAK-1, but again, this result was not affected by simvastatin (Figure 4C).

Inhibitory effects of simvastatin on TLR8 signaling are mediated by inhibition of NF-κB but not AP-1. (A) Inhibition of NF-κB-induced luciferase production in HEK Blue TLR8 cells in response to 16 h incubation with 5 µg/mL R-848 after a 30-min incubation with simvastatin. (B) HEK Blue TLR8 cells were treated with 10 µg/mL simvastatin for 30 min ± 10 µmol/L GGPP before treatment with 5 µg/mL R-848 for 16 h. Cell lysates were examined for phosphorylation of p38 by Western blotting. (C) Primary human monocytes were treated with 10 µg/mL simvastatin ± 10 µmol/L GGPP for 30 min before treatment with 2 µg/mL R-848 for 16 h. Cell lysates were examined for phosphorylation of p38, IKKα/β and TAK-1 by Western blotting. GAPDH was used as a loading control. These blots are representative of three independent experiments. (D) Densitometric analysis of Western blots for phosphorylated IKKα/β. Data are expressed as arbitrary units and are the mean ± SE (n = 3). *p < 0.05 versus R-848-treated samples by Student t test for paired data.
Simvastatin Inhibits Spontaneous TNF Production from Human RA Synovial Membrane Cultures
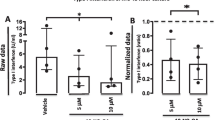
Synovial membrane cultures isolated from human RA joints spontaneously secrete cytokines. The importance of TLR8 in the production of TNF from these cultures has previously been demonstrated (32), so we hypothesized that simvastatin may also have antiinflammatory effects in this system. Therefore, rheumatoid synovial membrane cells harvested from elective joint replacement surgery were cultured with increasing concentrations of simvastatin for 24 h, and the secretion of TNF was measured. Incubation with 20 µg/mL simvastatin significantly reduced the concentrations of these molecules in the cell supernatants (Figure 5A). The effect of simvastatin was dose-dependent and was not reversed by addition of mevalonate to the cultures (Figure 5B). Cell viability was measured by MTT assay at the end of the 24-h incubation in each experiment and showed no difference in cell viability between any of the experimental conditions (Figure 5C).

Simvastatin inhibits spontaneous TNF production from RA synovial membrane cultures that is not reversed by mevalonate. (A) RA synovial membrane cells were cultured in medium alone or medium containing 20 µg/mL simvastatin (**p < 0.0001). Data are shown from seven RA patient samples from separate donors. (B) RA synovial membrane cells were cultured for 24 h in the presence of simvastatin ± 1 µmol/L mevalonate. Supernatants were analyzed for TNF. (C) Effects of simvastatin ± 1 mmol/L mevalonate on cell viability were measured by MTT assay. Data shown in (B) and (C) are from triplicate cultures shown as the mean ± SD and are representative of three separate experiments in unrelated donors.
Discussion
This study increases the understanding of the antiinflammatory mechanism of simvastatin by demonstrating an inhibitory effect on TLR8 signaling in primary human monocytes. This effect of simvastatin on TLR8 signaling is, to our knowledge, entirely novel. Antiinflammatory actions of statins that are independent of the reduction in cholesterol have been known for some time, such as inhibition of oxidative stress-induced NF-κB activation (6,33), leukocyte adhesion (34,35) and cell proliferation/apoptosis (36–38). Some of these antiinflammatory effects have been attributed to an alteration of protein prenylation. Blocking the cholesterol synthesis pathway prevents the downstream synthesis of isoprenoid intermediates such as farnesyl pyrophosphate and GGPP, which can be incorporated into proteins via covalent attachment to conserved cysteine residues in a process known as protein prenylation (13). RhoA prenylation in particular has been shown to be important in activation of NF-κB and secretion of proinflammatory cytokines from TNF-activated rheumatoid synoviocytes (39) and TLR2-activated monocytes from RA patients (23).
The inhibition of TLR signaling by simvastatin observed was unexpected in that it was not reversible by mevalonate or GGPP and was specific to TLR8. Instead the mechanism appeared to be through inhibition of downstream IKK phosphorylation and consequently NF-κB activation. However, phosphorylation of p38 MAPK that leads to AP-1 activation was unaffected. TAK1 acts upstream of both IKKβ and p38 (40) and was clearly phosphorylated upon treatment of monocytes with R-848. However, this was not inhibited by simvastatin, suggesting that simvastatin may act either directly on the IKK complex or via another upstream molecule necessary for IKK phosphorylation. Interestingly, TLR8-mediated phosphorylation of IKK was previously suggested to be independent of TAK1 instead of being phosphorylated by mitogen-activated protein kinase kinase kinase 3 (MEKK3) (41). This alternate pathway of IKK activation by TLR8 may explain why no effect of simvastatin was observed on TLR2-, TLR4- or TLR5-induced TNF production in monocytes in this study.
Conversely, there have been reports of modulation of TLR2 and TLR4 by simvastatin. In monocytes from RA patients, simvastatin inhibited activation of NF-κB and secretion of TNF and IL-1β in response to TLR2 stimulation (23). In monocytes from healthy donors, simvastatin has also been shown to inhibit the expression of TLR4 (24). However, these differences may be explained by the methodologies used, in particular, in the length of exposure to simvastatin before stimulation. A short 30-min incubation with simvastatin before adding TLR ligands was used in this study, whereas the other studies used a 24-h pre-incubation (23,24). Interestingly, the effects of simvastatin shown in both of these studies were reversible with mevalonate and/or GGPP, suggesting a possible reduction of prenylation of TLR signaling molecules with a longer pre-incubation period.
TLRs are suggested to contribute to the maintenance of inflammation in RA. In previous studies, we had identified a role for TLRs and, in particular, TLR8 in the production of spontaneous TNF in human RA synovial membrane cultures (32,42).
In agreement with a pathogenic role for TLR8 in RA, a recent study observed that expression of human TLR8 in transgenic mice resulted in increased susceptibility to collagen-induced arthritis (43). This same study also reported increased expression of TLR8 in the blood of patients with systemic arthritis (43). Because simvastatin is suggested to be antiinflammatory in RA (44) and was observed to inhibit TLR8 signaling in monocytes in this study, the effect of simvastatin on the production of TNF from human RA synovial cultures was investigated.
Treatment of human RA synovial cultures with simvastatin significantly decreased the spontaneous production of TNF, which was refractory to mevalonate treatment, as had been observed for TLR8-induced TNF production in monocytes. This result suggested that it was not due to effects on other TLRs, such as TLR2 and TLR4, which can be reversed with mevalonate. This antiinflammatory effect in the RA synovial membrane cultures is consistent with previous studies showing beneficial effects of statins in RA (45) and suggest that the effects of statins on lowering joint pain and swelling (44,46) may be due, at least in part, to the inhibition of TNF released from synovial tissue. TNF is known to be a key cytokine driving chronic inflammation in RA, as has been demonstrated by the clinical success of anti-TNF biological therapies (47). This reduction in TNF may in part be due to inhibition of TLR8; however, in RA patients, long-term simvastatin use may have multiple effects that together generate the clinical benefits observed, such as the reduction of expression and function of other TLRs, decreased cell proliferation and a reduction in inflammation initiated by other pathways (8–11,44).
Conclusion
Together, these data provide new insights into the intracellular signaling pathways downstream of TLR8 activation. The precise mechanism by which phosphorylation of IKK in response to TLR8 stimulation is inhibited by simvastatin remains to be fully elucidated. However, the data build upon our existing knowledge of the antiinflammatory effects of simvastatin and suggest a possible mechanism by which simvastatin contributes to a reduction in disease activity in RA.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Choy EH, Panayi GS. (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N. Engl. J. Med. 344:907–16.
Chaabo K, Kirkham B. (2015) Rheumatoid arthritis: anti-TNF. Int. Immunopharmacol. 27:180–4.
Gavrila BI, Ciofu C, Stoica V, Panaitescu E. (2015) The efficiency of biologic therapy in a group of patients with rheumatoid arthritis. J. Med. Life. 8:79–84.
Gonzalez-Gay MA, Gonzalez-Juanatey C, Martin J. (2005) Rheumatoid arthritis: a disease associated with accelerated atherogenesis. Semin. Arthritis Rheum. 35:8–17.
Van Doornum S, Jennings GL, Wicks IP. (2006) Reducing the cardiovascular disease burden in rheumatoid arthritis. Med. J. Aust. 184:287–90.
Ortego M, et al. (1999) Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis. 147:253–61.
Zelvyte I, Dominaitiene R, Crisby M, Janciauskiene S. (2002) Modulation of inflammatory mediators and PPARgamma and NFkappaB expression by pravastatin in response to lipoproteins in human monocytes in vitro. Pharmacol. Res. 45:147–54.
Arnaud C, Mach F. (2006) Potential antiinflammatory and immunomodulatory effects of statins in rheumatologic therapy. Arthritis Rheum. 54:390–2.
Costenbader KH, Coblyn JS. (2005) Statin therapy in rheumatoid arthritis. South Med. J. 98:534–540; quiz 541, 572.
Kanda H, et al. (2007) Effects of low-dosage simvastatin on rheumatoid arthritis through reduction of Th1/Th2 and CD4/CD8 ratios. Mod. Rheumatol. 17:364–8.
Tang TT, et al. (2011) Atorvastatin upregulates regulatory T cells and reduces clinical disease activity in patients with rheumatoid arthritis. J. Lipid Res. 52:1023–32.
Leung BP, et al. (2003) A novel anti-inflammatory role for simvastatin in inflammatory arthritis. J. Immunol. 170:1524–30.
Zhang FL, Casey PJ. (1996) Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 65:241–69.
Falgarone G, Jaen O, Boissier MC. (2005) Role for innate immunity in rheumatoid arthritis. Joint Bone Spine. 72:17–25.
Gierut A, Perlman H, Pope RM. (2010) Innate immunity and rheumatoid arthritis. Rheum. Dis. Clin. North Am. 36:271–96.
Huang QQ, Pope RM. (2009) The role of toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 11:357–64.
Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. (1997) MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 7:837–47.
Zhang FX, et al. (1999) Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J. Biol. Chem. 274:7611–4.
Fitzgerald KA, et al. (2001) Mal (MyD88-adapter-like) is required for toll-like receptor-4 signal transduction. Nature. 413:78–83.
Yamamoto M, et al. (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the toll-like receptor signaling. J. Immunol. 169:6668–72.
Fitzgerald KA, et al. (2003) LPS-TLR4 signaling to IRF-3/7 and NF-kappaB involves the toll adapters TRAM and TRIF. J. Exp. Med. 198:1043–55.
O’Neill LA, Golenbock D, Bowie AG. (2013) The history of toll-like receptors: redefining innate immunity. Nat. Rev. Immunol. 13:453–60.
Lin H, et al. (2011) HMG-CoA reductase inhibitor simvastatin suppresses toll-like receptor 2 ligand-induced activation of nuclear factor kappa B by preventing RhoA activation in monocytes from rheumatoid arthritis patients. Rheumatol. Int. 31:1451–8.
Methe H, Kim JO, Kofler S, Nabauer M, Weis M. (2005) Statins decrease toll-like receptor 4 expression and downstream signaling in human CD14+ monocytes. Arterioscler. Thromb. Vasc. Biol. 25:1439–1445.
Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. (1989) Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 2:244–7.
Brennan FM, Chantry D, Jackson AM, Maini RN, Feldmann M. (1989) Cytokine production in culture by cells isolated from the synovial membrane. J. Autoimmun. 2 Suppl:177–86.
Menck K, et al. (2014) Isolation of human monocytes by double gradient centrifugation and their differentiation to macrophages in teflon-coated cell culture bags. J. Vis. Exp. e51554.
Khoury M, et al. (2007) Inflammation-inducible anti-TNF gene expression mediated by intra-articular injection of serotype 5 adeno-associated virus reduces arthritis. J. Gene Med. 9:596–604.
Chinault SL, et al. (2012) Breast cancer cell targeting by prenylation inhibitors elucidated in living animals with a bioluminescence reporter. Clin. Cancer Res. 18:4136–44.
Mullen LM, Adams G, Chernajovsky Y. (2012) Increased disulphide dimer formation of latent associated peptide fusions of TGF-beta by addition of L-cystine. J. Biotechnol. 161:269–77.
Mosmann T. (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65:55–63.
Sacre SM, et al. (2008) Inhibitors of TLR8 reduce TNF production from human rheumatoid synovial membrane cultures. J. Immunol. 181:8002–9.
Hilgendorff A, et al. (2003) Statins differ in their ability to block NF-kappaB activation in human blood monocytes. Int. J. Clin. Pharmacol. Ther. 41:397–401.
Liu L, et al. (1999) Integrin-dependent leukocyte adhesion involves geranylgeranylated protein(s). J. Biol. Chem. 274:33334–40.
Yoshida M, et al. (2001) Hmg-CoA reductase inhibitor modulates monocyte-endothelial cell interaction under physiological flow conditions in vitro: involvement of Rho GTPase-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 21:1165–71.
Guijarro C, et al. (1998) 3-Hydroxy-3-methylglutaryl coenzyme a reductase and isoprenylation inhibitors induce apoptosis of vascular smooth muscle cells in culture. Circ. Res. 83:490–500.
Munro E, et al. (1994) Inhibition of human vascular smooth muscle cell proliferation by lovastatin: the role of isoprenoid intermediates of cholesterol synthesis. Eur. J. Clin. Invest. 24:766–72.
Schonbeck U, Libby P. (2004) Inflammation, immunity, and HMG-CoA reductase inhibitors: statins as antiinflammatory agents? Circulation. 109:II18–26.
Xu H, et al. (2006) RhoA-mediated, tumor necrosis factor alpha-induced activation of NF-kappaB in rheumatoid synoviocytes: inhibitory effect of simvastatin. Arthritis Rheum. 54:3441–51.
Ajibade AA, et al. (2012) TAK1 negatively regulates NF-kappaB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity. 36:43–54.
Qin J, et al. (2006) TLR8-mediated NF-kappaB and JNK activation are TAK1-independent and MEKK3-dependent. J. Biol. Chem. 281:21013–21.
Alzabin S, et al. (2012) Investigation of the role of endosomal toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res. Ther. 14:R142.
Guiducci C, et al. (2013) RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 210:2903–19.
Kanda H, et al. (2002) Antiinflammatory effect of simvastatin in patients with rheumatoid arthritis. J. Rheumatol. 29:2024–6.
Paraskevas KI. (2008) Statin treatment for rheumatoid arthritis: a promising novel indication. Clin. Rheumatol. 27:281–7.
Okamoto H, et al. (2007) Beneficial action of statins in patients with rheumatoid arthritis in a large observational cohort. J. Rheumatol. 34:964–8.
Radner H, Aletaha D. (2015) Anti-TNF in rheumatoid arthritis: an overview. Wien. Med. Wochenschr. 165:3–9.
Acknowledgments
This work was supported by funding from the European Union Seventh Framework Programme (integrated project Masterswitch; 223404) and by Brighton and Sussex Medical School.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Mullen, L., Ferdjani, J. & Sacre, S. Simvastatin Inhibits Toll-like Receptor 8 (TLR8) Signaling in Primary Human Monocytes and Spontaneous Tumor Necrosis Factor Production from Rheumatoid Synovial Membrane Cultures. Mol Med 21, 726–734 (2015). https://doi.org/10.2119/molmed.2015.00154
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2015.00154