
Abstract
The autonomic nervous system is an important modulator of cardiac signaling in both health and disease. In fact, the significance of altered parasympathetic tone in cardiac disease has recently come to the forefront. Both neuronal and nonneuronal cholinergic signaling likely play a physiological role, since modulating acetylcholine (ACh) signaling from neurons or cardiomyocytes appears to have significant consequences in both health and disease. Notably, many of these effects are solely due to changes in cholinergic signaling, without altered sympathetic drive, which is known to have significant adverse effects in disease states. As such, it is likely that enhanced ACh-mediated signaling not only has direct positive effects on cardiomyocytes, but it also offsets the negative effects of hyperadrenergic tone. In this review, we discuss recent studies that implicate ACh as a major regulator of cardiac remodeling and provide support for the notion that enhancing cholinergic signaling in human patients with cardiac disease can reduce morbidity and mortality. These recent results support the idea of developing large clinical trials of strategies to increase cholinergic tone, either by stimulating the vagus or by increased availability of Ach, in heart failure.
Similar content being viewed by others


Overview
Acetylcholine (ACh) is an ancient signaling molecule that regulates many physiological functions both in the central and peripheral nervous system. In the periphery, ACh is the major chemical neurotransmitter, regulating both parasympathetic and sympathetic tone. Importantly, the autonomic nervous system regulates the function of several different organ systems via activation of very specific pathways. However, for the purpose of this review, which focuses on its effects in the cardiovascular system, parasympathetic and sympathetic tone will refer to autonomic efferent outflow to the heart.
Genesis of ACh depends on the actions of the enzyme choline acetyltransferase (ChAT) (1), which converts free choline and acetyl-CoA into ACh, a process first described by Nachmansohn and Machado (2). ChAT is in kinetic excess in nerve terminals; therefore, minor reductions in ChAT activity have negligible impact on ACh content and release (3). ACh synthesis is exquisitely coupled to the high-affinity choline transporter (CHT1), which serves as the rate-limiting step for ACh production (4). This step is because of the importance of CHT1 in regulating the transport of sufficient amounts of free choline from the extracellular environment into the cholinergic presynaptic nerve terminal (5), where it can be used to synthesize ACh and maintain its sustained release. No other choline transporter can replace CHT1 in cholinergic nerve endings, since its genetic deletion leads to death shortly after birth because of respiratory failure (6).
The secretion of ACh into the extracellular environment depends on its packaging into exocytic vesicles via the vesicular acetylcholine transporter (VAChT) (7). VAChT is a 12-transmembrane domain protein that uses the electrochemical gradient of a proton ATPase to store ACh in synaptic vesicles (7). Elimination of the VAChT gene abolishes stimulated ACh release (8). In addition, decreased expression levels of VAChT lead to proportional decreases in ACh release (9). On the other hand, overexpression of VAChT in immature Xenopus neurons leads to increased synaptic responses (10). Furthermore, overexpression of VAChT in mice has been shown to increase ACh release (11,12), improve physical fitness and cause abnormalities in cognitive behavior (11). Hence, VAChT expression is unique in its ability to regulate ACh release.
The parasympathetic nervous system acts through the vagus nerves, which mainly innervate the atria of the heart, with some sparse ventricular innervation (13), and plays a crucial role in regulating several aspects of cardiac physiology. Cholinergic signaling leads to a reduction in heart rate, the contractile forces of the atria and the conduction velocity of both the sinoatrial and atrioventricular nodes. These actions are mediated by the binding of ACh to M2 muscarinic receptors in atrial myocytes (14,15). M2 receptors are coupled to Gi proteins, which mediate the decrease in inotropic and chronotropic responses through various downstream mechanisms. One mechanism involves direct inhibition of adenylyl cyclase, which leads to decreased production of adenosine 3′,5′-cyclic monophosphate (cAMP) and inhibition of protein kinase A (PKA) (16,17). Muscarinic receptor activation can also lead to indirect inhibition of L-type Ca2+ channels through a decrease in cAMP production (18). Furthermore, the negative inotropic and chonotropic effects observed after activation of M2 receptors are due to hyperpolarization of atrial cells following the direct activation of inwardly rectifying ACh-sensitive potassium channels (KACh) by the Giβγ subunit that is normally activated by the M2 receptor (19). In addition, activation of muscarinic receptors leads to negative chronotropic responses due to decreased cAMP levels, which results in downstream inhibition of the HCN (hyperpolarization activation, cyclic nucleotide-gated) channel-mediated “funny” current (If) (20,21).
The M2 receptor is the main muscarinic subtype in the heart (22,23); however, mounting evidence suggests that M3 muscarinic receptors also play a role in mediating physiological signaling in the heart. Indication of cholinergic signaling via the M3 receptor subtype came initially from studies showing that pretreatment with an M3-selective inhibitor attenuated the increase in inositol triphosphate (IP3) formation observed after treatment with carbachol (24). This study suggested that cholinergic signaling via the M3 muscarinic receptor in the heart is coupled with Gq-mediated activation of the phospholipase C (PLC)/IP3 pathway. Furthermore, M3 receptors were also shown to mediate inotropic signaling in the atria (25). The biphasic inotropic response observed in isolated atria has been attributed to both M2 and M3 receptor signaling, with the latter mediating the positive inotropic response (25). M3 muscarinic receptors also appear to regulate pathophysiological responses. It has been reported that cholinergic signaling via M3 receptors in the heart can lead to cardioprotection after myocardial ischemia, a process thought to occur through inhibition of miR-376b-5p (26). Furthermore, it was recently shown that upregulation of the M3 muscarinic receptor in mice attenuates angiotensin II (Ang II)-induced cardiac hypertrophic response (27).
In addition to its direct effects on the heart via muscarinic receptors, it is well known that ACh is also critical in regulating sympathetic signaling, since it mediates fast transmission through sympathetic ganglia by activating α-neurotoxin-sensitive postsynaptic nicotinic ACh receptors (28). In addition, ACh can mediate slow excitatory synaptic transmission at sympathetic ganglia via post-ganglionic M1 receptors (29). Furthermore, ACh can also regulate sympathetic signaling by binding to presynaptic receptors on sympathetic nerve terminals innervating the heart. Activation of M1 muscarinic receptors at presynaptic sympathetic nerves can increase norepinephrine (NE) release (30), whereas M2 muscarinic receptors appear to mediate ACh-induced inhibition of NE release at the atria (31,32).
Interestingly, muscarinic G-proteincoupled receptor (GPCR) signaling via downstream G proteins is partially modulated by the actions of the regulator of G protein signaling (RGS) proteins, which can terminate G protein signaling by increasing hydrolysis of bound guanosine-5′-triphosphate (GTP) (33). RGS2, in particular, has been shown to decrease both Gq and Gi protein signaling (34–36), although it acts on the latter with a lower potency and shows Gq selectivity (37). In addition, RGS2 has previously been shown to reduce adenylyl cyclase activity downstream of Gs signaling (38). Importantly, RGS2 can inhibit hypertrophy in response to β-adrenergic stimulation (39), an effect that may partially be due to its role in inhibiting eukaryotic initiation factor 2B (eIF2B)-mediated protein synthesis (40). Furthermore, RGS2 knockout (KO) mice display hypertension under basal conditions (41) and increased ventricular dysfunction and dilation after transverse aortic constriction, thus suggesting a role for RGS2 signaling in the cardiovascular response to pathological stimuli (42).
Evidence suggests that, under physiological conditions, parasympathetic tone is the main regulator of heart rate and cardiac activity (43,44). Therefore, it is likely that, under pathological conditions, cholinergic tone balances sympathetic signaling to prevent adverse effects associated with increased activation of β-adrenergic receptor-mediated signaling. This result may serve to prevent cardiomyocyte remodeling associated with overactivation of the sympathetic system. Thus, cholinergic activity modulation may serve as a new target for the treatment of cardiac disease. Here, recent findings highlighting the importance of altered autonomic regulation, especially cholinergic signaling, and its implications in cardiac disease will be highlighted.
Autonomic Nervous System in Heart Failure: Enhanced Sympathetic Activity
Heart failure is a progressive syndrome resulting from a number of different conditions characterized by significant damage and weakening of the cardiac muscle, leading to decreased cardiac output. The prevalence of heart failure is high, with more that 5.1 million people over the age of 20 suffering from heart failure in the United States alone (45). Despite significant advancements in clinical care of heart failure patients, this syndrome continues to be an important contributor to high morbidity (>1 million hospital discharges/year), mortality (>50,000 deaths/year) and economic burden ($30.7 billion/year) in the United States, according to the latest update on heart disease and stroke (45).
The autonomic nervous system is the main regulator of cardiac output (that is, the efficiency with which the heart pumps blood). It is accepted that chronic autonomic sympathetic/parasympathetic imbalance plays a crucial role in the development of heart failure (46–49). Increased sympathetic tone, coupled with decreased parasympathetic drive, is seen even in the early stages of chronic heart failure (50,51).
Several lines of research implicate over-activation of the sympathetic nervous system in patients as a major contributor to cardiac remodeling (52). This result is associated with poorer prognosis and higher morbidity and mortality (53). Although increased sympathetic tone may help to preserve cardiac function initially, it may also contribute to remodeling (48,54). The activation of neurohumoral mechanisms, including increased sympathetic nervous system signaling, is a hallmark of heart failure resulting from homeostatic regulation after the initial decrease in cardiac output (48,54). Sympathetic nervous system hyperactivity is observed in several cardiac diseases including hypertension. Increased sympathetic nervous system (SNS) activity in hypertensive patients seems to contribute to altered blood pressure regulation as well as left ventricular dysfunction (55). Importantly, decreasing SNS signaling through activation of the baroreflex has been shown to increase survival in an animal model of chronic heart failure (56), thus suggesting that enhanced sympathetic signaling plays a critical role in the progression of cardiac dysfunction.
Modulation of the autonomic nervous system has become an important target in decreasing morbidity and mortality in heart failure patients. Currently, β-adrenergic receptor blockers, in combination with angiotensin-converting enzyme inhibitors and angiotensin receptor blockers, are used as a first-line treatment for heart failure (57). β-Blockers work by inhibiting the effect of noradrenaline, released from sympathetic neurons, on the β-adrenergic receptors in both the heart and vasculature and can decrease heart rate and lower blood pressure through vasodilation. This step reduces the pressure placed on the heart and thus preserves cardiac function. The chronic use of β-blockers has been quite successful and has been shown to increase cardiac function and reduce left ventricular remodeling and mortality in patients, with third-generation nonselective β-blockers decreasing mortality more than second-generation β1-selective drugs (34% versus 40%, respectively) (58–60).
The importance of modulating sympathetic tone in left ventricular hypertrophy and cardiomyopathy is further highlighted by the fact that autoantibodies against the β1-adrenergic receptor are found in patients with both cardiac hypertrophy and dilated cardiomyopathy (61,62). The cause-and-effect relationship between the production of autoantibodies and cardiomyopathy has not yet been fully elucidated. However, the presence of these autoantibodies has been shown to activate signaling cascades downstream of β1-adrenergic activation in cultured myocytes and may contribute to the cardiac dysfunction observed in cardiomyopathy (63). Additionally, it is possible that the presence of these autoantibodies also leads to autoimmune-mediated cardiac damage.
In addition to the positive effects observed after inhibition of direct actions of sympathetic signaling in the heart, positive outcomes have been observed following renal sympathetic denervation in patients with heart failure (64,65). Activation of the renal sympathetic nerves in heart failure leads to increased peripheral vascular resistance and ventricular remodeling, partly due to higher levels of Ang II. Renal denervation has shown promise in human heart failure patients, since there was a trend toward improvement in exercise capacity and left ventricular hypertrophy (64,65). Furthermore, independent of its blood pressure-lowering effects, renal denervation has been shown to have a positive effect on atrial fibrillation (66) and heart rate reduction (67), both of which have been shown to play a critical role in morbidity and mortality in heart failure patients.
Autonomic Nervous System in Heart Failure: Reduced Parasympathetic Signaling
Hyperactivity of the sympathetic nervous system is coupled with decreased activation of the parasympathetic nervous system, leading to decreased signaling through ACh (68). A decrease in parasympathetic signaling is observed early after induction of cardiac remodeling, even before the onset of heart failure, and this cholinergic hypoactivity appears to contribute to the cardiac dysfunction observed in heart failure (68). The importance of cholinergic signaling in cardiac remodeling is further highlighted by previous data, suggesting a strong association between decreased vagal reflex, as measured through baroreceptor signaling, and ventricular arrhythmias after myocardial infarction (69,70). This decrease in cholinergic signaling may partially be related to the production of autoantibodies against M2 muscarinic receptors, which significantly reduce ligand binding in patients with idiopathic dilated cardiomyopathy and effectively inhibit downstream signaling (71).
Importantly, it was previously reported that pacing-induced left ventricular dysfunction and heart failure in dogs leads to changes in both sympathetic as well as vagal control of the heart (72). Furthermore, changes in vagus nerve-mediated regulation of heart rate have been observed during the early stages of left ventricular dysfunction, suggesting that alterations in cholinergic signaling in the heart may contribute to the heart failure phenotype (72). These changes in vagal control of heart rate appear to be due to both cardiac postganglionic signaling as well as alterations in parasympathetic ganglionic signaling (73). One additional mechanism that may contribute to decreased cholinergic signaling in the heart involves the activation of mechanoreceptors in dilated hearts, due to greater mechanical stretch of the cardiac muscle. This mechanical stretch activates sympathetic afferent fibers, which, in turn, favors reflex inhibition of intracardiac cholinergic signaling (74).
In agreement with the notion of a causal relationship between lower cholinergic signaling and cardiac dysfunction, M2 muscarinic receptor KO mice exhibit a greater impairment in ventricular function after an acute dose of phenylephrine, which increases cardiac afterload (75). Although these mice exhibited no difference in ventricular function under baseline conditions, chronic treatment with isoproterenol to induce cardiac remodeling led to a significant increase in matrix metalloproteinase (MMP) activity in M2 receptor KO mice, compared with wild-type mice (75). This result suggests that cholinergic tone may play a role in regulating MMP activity and, therefore, the progression of cardiac dysfunction. Importantly, increased MMP activity has been associated with ventricular dysfunction in patients with heart failure (76,77). In addition, we have previously reported that mice with reduced cholinergic neurotransmission, due to decreased expression of VAChT, exhibit cardiac remodeling and dysfunction (78,79). It has also been reported that mice with decreased expression of the presynaptic high-affinity choline transporter (CHT1; CHT1 heterozygous) presents age-dependent ventricular remodeling and dysfunction (80).
Parasympathetic System as a Target in Heart Failure
Both acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) modulate extracellular levels of ACh by regulating its hydrolysis. In fact, it has been shown that AChE KO mice are able to use BChE to breakdown ACh, suggesting a role for this enzyme in the regulation of cholinergic neurotransmission (81). Although the heart contains both AChE and BChE, the levels of BChE are much higher than that of AChE (82,83). Furthermore, both AChE and BChE are found in blood; however, the levels of circulating BChE are significantly greater than those of AChE (82) and likely serve to regulate ACh metabolism throughout the body. A previous report has suggested an inverse association between serum levels of BChE and overall mortality in middleaged and elderly individuals (84). Furthermore, the same report confirmed a positive association between BChE levels and several cardiovascular risk factors including albumin, cholesterol and triglycerides (84). An additional study suggested that increasing levels of BChE were associated with negative long-term outcomes, including mortality, in patients with stable coronary artery disease (85). In fact, the same group of researchers used cholinesterase activity as a bio-marker to predict mortality in coronary artery disease patients and found a strong inverse relationship between cholinesterase activity and prediction of mortality (86).
Although the aforementioned studies suggest the need for at least a minimal level of cholinesterase activity to reduce mortality in patients with cardiovascular disease, accumulating evidence suggests that restoring cholinergic activity after experimental heart failure can improve functional outcomes. In fact, in an animal model of heart failure, addition of vagal nerve stimulation to β-blockade therapy led to improvements in both cardiac contractility measured through left ventricular systolic and end-diastolic pressure as well as animal survival (87,88). Additionally, chronic treatment with the cholinesterase inhibitor donepezil was able to reduce cardiac remodeling and increase survival rates in animal models of heart failure (89,90). Furthermore, increasing ACh levels through administration of pyridostigmine, a peripheral cholinesterase inhibitor, led to greater vagal control of the heart and a reduction in ventricular dysfunction associated with heart failure in rats (91). Additionally, a recent study reported similar findings in two animal models of sympathetic hyperactivity where pyridostigmine administration prevented cardiac pathological remodeling and restored ventricular function (92).
Importantly, the positive effect of enhancing cholinergic tone is not limited to animal models of heart failure. In congestive heart failure patients, short-term administration of the cholinesterase inhibitor, pyridostigmine, led to a decrease in ventricular arrhythmias as well as an increase in heart rate variability, a parameter that serves as a marker for autonomic dysfunction in heart disease (93). Furthermore, a single dose of pyridostigmine reduced chronotropic responses during maximal exercise as well as improved heart rate recovery after maximal exercise in patients with chronic heart failure, suggesting that treatment with a cholinesterase inhibitor can enhance parasympathetic tone in heart failure patients (94,95). In addition, a clinical trial in which a small group of patients were subjected to vagal stimulation via an implantable system and followed for up to 6 months showed an increase in both their New York Heart Association class score as well as left ventricular end-systolic volume (96). It has also been shown that vagal nerve stimulation is tolerated well and improves quality of life and left ventricular function in chronic heart failure patients (97). Additionally, it was recently reported that the use of cholinesterase inhibitors in patients with Alzheimer’s disease led to a 34% reduction in risk of myocardial infarction and death (98). Finally, a phase II clinical trial is currently underway to study the safety of the cholinesterase inhibitor pyridostigmine when administered alone in heart failure patients (https://doi.org/clinicaltrials.gov identifier: NCT01415921). This study may serve as a precursor for additional clinical trials aimed at developing a novel therapeutic approach for the treatment of heart failure.
The mechanisms of action leading to the protective effects of cholinesterase inhibition have yet to be completely understood. One possibility is that cholinesterase inhibitors can reduce remodeling and cardiac dysfunction by directly enhancing the levels of ACh. However, it is also possible that quaternary cholinesterase inhibitors, such as pyridostigmine, can affect the progression of cardiac dysfunction indirectly via their ability to alter gene expression as previously shown in the brain (99,100), although this phenomenon has not yet been reported in the heart. Thus, evidence from the literature supports the fact that increasing systemic ACh levels through the use of cholinesterase inhibitors appears to decrease heart dysfunction after induction of cardiac disease. Additionally, oral administration of choline also appears to decrease the hypertrophic response in mice subjected to transverse aortic constriction, an effect that may depend on the actions of a specific miRNA, miR-133a, as well as calcineurin (101). Importantly, the antihypertrophic effects of orally administered choline are due to its direct actions on cardiomyocytes, since choline treatment prevents cellular hypertrophy in cultured cardiomyocytes treated with isoproterenol to induce remodeling (101).
The effect of choline is not limited to protection against afterload-induced hypertrophy. It has been reported that choline pretreatment also leads to a reduction in Ang II-induced remodeling through inhibition of cellular signaling. The mechanism seems to involve the reactive oxygen species (ROS)-mediated p38 mitogen-activated protein kinase (MAPK) pathway as well as the Ca2+-calcineurin signaling pathway, both of which are associated with Ang II-mediated cardiac dysfunction (102). Also in this example, the protective effect of choline appears to be due to its direct actions on cardiomyocytes, since pretreatment attenuated Ang II-induced hypertrophic response in cultured neonatal cardiomyocytes (102). However, it is unclear whether the protective effect of choline is due to its role in the production of ACh or because choline can act as a direct agonist of M3 muscarinic receptors, which have been linked to cardioprotection.
In agreement with the notion that cholinergic tone in the heart plays a role in heart failure, cardiac sympathetic nerves switch neurotransmitters and undergo cholinergic transdifferentiation during heart failure in both animal models and humans (103). Genetic inhibition of this transdifferentiation increases mortality of animal models, suggesting that this transdifferential switch is protective (103). This observation agrees with previous findings from our laboratory that genetic disturbance of cholinergic machinery in mice leads to cardiac dysfunction, a phenotype that can be reversed by treatment with peripheral cholinesterase inhibitors (78,79).
Nonneuronal Cholinergic System
For many years, ACh has been classified as the major neurotransmitter for parasympathetic signaling. The importance of ACh in the nervous system is indisputable; however, ACh can also play a significant role in cells other than neurons. A nonneuronal cholinergic system (NNCS) appears to be linked to proper function of various organ systems in both health and disease. ACh is a phylogenetically ancient molecule that can be produced and secreted by both prokaryotes (bacteria) and lower-order eukaryotes, including fungi and protozoa (1). Furthermore, several different cell types in higher-order eukaryotes, including humans, use ACh as a signaling molecule. These include keratinocytes, which have been shown to produce ACh at very high levels (104), as well as both oesophageal and upper respiratory tract epithelial cells (105,106). Furthermore, prototypical markers of the cholinergic system have also been detected in the kidney (107) as well as pancreatic α-cells, where nonneuronal ACh plays a crucial role in priming β-cells to secrete insulin in humans (108). Finally, the NNCS is active in regulating immune function as T cells secrete ACh, which can then act in an autocrine/paracrine fashion to inhibit inflammatory cytokine production (109,110).
A universally accepted principle in autonomic regulation of cardiac function is that parasympathetic innervation of the ventricles is very sparse (111,112). However, vagal stimulation has been shown to reduce ventricular contractility, in a mechanism independent of sympathetic signaling (113–115). These direct negative inotropic effects in response to vagal stimulation have led to further analysis of parasympathetic innervation in the ventricles. Importantly, although the level of cholinergic innervation in the ventricles is significantly less than adrenergic innervation, recent studies suggest that cholinergic nerve fibers are present at greater levels than previously thought. Immunostaining for AChE has revealed a dense innervation of both the endocardial and epicardial surfaces of the ventricles in several species, including human (116–118). However, immunostaining using the cholinergic marker CHT1 has revealed that the presence of parasympathetic nerve fibers in the ventricles is mainly restricted to the cardiac conduction system, although some staining was observed in the ventricular-free wall (111). In addition to vagal innervation of the ventricles, it was recently proposed that cardiomyocytes are also able to synthesize and release ACh at the cellular level. Cardiomyocytes posses the machinery (ChAT, VAChT and CHT1) for de novo production and storage of ACh (119,120). This nonneuronal ACh may then act in an autocrine/paracrine fashion to amplify neuronal cholinergic signaling and thereby protect the heart under stressful conditions, including heart failure (120). VAChT, which is an important component of the cholinergic machinery, was shown to be present in vesicles in cardiomyocytes (119), suggesting quantal release of ACh from these cells. Cardiomyocytes appear to differ from other nonneuronal cell types with regards to the mechanism of ACh secretion. Other tissues that secrete ACh, including the tracheal and colonic epithelium as well as human placenta, do not express the VAChT but rather use the organic cation transporters (OCT1/2/3) for ACh release (121–124). In contrast, VAChT-mediated secretion appears to be the sole mechanism of ACh release from cardiomyocytes (125,126). In addition, release of ACh by α-cells in the pancreas also requires VAChT (108).
Recently, we have shown that cholinesterase inhibition decreases cellular hypertrophy and remodeling induced by β-adrenergic stimulation in cultured cardiomyocytes (125). Specifically, we observed a decrease in the expression of cardiac stress markers, reduced nuclear factor of activated T-cells (NFAT) nuclear translocation and decreased peak systolic calcium after cholinesterase inhibition in isoproterenol-treated cardiomyocytes when compared with isoproterenol treatment alone (125). Furthermore, our in vivo work supports the notion that a nonneuronal cardiomyocyte-derived source of ACh plays a crucial role in regulating heart function (126). We have shown that inhibition of ACh secretion from cardiomyocytes through genetic deletion of VAChT selectively from cardiac cells, using the Cre/loxP system, leads to delayed heart rate recovery after exercise as well as significant ventricular remodeling (126). In addition, VAChT KO cardiomyocytes displayed increased hypertrophy as well as activation of the fetal gene program, increased ROS production and enhanced peak systolic calcium when compared with control mice (126). Furthermore, the molecular remodeling observed in cardiomyocyte-specific VAChT-KO mice led to abnormal ventricular hemodynamics after a bolus dose of isoproterenol, suggesting that the NNCS plays a role in regulating cardiac function under stress (126). Importantly, cardiomyocyte-specific ChAT-KO mice showed a similar phenotype, thus supporting the notion that the NNCS is critical in regulating physiological responses independent of vagal signaling.
Other studies have also highlighted the importance of the NNCS in regulating heart physiology. Recent work has shown that ChAT KO HL-1 cells, derived from murine atrial cardiac tissue, display lower levels of cellular ATP, which leads to decreased viability after induction of chemical hypoxia (127). In addition, ChAT KO HL-1 cells appear to generate greater levels of ROS than control cells after an acute challenge with norepinephrine, as measured using the ROS indicator aminophenyl fluorescein (APF) (127). This increase in ROS production in ChAT KO cells may contribute to the increased caspase-3 activation and apoptotic response observed after norepinephrine treatment (127). Conversely, the overexpression of choline acetyltransferase (ChAT-Tg), the enzyme responsible for the synthesis of ACh, specifically in cardiomyocytes, appears to decrease remodeling and enhance survival after myocardial infarction (128). These ChAT-Tg mice also appear to be less susceptible to ex vivo ischemia-reperfusion injury, induced by using a Langerdorff apparatus (128). The infarcted region was significantly decreased in the ChAT-Tg mice, and the time from onset of ischemia to beating arrest was significantly increased in the mutant mice (128). These data further highlight the importance of the cardiac NNCS after induction of stress, including ischemia. Together, these findings provide for an unanticipated mechanism by which nonneuronal ACh may play an important role in cardiac function.
Cholinergic Antiinflammatory Pathway
ACh secretion from nonneuronal cells is not unique to cardiomyocytes. Lymphocytes can also release ACh, which plays a crucial role in regulating the immune response by activating an inflammatory reflex, the cholinergic antiinflammatory pathway (109,110,129). This reflex depends on the peripheral actions of ACh released from the vagus (130,131); however, nerve fibers innervating the splenic nerve from the celiac ganglion are adrenergic rather than cholinergic (132). As such, the presence of ACh in the spleen is not due to neuronal cholinergic innervation but, rather, is due to activation of the efferent arc of the vagus nerve and downstream activation of splenic T cells, which then secrete ACh (109). This T cell-derived ACh can then activate α7 nicotinic acetylcholine receptors (nAChRs) on macrophages and thereby inhibit proinflammatory cytokine secretion, including tumor necrosis factor (TNF)-α, interleukin (IL)-1α and IL-6 (130,131). The antiinflammatory effects of ACh appear to be mediated specifically by its interaction with the α7 receptors on macrophages, since this effect can be blunted using selective nicotinic antagonists or α7 receptor KO mice (131,133–135).
This pathway appears to play a crucial role in the progression of several cardiovascular diseases. In fact, in a two-kidney one-clip animal model of hypertension, a significant downregulation of α7 receptors, coupled with an increase in serum levels of TNF-α, was previously observed (136). This occurred downstream of reduced vagal tone, thus suggesting that the cholinergic antiinflammatory pathway plays a role in the induction of secondary hypertension. Furthermore, increased expression of inflammatory cytokines, which may serve as an indicator of reduced activity of the cholinergic antiinflammatory reflex, has been observed in systemic circulation during chronic heart failure as well as in the failing myocardium (137,138).
Activation of the cholinergic anti-inflammatory pathway, which depends on both vagal as well as nonneuronal ACh, has been shown to be beneficial in heart failure (139) and may contribute to the cardioprotective effects of increased cholinergic tone observed in several animal models of heart failure, including volume overload, tachycardia and myocardial infarction (87–90). This cholinergic antiinflammatory pathway serves as an important determinant of the extent of cardiac remodeling after induction of cardiac disease. Notably, it appears to be altered in heart failure because of the decrease in parasympathetic tone and thus peripheral ACh secretion. In fact, it was recently shown that, after LPS treatment-induced endotoxemia in rats, vagal nerve stimulation partially attenuated myocardial inflammation and hemodynamic alterations (140). Furthermore, the inflammatory response in these mice was enhanced after vagotomy, thus suggesting that parasympathetic signaling via the vagus plays a critical role in reducing the local inflammatory response and decreasing cardiac dysfunction (140).
The exact relationship between levels of circulating inflammatory cytokines and cardiac dysfunction is still unclear. However, recent observations suggest the possibility that inflammation can have adverse effects on the progression of heart failure. In a canine high-rate pacing model of heart failure, chronic vagal nerve stimulation led to significant inhibition of heart failure development, which was associated with attenuated systemic inflammation (88), thus suggesting a possible beneficial effect of reduced inflammatory cytokines in heart failure. Additionally, spontaneously hypertensive rats, as well as those with abdominal aorta coarctation-induced hypertension, exhibited greater end organ damage, coupled with decreased levels of the α7nAChR (141). Chronic treatment of spontaneously hypertensive rats with an α7nAChR agonist led to decreased tissue levels of proinflammatory cytokines, which was associated with decreased end organ damage (141). Increased levels of inflammatory cytokines have been shown to be directly related to decreased left ventricular ejection fraction (142). Moreover, increased levels of the proinflammatory cytokine TNF-α can induce cardiac hypertrophy and promote cardiomyocyte apoptosis, thus contributing to the onset and progression of ventricular dysfunction and remodeling (137,143). Therefore, these data suggest an important role for the cholinergic inflammatory reflex in regulating cardiac function and attenuating the progression of heart failure, perhaps by regulating levels of circulating cytokines such as TNF-α.
Significant amounts of data suggest that vagal stimulation can induce an antiinflammatory response. However, it was recently reported that the reflex activation of the inflammatory system by the autonomic nervous system after LPS treatment is due to the actions of the sympathetic system (144). Furthermore, a previous study by the same group suggested that vagal nerve stimulation in anesthetized rats did not lead to activation of splenic nerves (145). As such, it is possible that sympathetic splanchnic nerves are responsible for the direct activation of postganglionic splenic nerves. Furthermore, although currently unclear, it is possible that vagal signaling can inhibit secretion of antiinflammatory cytokines via an unexplored, indirect pathway. Regardless of the specific mechanisms through which vagal signaling can elicit its effects, it is well established that the parasympathetic nervous system is important in regulating the innate immune response and can inhibit cytokine release and systemic inflammation (146). In fact, several studies have provided novel insight into the specific mechanisms through which ACh can act peripherally to control the immune response and regulate the extent of cardiac dysfunction (88,140,141).
Conclusion
Although the exact role of the para-sympathetic nervous system in cardiac dysfunction and heart failure has yet to be completely understood, a number of studies have recently highlighted the importance of modulating this system in heart disease. It is evident that increasing ACh levels, through either vagal stimulation or cholinesterase inhibitors, can act through a variety of pathways (Figure 1) and reduce cardiac remodeling in several different models of cardiac disease. Furthermore, recent data suggest that it may be possible to modulate the activity of the NNCS present in both cardiomyocytes, as well as T cells, to delay the progression of cardiac dysfunction. As such, the cholinergic system may serve as an important pharmacological target in patients with heart disease.

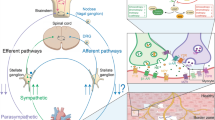
Autonomie regulation of cardiac function. (1) Baroreceptors in the carotid arteries and aortic arch monitor arterial pressure. Increased arterial pressure activates signaling to the nucleus of the tractus solitarius (NTS) and reduces sympathetic output and simultaneously activates the parasympathetic nervous system. A decrease in arterial pressure has the opposite effect. (2) Activation of sympathetic nerves leads to positive chronotropic and inotropic responses as well as arteriolar constriction. (3) Activation of the parasympathetic system via the vagus nerve leads to negative chronotropy and lusitropy. (4) The cardiac NNCS amplifies parasympathetic cholinergic signaling and regulates heart rate recovery after activity. In addition, the cardiomyocyte-derived ACh prevents cardiac remodeling and hypertrophy under physiological conditions. (5) Vagus and/or sympathetic splanchnic nerve stimulation leads to activation of the antiinflammatory pathway, which involves secretion of nonneuronal ACh from splenic T cells. This T cell-derived ACh activates α7 nicotinic acetylcholine receptors (α7nAChRs) on macrophages and inhibits the release of proinflammatory cytokines.
Recent efforts to repurpose drugs in cancer and neurological diseases are underway with the leadership of the National Institutes of Health (NIH) (147). The safety and tolerance of cholinesterase inhibitors are well known because of their wide use in Alzheimer’s disease. Therefore, it would be easy to repurpose this class of drugs for heart disease. Modulation of cholinergic signaling may serve as a novel, unexplored clinical avenue for the treatment of heart failure in humans. However, clinical data on the benefits of cholinesterase inhibitors in heart failure are still scant. Clinical trials are certainly warranted to test potential benefits of adding cholinesterase inhibitors to current therapies and evaluating the long-term effects of cholinesterase inhibitors at different stages of heart failure.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Wessler I, Kirkpatrick CJ, Racke K. (1999) The cholinergic ‘pitfall’: acetylcholine, a universal cell molecule in biological systems, including humans. Clin. Exp. Pharmacol. Physiol 26:198–205.
Nachmansohn D, Machado AL. (1943) The formation of acetylcholine: a new enzyme: choline acetylase. J. Neurophysiol 6:397–403.
Brandon EP, et al. (2004) Choline transporter 1 maintains cholinergic function in choline acetyltransferase haploinsufficiency. J. Neurosci. 24:5459–66.
Okuda T, Haga T. (2003) High-affinity choline transporter. Neurochem. Res. 28:483–8.
Kuhar MJ, Murrin LC. (1978) Sodium-dependent, high affinity choline uptake. J. Neurochem. 30:15–21.
Ferguson SM, et al. (2004) Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc. Natl. Acad. U. S. A. 101:8762–7.
Erickson JD, et al. (1994) Functional identification of a vesicular acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem. 269:21929–32.
de Castro BM, et al. (2009) The vesicular acetylcholine transporter is required for neuromuscular development and function. Mol. Cell. Biol. 29:5238–50.
Prado VF, et al. (2006) Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. 51:601–12.
Song H, et al. (1997) Expression of a putative vesicular acetylcholine transporter facilitates quantal transmitter packaging. Neuron. 18:815–26.
Kolisnyk B, et al. (2013) ChAT-ChR2-EYFP mice have enhanced motor endurance but show deficits in attention and several additional cognitive domains. J. Neurosci. 33:10427–38.
Nagy PM, Aubert I. (2012) Overexpression of the vesicular acetylcholine transporter increased acetylcholine release in the hippocampus. Neuroscience. 218:1–11.
Kent KM, Epstein SE, Cooper T, Jacobowitz DM. (1974) Cholinergic innervation of the canine and human ventricular conducting system: anatomic and electrophysiologic correlations. Circulation. 50:948–55.
Brodde OE, Bruck H, Leineweber K, Seyfarth T. (2001) Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res. Cardiol. 96:528–38.
Agostoni E, Chinnock JE, De Daly MB, Murray JG. (1957) Functional and histological studies of the vagus nerve and its branches to the heart, lungs and abdominal viscera in the cat. J. Physiol. 135:182–205.
Sunahara RK, Dessauer CW, Gilman AG. (1996) Complexity and diversity of mammalian adenylyl cyclases. Ann. Rev. Pharmacol. Toxicol. 36:461–80.
Ishikawa Y, Homcy CJ. (1997) The adenylyl cyclases as integrators of transmembrane signal transduction. Circ. Res. 80:297–304.
Osterrieder W, et al. (1982) Injection of subunits of cyclic AMP-dependent protein kinase into cardiac myocytes modulates Ca2+ current. Nature. 298:576–8.
Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. (1987) The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 325:321–6.
DiFrancesco D, Ducouret P, Robinson RB. (1989) Muscarinic modulation of cardiac rate at low acetylcholine concentrations. Science. 243:669–71.
DiFrancesco D, Tromba C. (1987) Acetylcholine inhibits activation of the cardiac hyperpolarizingactivated current, if. Pflugers Arch. 410:139–42.
Peralta EG, et al. (1987) Distinct primary structures, ligand-binding properties and tissue-specific expression of four human muscarinic acetylcholine receptors. EMBO J. 6:3923–9.
Caulfield MP. (1993) Muscarinic receptors: characterization, coupling and function. Pharmacol. Ther. 58:319–79.
Ponicke K, Heinroth-Hoffmann I, Brodde OE. (2003) Demonstration of functional M3-muscarinic receptors in ventricular cardiomyocytes of adult rats. Br. J. Pharmacol. 138:156–60.
Nishimaru K, Tanaka Y, Tanaka H, Shigenobu K. (2000) Positive and negative inotropic effects of muscarinic receptor stimulation in mouse left atria. Life Sci. 66:607–15.
Pan Z, et al. (2012) M3 subtype of muscarinic acetylcholine receptor promotes cardioprotection via the suppression of miR-376b-5p. PloS One. 7:e32571.
Liu Y, et al. (2013) Upregulation of M(3) muscarinic receptor inhibits cardiac hypertrophy induced by angiotensin II. J. Transl. Med. 11:209.
Chiappinelli VA, Dryer SE. (1984) Nicotinic transmission in sympathetic ganglia: blockade by the snake venom neurotoxin kappa-bungarotoxin. Neurosci. Lett. 50:239–44.
Galvan M. (1988) Modulation of synaptic transmission in autonomic ganglia mediated via the activation of postganglionic muscarinic M1 receptors. Pharmacology. 37 (Suppl. 1):11–6.
Habermeier-Muth A, Altes U, Forsyth KM, Muscholl E. (1990) A presynaptic excitatory M1 muscarine receptor at postganglionic cardiac noradrenergic nerve fibres that is activated by endogenous acetylcholine. Naunyn-Schmiedebergs Arch. Pharmacol. 342:483–9.
Trendelenburg AU, Gomeza J, Klebroff W, Zhou H, Wess J. (2003) Heterogeneity of presynaptic muscarinic receptors mediating inhibition of sympathetic transmitter release: a study with M2- and M4-receptor-deficient mice. Br. J. Pharmacol. 138:469–80.
Liu HR, Zhao RR, Jiao XY, Wang YY, Fu M. (2002) Relationship of myocardial remodeling to the genesis of serum autoantibodies to cardiac beta(1)-adrenoceptors and muscarinic type 2 acetylcholine receptors in rats. J. Am. Coll. Cardiol. 39:1866–73.
Hollinger S, Hepler JR. (2002) Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 54:527–59.
Heximer SP, Watson N, Linder ME, Blumer KJ, Hepler JR. (1997) RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc. Natl. Acad. U. S. A. 94:14389–93.
Chidiac P, Ross EM. (1999) Phospholipase C-beta1 directly accelerates GTP hydrolysis by Galphaq and acceleration is inhibited by Gbeta gamma subunits. J. Biol. Chem. 274:19639–43.
Heximer SP, et al. (1999) G protein selectivity is a determinant of RGS2 function. J. Biol. Chem. 274:34253–9.
Cladman W, Chidiac P. (2002) Characterization and comparison of RGS2 and RGS4 as GTPase-activating proteins for m2 muscarinic receptor-stimulated G(i). Mol. Pharmacol. 62:654–9.
Roy AA, et al. (2006) Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signaling in osteoblasts. J. Biol. Chem. 281:32684–93.
Nunn C, et al. (2010) RGS2 inhibits beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Cell. Sig. 22:1231–9.
Chidiac P, Sobiesiak AJ, Lee KN, Gros R, Nguyen CH. (2014) The eIF2B-interacting domain of RGS2 protects against GPCR agonist-induced hypertrophy in neonatal rat cardiomyocytes. Cell. Sig. 26:1226–1234.
Heximer SP, et al. (2003) Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J. Clin. Invest. 111:445–52.
Takimoto E, et al. (2009) Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J. Clin. Invest. 119:408–20.
Just A, Faulhaber J, Ehmke H. (2000) Autonomic cardiovascular control in conscious mice. Am. J. Physiol. 279:R2214–21.
Olshansky B, Sabbah HN, Hauptman PJ, Colucci WS. (2008) Parasympathetic nervous system and heart failure: pathophysiology and potential implications for therapy. Circulation. 118:863–71.
Go AS, et al. (2014) Heart disease and stroke statistics: 2014 update: a report from the American Heart Association. Circulation. 129:e28–e292.
Mann DL. (1999) Mechanisms and models in heart failure: a combinatorial approach. Circulation. 100:999–1008.
Braunwald E, Bristow MR. (2000) Congestive heart failure: fifty years of progress. Circulation. 102:IV14–23.
Floras JS. (2009) Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J. Am. Coll. Cardiol. 54:375–85.
Massie BM. (1988) Is neurohormonal activation deleterious to the long-term outcome of patients with congestive heart failure? I. Introduction. J. Am. Coll. Cardiol. 12:547–550.
Grassi G, et al. (2001) Sympathetic and reflex abnormalities in heart failure secondary to ischaemic or idiopathic dilated cardiomyopathy. Clin. Sci. (Lond). 101:141–6.
Grassi G, et al. (1995) Sympathetic activation and loss of reflex sympathetic control in mild congestive heart failure. Circulation. 92:3206–11.
Rockman HA, Koch WJ, Lefkowitz RJ. (2002) Seven-transmembrane-spanning receptors and heart function. Nature. 415:206–12.
DiPiro JT, et al. (2008) Pharmacotherapy: A Pathophysiological Approach. 7th ed. New York: McGraw-Hill Medical. 2559 pp.
Triposkiadis F, et al. (2009) The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 54:1747–62.
Grassi G, et al. (2009) Sympathetic and baroreflex cardiovascular control in hypertension-related left ventricular dysfunction. Hypertension. 53:205–9.
Zucker IH, et al. (2007) Chronic baroreceptor activation enhances survival in dogs with pacing-induced heart failure. Hypertension. 50:904–10.
Yancy CW, et al. (2013) 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 62:e147–239.
Hjalmarson A, et al. (1999) Effect of metoprolol CR XL in chronic heart failure: Metoprolol CR XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet. 353:2001–7.
Poole-Wilson PA, et al. (2003) Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol, European Trial (COMET): randomised controlled trial. Lancet. 362:7–13.
Wollert KC, Drexler H. (2002) Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial: carvedilol as the sun and center of the beta-blocker world? Circulation. 106:2164–66.
Iwata M, et al. (2001) Autoantibodies against the second extracellular loop of beta1-adrenergic receptors predict ventricular tachycardia and sudden death in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 37:418–24.
Peukert S, et al. (1999) The frequency of occurrence of anti-cardiac receptor autoantibodies and their correlation with clinical manifestation in patients with hypertrophic cardiomyopathy. Autoimmunity. 29:291–7.
Magnusson Y, Wallukat G, Waagstein F, Hjalmarson A, Hoebeke J. (1994) Autoimmunity in idiopathic dilated cardiomyopathy: characterization of antibodies against the beta(1)-adrenoceptor with positive chronotropic effect. Circulation. 89:2760–7.
Davies JE, et al. (2013) First-in-man safety evaluation of renal denervation for chronic systolic heart failure: primary outcome from REACH-Pilot study. Int. J. Cardiol. 162:189–92.
Brandt MC, et al. (2012) Renal sympathetic denervation reduces left ventricular hypertrophy and improves cardiac function in patients with resistant hypertension. J. Am. Coll. Cardiol. 59:901–9.
Linz D, et al. (2012) Renal sympathetic denervation suppresses postapneic blood pressure rises and atrial fibrillation in a model for sleep apnea. Hypertension. 60:172–8.
Ukena C, et al. (2013) Effects of renal sympathetic denervation on heart rate and atrioventricular conduction in patients with resistant hypertension. Int. J. Cardiol. 167:2846–51.
Binkley PF, Nunziata E, Haas GJ, Nelson SD, Cody RJ. (1991) Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J. Am. Coll. Cardiol. 18:464–72.
Vanoli E, et al. (1991) Vagal stimulation and prevention of sudden death in conscious dogs with a healed myocardial infarction. Circ. Res. 68:1471–81.
Billman GE, Schwartz PJ, Stone HL. (1982) Baroreceptor reflex control of heart rate: a predictor of sudden cardiac death. Circulation. 66:874–80.
Fu LX, et al. (1993) Localization of a functional autoimmune epitope on the muscarinic acetylcholine receptor-2 in patients with idiopathic dilated cardiomyopathy. J. Clin. Invest. 91:1964–8.
Kinugawa T, Dibner-Dunlap ME. (1995) Altered vagal and sympathetic control of heart rate in left ventricular dysfunction and heart failure. Am. J. Physiol. 268:R310–6.
Bibevski S, Dunlap ME. (1999) Ganglionic mechanisms contribute to diminished vagal control in heart failure. Circulation. 99:2958–63.
Schwartz PJ, De Ferrari GM. (2011) Sympathetic-parasympathetic interaction in health and disease: abnormalities and relevance in heart failure. Heart Fail. Rev. 16:101–7.
LaCroix C, Freeling J, Giles A, Wess J, Li YF. (2008) Deficiency of M2 muscarinic acetylcholine receptors increases susceptibility of ventricular function to chronic adrenergic stress. Am. J. Physiol. 294:H810–20.
Martos R, et al. (2007) Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 115:888–895.
Yan AT, et al. (2006) Plasma matrix metallopro-teinase-9 level is correlated with left ventricular volumes and ejection fraction in patients with heart failure. J. Card. Fail. 12:514–9.
Lara A, et al. (2010) Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol. Cell. Biol. 30:1746–56.
Roy A, et al. (2012) An analysis of the myocardial transcriptome in a mouse model of cardiac dysfunction with decreased cholinergic neurotransmission. PLoS One. 7:e39997.
English BA, et al. (2010) Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am. J. Physiol. Heart Circ. Physiol. 299:H799–810.
Mesulam MM, et al. (2002) Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 110:627–39.
Chatonnet A, Lockridge O. (1989) Comparison of butyrylcholinesterase and acetylcholinesterase. Biochem. J. 260:625–34.
Li B, et al. (2000) Abundant tissue butyrylcholinesterase and its possible function in the acetylcholinesterase knockout mouse. J. Neurochem. 75:1320–31.
Calderon-Margalit R, Adler B, Abramson JH, Gofin J, Kark JD. (2006) Butyrylcholinesterase activity, cardiovascular risk factors, and mortality in middle-aged and elderly men and women in Jerusalem. Clin. Chem. 52:845–52.
Goliasch G, et al. (2012) Butyrylcholinesterase activity predicts long-term survival in patients with coronary artery disease. Clin. Chem. 58:1055–8.
Goliasch G, et al. (2012) Routinely available biomarkers improve prediction of long-term mortality in stable coronary artery disease: the Vienna and Ludwigshafen Coronary Artery Disease (VILCAD) risk score. Eur. Heart J. 33:2282–9.
Li M, et al. (2004) Vagal nerve stimulation markedly improves long-term survival after chronic heart failure in rats. Circulation. 109:120–4.
Zhang Y, et al. (2009) Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high-rate pacing model. Circ. Heart Fail. 2:692–9.
Handa T, et al. (2009) Anti-Alzheimer’s drug, donepezil, markedly improves long-term survival after chronic heart failure in mice. J. Card. Fail. 15:805–11.
Li M, et al. (2013) Donepezil markedly improves long-term survival in rats with chronic heart failure after extensive myocardial infarction. Circ. J. 77:2519–25.
Lataro RM, et al. (2013) Increase in parasympathetic tone by pyridostigmine prevents ventricular dysfunction during the onset of heart failure. Am. J. Physiol. 305:R908–16.
Gavioli M, et al. (2014) Cholinergic signaling exerts protective effects in models of sympathetic hyperactivity-induced cardiac dysfunction. PLoS One. 9:e100179.
Behling A, et al. (2003) Cholinergic stimulation with pyridostigmine reduces ventricular arrhythmia and enhances heart rate variability in heart failure. Am. Heart J. 146:494–500.
Androne AS, Hryniewicz K, Goldsmith R, Arwady A, Katz SD. (2003) Acetylcholinesterase inhibition with pyridostigmine improves heart rate recovery after maximal exercise in patients with chronic heart failure. Heart. 89:854–8.
Serra SM, Costa RV, Teixeira De Castro RR, Xavier SS, Nobrega AC. (2009) Cholinergic stimulation improves autonomic and hemodynamic profile during dynamic exercise in patients with heart failure. J. Card. Fail. 15:124–9.
Schwartz PJ, et al. (2008) Long term vagal stimulation in patients with advanced heart failure: first experience in man. Eur. J. Heart Fail. 10:884–91.
De Ferrari GM, et al. (2011) Chronic vagus nerve stimulation: a new and promising therapeutic approach for chronic heart failure. Eur. Heart J. 32:847–55.
Nordstrom P, Religa D, Wimo A, Winblad B, Eriksdotter M. (2013) The use of cholinesterase inhibitors and the risk of myocardial infarction and death: a nationwide cohort study in subjects with Alzheimer’s disease. Eur. Heart J. 34:2585–91.
Friedman A, et al. (1996) Pyridostigmine brain penetration under stress enhances neuronal excitability and induces early immediate transcriptional response. Nat. Med. 2:1382–5.
Dillman JF 3rd, et al. (2009) Gene expression profiling of rat hippocampus following exposure to the acetylcholinesterase inhibitor soman. Chem. Res. Toxicol. 22:633–8.
Zhao Y, et al. (2013) Choline protects against cardiac hypertrophy induced by increased afterload. Int. J. Biol. Sci. 9:295–302.
Wang S, et al. (2012) Choline inhibits angiotensin II-induced cardiac hypertrophy by intracellular calcium signal and p38 MAPK pathway. Naunyn-Schmiedebergs Arch. Pharmacol. 385:823–31.
Kanazawa H, et al. (2010) Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J. Clin. Invest. 120:408–21.
Grando SA, Kist DA, Qi M, Dahl MV. (1993) Human keratinocytes synthesize, secrete, and degrade acetylcholine. J. Invest. Dermatol. 101:32–6.
Reinheimer T, et al. (1998) Glucocorticoids mediate reduction of epithelial acetylcholine content in the airways of rats and humans. Euro. J. Pharmacol. 349:277–84.
Nguyen VT, et al. (2000) Choline acetyltransferase, acetylcholinesterase, and nicotinic acetylcholine receptors of human gingival and esophageal epithelia. J. Dent. Res. 79:939–49.
Maeda S, et al. (2011) Non-neuronal expression of choline acetyltransferase in the rat kidney. Life Sci. 89:408–14.
Rodriguez-Diaz R, et al. (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat. Med. 17:888–92.
Rosas-Ballina M, et al. (2011) Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 334:98–101.
Fujii T, et al. (1996) Localization and synthesis of acetylcholine in human leukemic T cell lines. J. Neurosci. Res. 44:66–72.
Hoover DB, Ganote CE, Ferguson SM, Blakely RD, Parsons RL. (2004) Localization of cholinergic innervation in guinea pig heart by immunohistochemistry for high-affinity choline transporters. Cardiovasc. Res 62:112–21.
Crick SJ, et al. (1994) Innervation of the human cardiac conduction system: a quantitative immunohistochemical and histochemical study. Circulation. 89:1697–708.
Gatti PJ, et al. (1997) Vagal control of left ventricular contractility is selectively mediated by a cranioventricular intracardiac ganglion in the cat. J. Auton. Nerv. Syst. 66:138–44.
Nakano H, et al. (1998) Right ventricular responses to vagus stimulation of fibers to discrete cardiac regions in dog hearts. J. Auton. Nerv. Syst. 74:179–88.
Lewis ME, et al. (2001) Vagus nerve stimulation decreases left ventricular contractility in vivo in the human and pig heart. J. Physiol. 534:547–52.
Rysevaite K, et al. (2011) Immunohistochemical characterization of the intrinsic cardiac neural plexus in whole-mount mouse heart preparations. Heart Rhythm. 8:731–8.
Zang WJ, Chen LN, Yu XJ. (2005) Progress in the study of vagal control of cardiac ventricles. Sheng Li Xue Bao. 57:659–72.
Kawano H, Okada R, Yano K. (2003) Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels. 18:32–9.
Rana OR, et al. (2010) Acetylcholine as an age-dependent non-neuronal source in the heart. Auton. Neurosci. 156:82–9.
Kakinuma Y, Akiyama T, Sato T. (2009) Cholinoceptive and cholinergic properties of cardiomyocytes involving an amplification mechanism for vagal efferent effects in sparsely innervated ventricular myocardium. FEBS J. 276:5111–25.
Kummer W, et al. (2006) Role of acetylcholine and polyspecific cation transporters in serotonin-induced bronchoconstriction in the mouse. Respir. Res. 7:65.
Lips KS, et al. (2005) Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am. J. Respir. Cell Mol. Biol. 33:79–88.
Wessler I, et al. (2001) Release of non-neuronal acetylcholine from the isolated human placenta is mediated by organic cation transporters. Br. J. Pharmacol. 134:951–6.
Bader S, Klein J, Diener M. (2014) Choline acetyltransferase and organic cation transporters are responsible for synthesis and propionate-induced release of acetylcholine in colon epithelium. Eur. J. Pharmacol. 733:23–33.
Rocha-Resende C, et al. (2012) Non-neuronal cholinergic machinery present in cardiomyocytes offsets hypertrophic signals. J. Mol. Cell. Cardiol. 53:206–16.
Roy A, et al. (2013) Cardiomyocyte-secreted acetylcholine is required for maintenance of homeostasis in the heart. FASEB J. 27:5072–82.
Kakinuma Y, et al. (2012) A non-neuronal cardiac cholinergic system plays a protective role in myocardium salvage during ischemic insults. PLoS One. 7:e50761.
Kakinuma Y, et al. (2013) Heart-specific overexpression of choline acetyltransferase gene protects murine heart against ischemia through hypoxia-inducible factor-1alpha-related defense mechanisms. J. Am. Heart Assoc. 2:e004887.
Rinner I, Schauenstein K. (1993) Detection of choline-acetyltransferase activity in lymphocytes. J. Neurosci. Res. 35:188–91.
Tracey KJ. (2002) The inflammatory reflex. Nature. 420:853–9.
Wang H, et al. (2003) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 421:384–8.
Bellinger DL, Lorton D, Hamill RW, Felten SY, Felten DL. (1993) Acetylcholinesterase staining and choline acetyltransferase activity in the young adult rat spleen: lack of evidence for cholinergic innervation. Brain Behav. Immun. 7:191–204.
Guarini S, et al. (2003) Efferent vagal fibre stimulation blunts nuclear factor-kappaB activation and protects against hypovolemic hemorrhagic shock. Circulation. 107:1189–94.
Shytle RD, et al. (2004) Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 89:337–43.
Borovikova LV, et al. (2000) Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 405:458–62.
Chen JK, et al. (2012) Downregulation of alpha7 nicotinic acetylcholine receptor in two-kidney one-clip hypertensive rats. BMC Cardiovasc. Disord. 12:38.
Levine B, Kalman J, Mayer L, Fillit HM, Packer M. (1990) Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 323:236–41.
Shan K, et al. (1997) The role of cytokines in disease progression in heart failure. Curr. Opin. Cardiol. 12:218–23.
Mann DL. (2002) Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ. Res. 91:988–98.
Schulte A, Lichtenstern C, Henrich M, Weigand MA, Uhle F. (2014) Loss of vagal tone aggravates systemic inflammation and cardiac impairment in endotoxemic rats. J. Surg. Res. 188:480–8.
Li DJ, et al. (2011) Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension. Hypertension. 57:298–307.
Aukrust P, et al. (1998) Elevated circulating levels of C-C chemokines in patients with congestive heart failure. Circulation. 97:1136–43.
Kubota T, et al. (1997) Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ. Res. 81:627–35.
Martelli D, Yao ST, McKinley MJ, McAllen RM. (2014) Reflex control of inflammation by sympathetic nerves, not the vagus. J. Physiol. 592:1677–86.
Bratton BO, et al. (2012) Neural regulation of inflammation: no neural connection from the vagus to splenic sympathetic neurons. Exp. Physiol. 97:1180–5.
Borovikova LV, et al. (2000) Role of vagus nerve signaling in CNI-1493-mediated suppression of acute inflammation. Auton. Neurosci. 85:141–7.
Collins FS. (2011) Mining for therapeutic gold. Nat. Rev. Drug Discov. 10:397.
Acknowledgments
The authors thank Hermona Soreq for comments on an early version of this review and Ciric To for preparation of the figure in this paper. Research by the authors is supported by the Heart & Stroke Foundation, Canadian Foundation for Innovation, Ontario Research Trust, Canadian Institutes of Health Research, The Weston Brain Institute and a multiinvestigator grant from Brain Canada (Canada). S Guatimosim was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) (Brazil) and the NIH-Fogarty Center. A Roy was the recipient of an Ontario Graduate Scholarship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Roy, A., Guatimosim, S., Prado, V.F. et al. Cholinergic Activity as a New Target in Diseases of the Heart. Mol Med 20, 527–537 (2014). https://doi.org/10.2119/molmed.2014.00125
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2014.00125