
Abstract
High-mobility group box 1 (HMGB1) protein is a highly abundant protein that can promote the pathogenesis of inflammatory and autoimmune diseases once it is in an extracellular location. This translocation can occur with immune cell activation as well as cell death, with the conditions for release associated with the expression of different isoforms. These isoforms result from post-translational modifications, with the redox states of three cysteines at positions 23, 45 and 106 critical for activity. Depending on the redox states of these residues, HMGB1 can induce cytokine production via toll-like receptor 4 (TLR4) or promote chemotaxis by binding the chemokine CXCL12 for stimulation via CXCR4. Fully oxidized HMGB1 is inactive. During the course of inflammatory disease, HMGB1 can therefore play a dynamic role depending on its redox state. As a mechanism to generate alarmins, cell death is an important source of HMGB1, although each major cell death form (necrosis, apoptosis, pyroptosis and NETosis) can lead to different isoforms of HMGB1 and variable levels of association of HMGB1 with nucleosomes. The association of HMGB1 with nucleosomes may contribute to the pathogenesis of systemic lupus erythematosus by producing nuclear material whose immunological properties are enhanced by the presence of an alarmin. Since HMGB1 levels in blood or tissue are elevated in many inflammatory and autoimmune diseases, this molecule can serve as a unique biomarker as well as represent a target of novel therapies to block its various activities.
Similar content being viewed by others

Introduction
High mobility group box 1 (HMGB1) protein is a highly abundant and conserved protein that has important biological activities inside as well as outside the cell. Inside the nucleus, HMGB1 interacts with DNA and histones to determine chromatin structure and regulate key processes such as transcription. HMGB1 has other intracellular actions since, when translocated to the cytoplasm, it can mediate autophagy. Outside the cell, HMGB1 acquires a new identity to serve as an alarmin or a damage-associated molecular pattern (DAMP) (1,2). In addition to the intrinsic activity of HMGB1, the extracellular actions of this molecule are extended by interaction with pathogen-associated molecular patterns (PAMPs), cytokines and chemokines (3). As such, HMGB1 can play multiple roles in the pathogenesis of inflammatory and autoimmune disease and mediate processes that range from inflammation to repair (4).
The biological actions of HMGB1 are striking in their diversity, reflecting the unique biochemistry of this protein and its propensity for posttranslational modification. Therefore, to elucidate the role of HMGB1 as a mediator of autoimmune and inflammatory disease, in this review, we emphasize that (a) HMGB1 is an ensemble of molecules and not a single species with a fixed structure; (b) the function of HMGB1 can evolve over time because of posttranslational modification; and (c) cell death, a major source of extracellular HMGB1 in immune-mediated disease, has multiple forms whose immunological activities vary.
Expression of HMGB1 in Inflammatory and Autoimmune Disease
HMGB1 was originally defined in some of the most acute settings of medicine (that is, sepsis, shock) (5), but its intriguing immunological properties rapidly prompted the study of its role in chronic inflammatory and autoimmune conditions, with rheumatoid arthritis (RA) receiving the most attention. Evidence for a key role of HMGB1 in immune-mediated disease is compelling and has come from studies demonstrating (a) increased HMGB1 levels in the blood or other biological fluid (for example, synovial fluid, urine); (b) increased HMGB1 expression in diseased tissue, especially outside the cell; (c) induction of characteristic tissue pathology (for example, arthritis) or functional disturbance (for example, muscle weakness in myositis) by HMGB1 in in vitro or in vivo models; and (d) effectiveness of agents directed at HMGB1 in animal models (6–19). Table 1 lists diseases in which studies implicate a role of HMGB1 in pathogenesis.
Given the function of HMGB1 as an alarmin, its role in pathogenesis has been attributed to its ability to induce the production of cytokines, promote chemotaxis, activate cells (immune, endothelial, fibroblast) or serve as an adjuvant to stimulate the production of autoantibodies. As such, HMGB1 would serve a role in pathogenesis similar to that of conventional cytokines or chemokines. The main difference between conventional cytokines and HMGB1 relates to doses required for these activities; as shown in in vitro studies, HMGB1 stimulation requires higher concentrations than cytokines for activity. Such dose dependency could suggest a more significant role of HMGB1 operating at local sites of inflammation in contrast to systemically.
While studies on the role of HMGB1 in inflammatory and autoimmune disease have been enormously informative, they have been based on a paradigm that is currently undergoing rapid change. In this review we consider the changing paradigm, relating findings from in vitro and in vivo systems into concepts relevant to the immunopathogenesis of inflammatory and autoimmune disease as well as the use of HMGB1 as a biomarker.
Biochemistry and Cell Biology of HMGB1
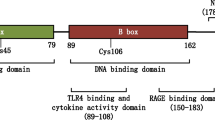
HMGB1 is 215 amino acids long and consists of two DNA binding boxes (A box and B box) as well as a C-terminal tail that is highly enriched in anionic residues. Reflecting this structure, HMGB1 can bind to DNA and histones, with these interactions resulting from distinct protein sequences. Whereas the A and B boxes can bind DNA, the C-terminal region can bind to histones; the C-terminal region can also interact with the A and B boxes, modifying the 3-dimensional structure of HMGB1 and its molecular interactions. In its binding to DNA, HMGB1 shows preferences for certain DNA structures such as bends or cruciforms, consistent with a role in modifying nucleosomal structure to regulate transcription, recombination or repair (20,21).
In the nucleus, HMGB1 appears fleeting in its binding to nucleosomes as HMGB1 travels up and down chromatin in a “hit-and-run” pattern. In this property, HMGB1 resembles the H1 histone that can bind linker DNA (22). While commonly considered a nuclear molecule, HMGB1 can also exist in the cytoplasm, with the relative distribution in these locations varying with cell type and state of differentiation or activation.
Translocation of HMGB1
HMGB1 translocation results from two main processes that commonly occur together in inflammatory or autoimmune disease. The first is activation of inflammatory cells such as macrophages, monocytes and dendritic cells. As shown in the initial observations on alarmin activity, HMGB1 translocation is a late consequence of stimulation by LPS, reflecting a nonconventional secretory process (5,23,24). With macrophages, LPS stimulation leads to the acetylation of lysines that represent nuclear localization signals. With these lysines acetylated, HMGB1 moves to the cytoplasm, where it enters secretory lysosomes for secretion. In addition to LPS, cytokines such as interferon γ (IFN-γ) can stimulate this process.
In contrast to the effects of immune cell activation, the effects of cell death on HMGB1 translocation have been more uncertain and sometimes controversial, relating in part to an initial focus on cell death in terms of a binary categorization as necrosis and apoptosis. Of relevance to the HMGB1’s function as an alarmin in inflammatory and autoimmune disease, necrosis has been considered as proinflammatory, whereas apoptosis has been considered to be noninflammatory or even antiinflammatory. These studies suggested a critical role of HMGB1 translocation in immunity, positing that the location of HMGB1 could serve essentially as an on-off switch for inflammation when cells die (25,26).
In the past few years, the picture of HMGB1 translocation and activity has grown more nuanced with the recognition that cell death can lead to posttranslational modifications that impact significantly on the structure of HMGB1. These structural variants, whose expression also depends on the ambient environment at a site of inflammation (e.g., joint) in turn determine functional activity. Furthermore, the classification of cell death has also expanded to encompass an ever growing variety of distinct forms (27). As a framework to understand the variants that can derive from the HMGB1 platform, we will review the role of redox changes on the immune activity of HMGB1 and their generation during cell death.
Effects of Redox on the Immune Activity of HMGB1
Studies to define the action of HMGB1 in both acute and chronic inflammation led to many interesting, albeit sometimes confusing, findings. Thus, these studies showed that HMGB1 can interact with multiple immune sensors and receptors. These receptors included receptors for advanced glycation end products (RAGE) as well as toll-like receptor 2 (TLR2), TLR4 and TLR9, among others. The binding to TLR4 in particular can lead to activation of nuclear factor κB (NF-κB) and production of cytokines such as interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) by macrophages (28–32). In addition, these studies showed that HMGB1 can induce chemotaxis, a process which involves a distinct receptor set (3).
As noted, HMGB1 has a unique regional structure in which different sequences allow interaction with biochemically disparate molecules (33). This property suggests that extracellular HMGB1 could be less discreet in its interaction with receptors in comparison to a conventional cytokine. Furthermore, studies indicated that HMGB1 can bind to other molecules, such as PAMPs such as lipopolysaccharide (LPS), double-stranded RNA or DNA, or a cytokine such as IL-1β, and act through the partner’s receptor. This synergism could induce expression of mediators like the prostaglandins to promote pain and inflammation in arthritis (34–37).
A particularly curious aspect of these studies concerned observations that HMGB1, especially in purified form, could be immunologically inactive, in contrast to original findings on its alarmin activity. Ultimately, this situation was resolved by elegant experiments showing that the immune activity of HMGB1 is mutable and depends on the redox state of three cysteine residues at positions 23, 45 and 106 (38,39). As these studies showed, with a disulfide bond at positions 23 and 45 and a free thiol at position 106, HMGB1 can interact with TLR4 and induce cytokine production. In contrast, HMGB1 with all sulfydryls in the thiol state cannot stimulate TLR4 but rather can bind to CXCL12 to induce chemotaxis via CXCR4. With terminal oxidation of the sulfydryl groups to sulfonates, HMGB1 loses the ability to induce cytokine production or chemotaxis (40).
Given the nature of redox modifications caused by reactive oxygen species (ROS), the immune activities of HMGB1 at any location can vary over time and space and result from events inside as well as outside the cells. Thus, the redox state can vary depending on cell stress amounts and intercurrent events such as ischemia-reperfusion; the redox state in turn can affect HMGB1 activity to mediate a transition from chemotaxis to cytokine production to inactivity during the course of an inflammatory response (41). Because of the impact of redox changes, the demonstration of extracellular HMGB1 in the joint or other tissue does not specify its activity or role in disease. Indeed, it could be nil, if levels of reactive oxygen oxidize the three cysteines, a terminal event.
Dynamics of HMGB1 Translocation During Cell Death
Recent studies have expanded categorization of immunologically relevant cell death forms, although all likely lead to HMGB1 release and impact the pathogenesis of inflammatory and autoimmune disease. The recognition of the nature of these cell death forms is key to understanding the origin of HMGB1 at sites of local inflammation (e.g., synovium, salivary gland, kidney, muscle) since it enlarges beyond apoptosis and necrosis the range of processes that may lead to histopathologic findings, such as loss of nuclear HMGB1, cytoplasmic expression of HMGB1 or actual extracellular release of HMGB1. These cell death forms will now be reviewed.
Necrosis
As commonly understood, necrosis is a form of accidental cell death induced by chemical or physical trauma. While necrosis can be modeled as explosive and instantaneous, the process of necrosis can actually be gradual, with the type of inducing agent determining biochemical changes in the cell and the ultimate translocation of molecules like HMGB1. The hallmark of necrosis is cellular destruction and diffusion of the intracellular contents away from the scene of cell death.
In a seminal study, Scaffidi and colleagues elegantly investigated HMGB1 translocation during cell death (25). Using sensitive biophysical assays, these investigators demonstrated that HMGB1 in living cells shows rapid intranuclear mobility distinct from that of histones, which is more fixed. Because of its weak interaction with chromatin, HMGB1 can readily move outside the cell, when membrane integrity is lost during necrosis. In contrast, in these experiments, HMGB1 showed nuclear retention and decreased intranuclear mobility in apoptosis. Importantly, since necrotic cells lacking HMGB1 fail to induce cytokine production, these studies suggested that HMGB1 is a dominant immune player during cell death (26).
Since necrosis is heterogeneous, a subsequent in vitro study explored HMGB1 translocation in various models of this form of cell death. These treatments included freeze-thaw, heat, ethanol and hydrogen peroxide (H2O2) and used Jurkat cells as a model. While each of these treatments killed cells effectively, the extent of HMGB1 released during cell death varied widely, as assessed by Western blotting of culture supernatants. Whereas freeze-thaw produced an immediate and robust release of HMGB1, release was more gradual with the other treatments and, with ethanol, very limited (42). These observations indicate that HMGB1 release is not an invariable feature of necrosis, a finding which should be considered in experiments to assess the role of this molecule during cell death.
It can be difficult to extrapolate the properties of dead cells and their products from the in vitro to the in vivo situation, because cell death in vivo may be affected by conditions such as hypoxia, acidosis or oxidation. Thus, Venereau and colleagues showed, in a mouse model of muscle injury, that HMGB1 is reduced inside the cell nucleus. When released from necrotic cells following treatment of muscle with cardiotoxin, however, it can be oxidized extracellularly to the disulfide-HMGBl form by the oxidative milieu created by infiltrating leukocytes (38). Thus, the extracellular environment during necrosis may impact the biochemical and functional properties of any released HMGB1.
Apoptosis
As a general mechanism for somatic cell death, apoptosis is the main counterpart to necrosis. Apoptosis is classically defined as a form of programmed cell death that can occur in both physiological and pathological settings; necrosis is likely always pathological. In apoptosis, enzyme cascades lead to nucleolytic and proteolytic cleavage events that cause profound morphological changes in the cell. Despite these disruptions, cell membrane integrity persists until the late stage. Furthermore, apoptotic cells are swiftly recognized and removed by phagocytic cells such as macrophages. As a result, intracellular material including HMGB1 can be shielded from the immune system, making apoptosis immunologically “silent” (43,44).
If left unperturbed or uncleared, apoptotic cells can enter a phase called late apoptosis or secondary necrosis that includes membrane breakdown and the release of intracellular content. This transition has been closely linked to the pathogenesis of the autoimmune disease systemic lupus erythematosus (SLE), since genetic abnormalities such as deficiency of complement or other cellular or humoral clearance system can lead to autoantibody production in patients and in animal models (43,44). The most characteristic antibodies in these settings are directed to DNA or histones, the partners of HMGB1 in the nucleus, and are considered the result of an immune response to persistent dead cell remains, with DAMPs serving as autoadjuvants.
As noted, initial reports suggested a marked difference in the movement of HMGB1 during necrosis and apoptosis (25). Importantly, these studies indicated that, during apoptosis, HMGB1 loses its free intranuclear mobility. By binding tightly to chromatin, HMGB1 can remain inside the nucleus as membrane integrity breaks down. Given the interaction of HMGB1 with DNA or histones, these findings suggested posttranslational modification to change the ordinary pattern of intermolecular binding. Since trichostatin A, an inhibitor of histone acetylation, could alter the location of HMGB1, these findings pointed to histone as at least one molecule undergoing modification.
The pattern of intranuclear retention of HMGB1 illustrated in these initial in vitro studies differs from that observed in the other studies on extracellular translocation of DNA, histones or nucleosomes during apoptosis. As studies in both in vitro and in vivo systems indicate, during apoptotic cell death, DNA can move to an extracellular location, and indeed it can rise to high levels in the blood of mice following treatment with a specific apoptotic stimulus (e.g., anti-Fas treatment). Furthermore, levels of DNA in the blood are elevated in many of the same conditions as is HMGB1, including SLE (45).
To reconcile these divergent results of nuclear molecule translocation, additional model systems were investigated. Using this approach, Bell et al. showed that, like necrotic cells, apoptotic cells can release HMGB1 in vitro in a time-dependent manner (46). Other studies have explored the role of redox modification of HMGB1 during apoptosis, seeking to identify the role of this molecule in tolerogenic state-induced apoptotic cells. As shown by Kazama et al., HMGB1 is completely reduced inside the nucleus of live cells. The situation changes during apoptosis, however, as high ROS levels (resulting from caspase effects on mitochondria) can lead to terminal oxidation of the critical cysteines in HMGB1 to sulfonates, abrogating the immunostimulatory activity. As these studies suggest, the abrogation of HMGB1 activity by ROS could contribute to the ability of apoptotic cells to induce tolerance as opposed to an adaptive response (47).
Among events that can occur during apoptosis, autophagy can change the pattern of HMGB1 expression and extracellular release in dying cells. As shown in in vitro models, treatment of cancer cells with certain cytotoxic agents can induce autophagy. In this situation, with autophagy induced, cancer cells can actively release HMGB1 to the extracellular space (48,49); with autophagy blocked, caspase activation and nuclear retention of HMGB1 can occur. The interplay between autophagy and apoptosis is complex, however, with studies by Tang and colleagues demonstrating that HMGB1 plays a crucial role in this cross-talk. These investigators showed that treatment of cancer cells with reducible HMGB1 can induce autophagy by binding to RAGE and promote resistance to cytotoxic agents; in contrast, oxidized HMGB1 can increase cytotoxicity and promote apoptosis (49). Together, these studies point to autophagy as a key determinant of HMGB1 release during cytotoxicity, with released HMGB1 in turn regulating the death mechanism.
While HMGB1 release may occur during apoptosis, the immunological consequences may differ from necrosis not just because of the timing and extent of release but also because of redox changes (47,50). While this conclusion is consistent with studies on the tolerogenic activity of apoptotic cells, its incorporation into a model for SLE may be problematic. Thus, as oxidation causes an irreversible loss of HMGB1 immunological activity from sulfonation, a transition from apoptosis to late apoptosis or secondary necrosis should not lead to the emergence of new activity that can drive autoimmunity, unless the oxidation was incomplete. Apoptosis as a source of nuclear material in SLE is a matter of conjecture. It is thus possible that the most relevant cell death in this setting occurs through one of the following two pathways.
Pyroptosis
Two additional forms of cell death—pyroptosis and NETosis—relevant to inflammatory and autoimmune disease have been recently defined, expanding the potential sources of extracellular HMGB1. While all cells can undergo programmed cell death, pyroptosis is considered to be a specialized form of regulated cell death of macrophages and dendritic cells (51). Like apoptosis, pyroptosis involves nuclear changes, with DNA condensation and cleavage resulting from the activity of an unidentified nuclease. Pyroptosis follows the activation of inflammasomes, leading to the expression of caspase-1 and its downstream effects, including generation of the cytokines IL-1β and IL-18 by the cleavage of their precursors. The inflammasome can be triggered by a variety of molecules, likely resulting from some type of cell stress.
Given the wide array of molecules displayed by bacteria (i.e., PAMPs), infectious agents likely can stimulate TLR and nucleotide-binding oligomerization domain receptor (NLR) signaling simultaneously. While this dual activation makes sense in innate immunity, it can complicate interpretation of the mechanism of HMGB1 release. To investigate the role of different signaling events on HMGB1 translocation and posttranslational modification, Nystrom and colleagues engineered macrophage cells in which inflammasome signaling via NLRC4 can be induced by intracellular expression of a flagellin sequence. These cells allow study of the properties of HMGB1 released during pyroptosis in vitro with and without TLR priming (52).
Using this system, Nystrom and colleagues showed that cells activated via NLRC4 to undergo pyroptosis release HMGB1 in the all-thiol redox form; pyroptosis itself does not induce ROS production. In contrast, TLR stimulation can initiate mitochondrial dysfunction and ROS release. As a result, NLRC4-induced cells, primed with cell surface TLR ligands, can release two different redox isoforms of HMGB1: all-thiol HMGB1 and the disulfide-Cys106-thiol form. Of note, HMGB1 showed acetylation with or without LPS priming. Together, these results suggest that immune activation can lead to different forms of HMGB1 depending upon which pathway—TLR or inflammasome—is activated (52,53). Figure 1 illustrates these mechanisms.

The relationship between HMGB1 release mechanism and HMGB1 isoforms. As this figure illustrates, the various release mechanisms (necrosis, pyroptosis, macrophage activation and apoptosis) can lead to the release of different redox forms of HMGB1. Necrotic cells and pyroptotic cells release the all-thiol or completely reduced form; this form can bind the chemokine CXC12 and signal through the CXCR4 receptor to induce chemotaxis. Pyroptosis, in association with stimulation by a TLR4 ligand, causes release of both fully reduced HMGB1 and HMGB1 with a disulfide bond between C23 and C45 and C106 in the thiol form. This form of HMGB1 can induce cytokine production by signaling through TLR4. Activated macrophages also release the cytokine-inducing form of HMGB1 upon TLT4 activation. Apoptotic cells release HMGB1 that is partially oxidized or completely oxidized at the critical cysteine residues. Completely oxidized HMGB1, with cysteines in the form of sulfonates, is unable to stimulate cytokines or induce chemotaxis; apoptotic cells expressing this form of oxidized HMGB1 can induce tolerance.
NETosis
NETosis is another form of regulated cell death that occurs primarily with neutrophils (54). Ordinarily, during inflammation, neutrophils are recruited to the site of inflammation to phagocytose and degrade pathogens and cell debris. Following the death of neutrophils by apoptosis, macrophages can clear their remains by a process called efferocytosis. In addition to apoptosis, neutrophils can display another, more dramatic response to stimuli such as bacteria, LPS, and cytokines, undergoing of a process called NETosis. This process culminates in cell death, releasing structures called neutrophil extracellular traps (NETs).
NETs resemble a mesh and are composed of DNA and histones as well as proteins from cytoplasmic granules such as neutrophil elastase (NE) and myeloperoxidase (MPO) that have been mixed during this process (54). Not surprisingly, HMGB1 is a component of NETs (14). NET formation requires ROS production and the activation of NADPH oxidase (54). The presence of NE and MPO as well as nuclear molecules like HMGB1 and histones in NETs underlie antibacterial action, providing both a physical trapping mechanism and localized killing. With the use of immunofluorescence microscopy and biochemical assays, NETosis can be identified in synovium, synovial fluid and sites of vascular inflammation in vasculitis (55).
The poise between apoptosis and NETosis in autoimmunity is illustrated in a study by Garcia-Romeo and colleagues (56). These investigators showed that neutrophils from patients with SLE die by apoptosis in vitro; these cells, however, switch to NETosis upon stimulation with anti-RNP antibodies, a common antinuclear autoantibody found in patients. In this regard, the term NETosis refers to a pathway of neutrophil cell death in which NETs are released. Recently, Yipp and colleagues demonstrated that, in response to Gram-positive organisms, neutrophils are able to extrude NETs and, despite enucleation, remain viable for some time, able to migrate and display phagocytic activity (57).
The biochemistry of HMGB1 released during NETosis has not yet been analyzed in detail. Since an environment filled with neutrophils is likely to be highly oxidant, however, the activity of HMGB1 is uncertain. Nevertheless, the process of NET release, with or without cell death, can be a source of extracellular HMGB1 that appears in tissue or can serve as a biomarker. This material can also be a source of autoantigen to simulate antibody production or form immune complexes. Figure 2 depicts nuclear molecule release during cell death forms, recognizing that HMGB1 can emanate as a component of NETs without cell death.

The pattern of HMGB1 translocation during cell death. Apoptotic cells can retain HMGB1 tightly bound to chromatin in the nucleus, but, during late apoptosis or secondary necrosis, can release this protein; this isoform is oxidized and lacks immunological activity (depicted by red dots). During necrosis, the plasma membrane and nuclear membrane lose integrity, releasing a proinflammatory form of HMGB1 (red stars). During pyroptosis, following inflammasome activation, the plasma membrane opens and HMGB1 release occur (red stars). If, with pyroptosis, TLR activation occurs, a cytokine-inducing form can also be released (not shown). While NETosis also induces inflammation and releases nuclear material, the redox state of the released HMGB1 in this type of cell death is yet unknown (depicted by red circles). The helical symbols indicate DNA. In NETosis, DNA is released in the form of strands or meshes; while the DNA has a high molecular weight with necrosis, it is not organized or associated with cytoplasmic proteins. The blue dots indicate a nucleosome structure. In apoptosis and pyroptosis, the DNA is cleaved (not shown); laddering occurs with apoptosis but not pyroptosis.
Extracellular Environment During Cell Death
In the extracellular environment during inflammation and cell death, the trio of DNA, HMGB1 and histones occur prominently, with this concomitant expression notable because of their capacity for mutual interaction to produce novel immunostimulatory structures. As shown recently, histones are potent immune stimulators that can mediate responses in settings such as sepsis. Histones are also antibacterial (58). DNA, the partner of both histones and HMGB1, has immune properties, but these properties are context dependent. Whereas free mammalian DNA fails to induce TLR9 activation, DNA can acquire activity when bound to another molecule such as HMGB1 (59). Thus, three main components of the nucleus can all have an impact on the immune response.
Given the high extracellular expression of mutually interacting nuclear molecules during inflammation, it is pertinent to ask whether the HMGB1 is ever free (in an unbound state) to act alone or whether it acts together with its usual partners in the nucleus or cytokines and chemokines in the local environment. Studies in the context of SLE point to a conjoint activity of HMGB1 with other nuclear molecules (60–62). Thus, in SLE, immune complexes formed with antinuclear antibodies can deposit in the kidney to induce nephritis or stimulate plasmacytoid dendritic cell to produce interferon. While the key antibodies forming these complexes are directed to DNA and histones, the complexes contain HMGB1, which can augment their activity. The HMGB1 in immune complexes likely reflects its nucleosomal binding, perhaps enhanced during apoptosis. In this regard, Urbonaviciute et al. showed in a mouse model that nucleosomes purified from apoptotic, but not living, cells can induce antinuclear antibody production, with HMGB1 a key component (61). Interestingly, in this model, autoantibody production involves TLR2, one of the receptors for HMGB1. In addition to these activities, complexes can also interact with RAGE on endothelium to create the vasculopathy that marks the course of SLE (63).
Given the range of cell death forms that operate during immune responses, it will be important in thinking about models for SLE and other autoimmune and inflammatory diseases to determine which cell death form(s) are most relevant. This determination is important for interpreting the role of disturbances in dead cell clearance in generating conditions for autoreactivity. The clearance systems for the remains of necrotic, pyroptotic and NETotic cells all need explication, although there is evidence that some of the same systems for apoptotic cell clearance mediate that for pyroptotic cells (64). Linking SLE to impaired clearance of pyroptotic cells, for example, would simplify the current models for SLE that put the apoptotic cell at center stage despite its ambiguous immune activity.
HMGB1 as a Biomarker of Inflammatory and Autoimmune Disease
Together, these findings point to remarkable plasticity in HMGB1 biochemistry and function and suggest that the use of HMGB1 as a biomarker optimally should include assessment of the acetylation and redox state to determine its origin (inflammation versus cell death) as well as its capacity to induce cytokine production or chemotaxis. Analyses of this kind have been performed in the setting of drug-induced liver injury with agents such as acetaminophen or heparins (65,66). These studies have demonstrated the dynamic nature of posttranslational modification of HMGB1, indicative of shifts in cellular mechanisms that lead to HMGB1 expression. At present, assay of various molecular forms of HMGB1 entails mass spectrometry, which is technically difficult and limits throughput.
Implications for Therapy
Studies on in in vitro systems and animal models clearly demonstrate the potential for agents directed at HMGB1 to inhibit disease, with shock and inflammatory arthritis being the two most well-studied examples (1,2). Given the current understanding of the diversity of HMGB1 isoforms and actions, it is important to ask whether the activity of HMGB1 to be targeted is induction of cytokine production or stimulation of chemotaxis, or both. This question is relevant to efforts to develop, for example, monoclonal antibodies that bind selectively to HMGB1 isoforms depending on redox state, acetylation and other posttranslational modifications. In this regard, as there are now approaches to block specific forms of cell death (e.g., caspase inhibitors for apoptosis, necrostatin for necrosis), agents that prevent pyroptosis or NETosis can also be considered, but the design and application of these agents require delineation of the processes leading to HMGB1 release (67,68).
An interesting situation arises with respect to the therapeutic actions of antioxidants whose use in arthritis and other inflammatory diseases has long been proposed to reduce the tissue-destructive effects of ROS. As the effects of redox modification of HMGB1 indicate, the alarmin action of this molecule depends on a critical balance in ROS levels. Since an antioxidant could limit sulfydryl oxidation to terminate HMGB1 activity, any maintenance of the disulfide bond could perpetuate inflammation and hinder resolution (69,70).
Conclusion
As this review indicates, the actions of HMGB1 in inflammatory and autoimmune diseases are diverse, complicated and time dependent, reflecting events inside the cell as well as outside the cell. Given the nonoverlapping activity of the reduced and disulfide-linked isoforms of HMGB1, the term DAMPs may be more appropriate. Furthermore, to the extent that HMGB1 helps resolve inflammation, the term “danger” molecule, which is sometimes applied to this molecule, may be misleading. Practically, the diverse isoforms of HMGB1 complicate its use as a biomarker, pending availability of more direct assays of the different isoforms. At the level of tissue histopathology, the finding of HMGB1 in either a cytoplasmic or extracellular location, while demonstrating activation or cell death, cannot specify either the cell death mechanism or its consequences.
Few molecules in all of biology have as many structures and functions as HMGB1. The process of gaining understanding of this plasticity and multiplicity should make HMGB1 a fascinating focus of investigation now and well into the future and a prototype for developing new antiinflammatory and immunosuppressive therapies.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Andersson U, Tracey KJ. (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–62.
Harris HE, Andersson U, Pisetsky DS. (2012) HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8:195–202.
Yang H, Antoine DJ, Andersson U, Tracey KJ. (2013) The many faces of HMGB1: a molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 93:856–73.
Andersson U, Harris HE. (2010) The role of HMGB1 in the pathogenesis of rheumatic disease. Biochim. Biophys. Acta. 1799:141–8.
Wang H, et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–51.
Pullerits R, et al. (2003) High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum. 48:1693–700.
Taniguchi N, et al. (2003) High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 48:971–81.
Kokkola R, et al. (2003) Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 48:2052–8.
Ulfgren AK, et al. (2004) Down-regulation of the aberrant expression of the inflammation mediator high mobility group box chromosomal protein 1 in muscle tissue of patients with polymyositis and dermatomyositis treated with corticosteroids. Arthritis Rheum. 50:1586–94.
Popovic K, et al. (2005) Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. 52:3639–45.
Ek M, Popovic K, Harris HE, Nauclér CS, Wahren-Herlenius M. (2006) Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjögren’s syndrome. Arthritis Rheum. 54:2289–94.
Grundtman C, et al. (2010) Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J. 24:570–8.
Ahn JK, Cha HS, Bae EK, Lee J, Koh EM. (2011) Extracellular high-mobility group box 1 is increased in patients with Behcet’s disease with intestinal involvement. J. Korean Med. Sci. 26:697–700.
Mitroulis I, et al. (2011) Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PLoS One. 6:e29318.
Abdulahad DA, et al. (2012) Urine levels of HMGB1 in systemic lupus erythematosus patients with and without renal manifestations. Arthritis Res. Ther. 14:R184.
Maugeri N, et al. (2012) Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity. 45:584–7.
Abdulahad DA, et al. (2013) High mobility group box 1 (HMGB1) in relation to cutaneous inflammation in systemic lupus erythematosus (SLE). Lupus. 22:597–606.
Oktayoglu P, et al. (2013) Elevated serum levels of high mobility group box protein 1 (HMGB1) in patients with ankylosing spondylitis and its association with disease activity and quality of life. Rheumatol. Int. 33:1327–31.
Schierbeck H, et al. (2013) HMGB1 levels are increased in patients with juvenile idiopathic arthritis, correlate with early onset of disease, and are independent of disease duration. J. Rheumatol. 40:1604–13.
Cato L, Stott K, Watson M, Thomas JO. (2008) The interaction of HMGB1 and linker histones occurs through their acidic and basic tails. J. Mol. Biol. 384:1262–72.
Štros M. (2010) HMGB proteins: interactions with DNA and chromatin. Biochim. Biophys. Acta. 1799:101–13.
Thomas JO, Stott K. (2012) H1 and HMGB1: modulators of chromatin structure. Biochem. Soc. Trans. 40:341–346.
Gardella S, et al. (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3:995–1001.
Bonaldi T, et al. (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 22:5551–60.
Scaffidi P, Misteli T, Bianchi ME. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–5.
Rovere-Querini P, et al. (2004) HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 5:825–30.
Galluzzi L, et al. (2012) Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19:107–20.
Hori O, et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270:25752–61.
Park JS, et al. (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279:7370–77.
Park JS, et al. (2006) High mobility group box 1 protein interacts with multiple toll-like receptors. Am. J. Physiol. Cell Physiol. 290:C917–24.
Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. (2005) Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur. J. Immunol. 35:2184–90.
Ivanov S, et al. (2007) A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 110:1970–81.
Dintilhac A, Bernués J. (2002) HMGB1 interacts with many apparently unrelated proteins by recognizing short amino acid sequences. J. Biol. Chem. 277:7021–8.
Bianchi ME. (2009) HMGB1 loves company. J. Leukoc. Biol. 86:573–6.
Wähämaa H, et al. (2011) High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res. Ther. 13:R136.
Hreggvidsdóttir HS, et al. (2012) High mobility group box protein 1 (HMGB1)-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol. Med. 18:224–30.
Leclerc P, et al. (2013) IL-1β/HMGB1 complexes promote the PGE2 biosynthesis pathway in synovial fibroblasts. Scand. J. Immunol. 77:350–60.
Venereau E, et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209:1519–28.
Yang H, et al. (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18:250–9.
Venereau E, Schiraldi M, Uguccioni M, Bianchi ME. (2013) HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 55:76–82.
Valdés-Ferrer SI, et al. (2013) HMGB1 mediates splenomegaly and expansion of splenic CD11b+ Ly-6C (high) inflammatory monocytes in murine sepsis survivors. J. Intern. Med. 274:381–90.
Beyer C, et al. (2012) The extracellular release of DNA and HMGB1 from Jurkat T cells during in vitro necrotic cell death. Innate Immun. 18:727–37.
Nagata S, Hanayama R, Kawane K. (2010) Autoimmunity and the clearance of dead cells. Cell. 140:619–30.
Kruse K, et al. (2010) Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis. 15:1098–113.
Pisetsky DS. (2014) The translocation of nuclear molecules during inflammation and cell death. Antioxid. Redox Signal. 20:1117–25.
Bell CW, Jiang W, Reich CF 3rd, Pisetsky DS. (2006) The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 291:C1318–25.
Kazama H, et al. (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 29:21–32.
Thorburn J, et al. (2009) Autophagy regulates selective HMGB1 release in tumor cells that are destined to die. Cell Death Differ. 16:175–83.
Tang D, et al. (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 29:5299–310.
Urbonaviciute V, et al. (2009) Oxidation of the alarmin high-mobility group box 1 protein (HMGB1) during apoptosis. Autoimmunity. 42:305–7.
Miao EA, Rajan JV, Aderem A. (2011) Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243:206–14.
Nyström S, et al. (2013) TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 32:86–99.
Lamkanfi M, et al. (2010) Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 185:4385–92.
Brinkmann V, Zychlinsky A. (2012) Neutrophil extracellular traps: is immunity the second function of chromatin? J. Cell Biol. 198:773–83.
Kessenbrock K, et al. (2009) Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15:623–5.
Garcia-Romo GS, et al. (2011) Netting neutrophils are major inducers of Type I IFN production in pediatric systemic lupus erythematosus. Sci. Transl. Med. 3:73ra20.
Yipp BG, et al. (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 18:1386–93.
Xu J, et al. (2009) Extracellular histones are major mediators of death in sepsis. Nat. Med. 15:1318–21.
Pisetsky DS. (2012) The origin and properties of extracellular DNA: from PAMP to DAMP. Clin. Immunol. 144:32–40.
Tian J, et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 8:487–96.
Urbonaviciute V, et al. (2008) Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J. Exp. Med. 205:3007–18.
Wen Z, et al. (2013) Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J. Immunol. 190:5411–22.
Sun W, et al. (2013) Immune complexes activate human endothelium involving the cell-signaling HMGB1-RAGE axis in the pathogenesis of lupus vasculitis. Lab. Invest. 93:626–38.
Wang Q, et al. (2013) Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int. Immunol. 25:363–72.
Antoine DJ, et al. (2012) Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Heptaol. 56:1070–9.
Harrill AH, et al. (2012) The effects of heparins on the liver: application of mechanistic serum biomarkers in a randomized study in healthy volunteers. Clin. Pharmacol. Ther. 92:214–20.
Musumeci D, Roviello GN, Montesarchio D. (2013) An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol. Ther. 141:347–57.
Zhou Y, et al. (2013) Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013:706156.
Peter ME. (2008) ROS eliminate danger. Immunity. 29:1–2.
Nathan C, Ding A. (2010) Nonresolving inflammation. Cell. 140:871–82.
Acknowledgments
This work was supported by a VA Merit Review Grant, a grant from the Alliance for Lupus Research (ALR) and National Institutes of Health grant 5U19-AI056363.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Magna, M., Pisetsky, D.S. The Role of HMGB1 in the Pathogenesis of Inflammatory and Autoimmune Diseases. Mol Med 20, 138–146 (2014). https://doi.org/10.2119/molmed.2013.00164
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2013.00164