
Abstract
X-linked protoporphyria (XLP) (MIM 300752) is a recently recognized erythropoietic porphyria due to gain-of-function mutations in the erythroid-specific aminolevulinate synthase gene (ALAS2). Previously, two exon 11 small deletions, c.1699_1670ΔAT (ΔAT) and c.1706_1709ΔAGTG (ΔAGTG), that prematurely truncated or elongated the ALAS2 polypeptide, were reported to increase enzymatic activity 20- to 40-fold, causing the erythroid accumulation of protoporphyrins, cutaneous photosensitivity and liver disease. The mutant ΔAT and ΔAGTG ALAS2 enzymes, two novel mutations, c.1734ΔG (ΔG) and c.1642C>T (p.Q548X), and an engineered deletion c.1670–1671TC>GA p.F557X were expressed, and their purified enzymes were characterized. Wild-type and ΔAGTG enzymes exhibited similar amounts of 54- and 52-kDa polypeptides on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), whereas the ΔAT and p.F557X had only 52-kDa polypeptides. Compared to the purified wild-type enzyme, ΔAT, ΔAGTG and Q548X enzymes had increased specific activities that were only 1.8-, 3.1- and 1.6-fold, respectively. Interestingly, binding studies demonstrated that the increased activity Q548X enzyme did not bind to succinyl-CoA synthetase. The elongated ΔG enzyme had wild-type specific activity, kinetics and thermostability; twice the wild-type purification yield (56 versus 25%); and was primarily a 54-kDa form, suggesting greater stability in vivo. On the basis of studies of mutant enzymes, the maximal gain-of function region spanned 57 amino acids between 533 and 580. Thus, these ALAS2 gain-of-function mutations increased the specific activity (ΔAT, ΔAGTG and p.Q548X) or stability (ΔG) of the enzyme, thereby leading to the increased erythroid protoporphyrin accumulation causing XLP.
Similar content being viewed by others
Introduction
X-linked protoporphyria (XLP) is a recently described erythropoietic porphyria due to gain-of-function mutations in the erythroid-specific 5-aminolevulinate synthase gene (ALAS2) (1) that encodes the first enzyme in erythroid heme biosynthesis (2). ALAS2 condenses glycine and succinyl-CoA to form 5-aminolevulinic acid in the presence of its vitamin B6 co-factor, pyridoxal 5′-phosphate. The increased ALAS2 activity, originally estimated to be 20–40 times normal (1), leads to the erythroid accumulation of free and zinc-chelated protoporphyrin. The pathogenesis and biochemical and clinical manifestations are similar to those of autosomal recessive erythropoietic protoporphyria (EPP), which results from loss-of-function mutations in the ferrochelatase (FECH) gene (3–5) that reduce FECH enzymatic activity to <35% of normal, accounting for the erythroid accumulation of free protoporphyrin (2).
Patients with XLP and EPP have a cutaneous photosensitivity, and shortly after exposure to sun or ultraviolet (UV) light, they develop a tingling, burning sensation in the exposed areas that may lead to severe, excruciating pain crises that may last several days. Because the accumulated insoluble protoporphyrin is excreted by the hepatic biliary system, bile stones can form that cause liver dysfunction and failure in about 5% of patients (6). Because there is no approved treatment available other than protection from sunlight, affected individuals should stay indoors, or when outdoors, should wear protective clothing and use UV-blocking sunscreens that are typically zinc oxide based. Currently, clinical trials are underway to evaluate the safety and effectiveness of an α-melanocyte stimulating hormone analog to increase melanin and protect the skin (7). Patients with liver failure have received orthotopic liver transplants or liver transplants combined with hematopoietic transplants to protect the transplanted liver (8).
XLP patients were initially diagnosed as EPP based on their cutaneous photosensitivity and markedly elevated erythrocyte protoporphyrins, of which the zinc-chelated form represented up to 50%. However, the absence of FECH mutations and X-linked inheritance ultimately led to the identification of their ALAS2 mutations. Two ALAS2 small deletions, c.1699_1700delAT (p.M567EfsX2, designated AAT) and c.1706_1709delAGTG (p.E569GfsX24, designated ΔAGTG), were originally identified as causing XLP (1). These mutations resulted in frameshift lesions that prematurely truncated or abnormally elongated the wild-type 587-amino acid polypeptide, respectively (Figure 1). The ΔAT mutation had a wild-type amino acid sequence up to residue 566, added a glutamate at residue 567 and deleted the 20 terminal wild-type residues, whereas the ΔAGTG mutation altered the last 19 carboxy-terminal wild-type amino acids and elongated the polypeptide of the mutant enzyme by four residues.

Purified wild-type and mutant recombinant ALAS2 enzymes. Shown are exon 11 predicted carboxy-terminal amino acid sequences for the ALAS2 mutations. Residues in red are novel resulting from the mutation or the frameshifts caused by the deletions. The boxed squence is a predicted helical region and the underlined sequences are identical regions in the two different frameshift mutation sequences.
Recently, two additional ALAS2 mutations causing XLP. c.1642C>T (designated p.Q548X) and c.1737delG (p.Q581SfsX13, designated ΔG) were identified among 155 North American patients originally diagnosed as EPP (9). The novel p.Q548X nonsense mutation prematurely truncates the enzyme subunit at glutamine 548, thereby deleting 40 carboxy-terminal residues. The novel ΔG mutation causes a frameshift that alters the wild-type enzyme polypeptide after proline 580 by substituting seven amino acids (SMSPPMP) and then elongating the polypeptide by five additional residues (EKPAA) (Figure 1).
To investigate the biochemical bases of these gain-of-function mutations causing XLP, we expressed wild-type ALAS2 and the four known exon 11 ALAS2 mutations, purified the recombinant enzymes and characterized their kinetic and thermostability properties. In contrast to the initial report that the ΔAT and ΔAGTG enzymes had a 20- to 40-fold increased ALAS2 activity (1), the ΔAT, ΔAGTG and p.Q548X purified recombinant mutant enzymes had only 1.8- and 3.1-fold increased specific activities, accounting for the increased erythroid protoporphyrins. Notably, the ΔG recombinant enzyme had wild-type specific activity, kinetics and thermostability but approximately two-fold greater purification yield and increased resistance to proteolysis during purification. These results indicate that the ALAS2 gain-of-function mutant enzymes causing XLP had modestly increased specific activities or stability.
Materials and Methods
Reagents
Compounds used in the experiments included pyridoxal 5′-phosphate, succinyl-CoA sodium salt, phenylmethylsulfonylfluoride, pepstatin, leupeptin and aprotinin (Sigma-Aldrich, St. Louis, MO, USA); unstained Precision Plus Protein standards (Bio-Rad, Hercules, CA, USA); enzyme grade HEPES (Fisher Biotech, Pittsburgh, PA, USA); ethylacetoacetate (Sigma-Aldrich); the pMAL-c2 and c4X prokaryotic expression vectors, maltose binding protein (MBP), factor Xa, amylose resin, T4-DNA ligase, the Quick Ligation Kit and restriction enzymes (New England Biolabs, Ipswich, MA, USA); QIAprep Spin Miniprep Kit, QIAfilter Plasmid Midi Kit, QIAquick Gel Extraction Kit, HotStarTaq® Master Mix Kit (Qiagen, Germantown, MD, USA); QuikChange XL Site-Directed Mutagenesis Kit and BL21 CodonPlus-RP Competent Cells (Stratagene, La Jolla, CA, USA) and Top10F′ competent cells (Invitrogen; Life Technologies, Carlsbad, CA, USA).
Preparation of ALAS2 Expression Constructs
The ALAS2 mutations were introduced into the prokaryotic expression vector, pMALc2-AE2 (10). Briefly, plasmid DNA was purified from XL1-Blue transformed cells, and site-directed mutagenesis was performed by using the Stratagene XL Site-Directed Mutagenesis Kit protocol with the primers listed in Supplementary Table S1. Positive clones for XLP mutations (ΔAGTG, ΔAT, ΔG and p.Q548X) were identified by restriction analysis. A construct was made that deleted the last 93 coding nucleotides of ALAS2 exon 11; c.1670–1671TC>GA (designated p.F557X), which expressed only the lower-molecular-weight ∼52-kDa band on SDS-PAGE. Another construct was made that deleted the last 21 coding nucleotides; c.1741C>T (designated p.Q581X) that expressed only the upper-molecular-weight ∼54-kDa band on SDS-PAGE. Each expression construct was confirmed by sequence analysis of the XhoI to EcoRI fragment at the 3′ end of the ALAS2 construct. This region was then excised and recloned into the XhoI/EcoRI-digested wild-type pMALc2-AE2 vector, and the junction sequences were confirmed by sequencing by using the primers listed in Supplementary Table S1.
Expression and Purification of Human Recombinant ALAS2 Enzymes
Recombinant wild-type and mutant ALAS2 proteins were purified as previously described (11). Briefly, an overnight culture of the expression vector was used to seed 1 liter of Luria broth (LB) media containing 0.2% glucose, 100 µg/mL ampicillin and 10 µmol/L pyridoxal 5′-phosphate (PLP), grown at 37°C with shaking to a density of 0.6–0.8 A. Recombinant ALAS2 expression was induced with 1 mmol/L isopropyl β-d-1-thiogalactopyranoside (IPTG) (Sigma-Aldrich) for 3 h. The cells were harvested by centrifugation and suspended in 50 mL lysis buffer containing 200 mmol/L NaCl, 50 mmol/L potassium 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (KHEPES), pH 7.4, 5 mmol/L dithiothreitol (DTT), 1 mmol/L EDTA, 0.4 mmol/L phenylmethylsulfonyl fluoride (PMSF), 200 µg/mL lysozyme, 10 µmol/L PLP, 0.02% sodium azide, including additives: 1.0 µg/mL pepstatin, 0.5 µg/ mL leupeptin, 50 µL of 1 mol/L MgCl2, 200 µL of 5 mg/mL DNase and 100 µL of 5 mg/mL RNase. Cells were lysed by freeze/thaw, and the cell debris was removed after centrifugation at 27,000g. The supernatant (crude extract) was purified by affinity chromatography on an amylose resin column (2.8 diameter × 6 cm), which was equilibrated with lysis buffer excluding the above additives. After collection of the flow-through and washing, the column was eluted with 10 mmol/L maltose in 1× modified lysis buffer, and the elute was concentrated by ammonium sulfate precipitation between 25% and 55% saturation. The precipitate was dissolved in cleavage buffer containing 100 mmol/L sodium hydroxide (NaCl), 50 mmol/L KHEPES, 2 mmol/L CaCl2, 0.5 mmol/L DTT and 10 µmol/L PLP, adjusted to pH 8.0, with 1 N NaOH, and the maltose binding protein (MBP)-ALAS2 fusion protein was cleaved overnight by Factor Xa (2.5 µg Factor Xa per 1 mg MBP-ALAS2) at room temperature, followed by removal of MBP by a second amylose affinity step, resulting in ALAS2 enzymes for which the amino-terminus was Asp79, the amino-terminal residue in the mature human mitochondrial enzyme (12). The native recombinant ALAS2 enzyme in the flow-through was concentrated by ammonium sulfate precipitation at 55% saturation, dissolved in 2 mL gel filtration buffer (50 mmol/L NaCl, 50 mmol/L KHEPES, 5 mmol/L DTT, 0.4 mmol/L PMSF, 10 µmol/L PLP and 0.02% NaN3, adjusted to pH 7.4) and purified over two tandem (1.6 × 51 cm) fast protein liquid chromatography (FPLC) columns containing Superose 12 gel filtration media (GE Healthcare, Piscataway, NJ, USA). Protein purity was assessed by SDS-PAGE (13) of protein from each purification step. Molecular masses were compared with Precision Plus Protein™ Standards from Bio-Rad.
Enzyme and Protein Assays
Recombinant ALAS2 enzymatic activity was determined colorimetrically by using succinyl-CoA and glycine as substrates, as previously described (11). Briefly, the 0.5 mL reaction mixture contained 100 mmol/L glycine, 50 mmol/L potassium HEPES, pH 7.4, 10 mmol/L MgC12, 100 µmol/L succinyl-CoA, 10 µmol/L PLP and 10–200 units ALAS2 activity. After incubation at 37°C for 5 min, the reaction was terminated with trichloroacetic acid and the 5-aminolevulinic acid (ALA) in the supernatants quantitated with fresh Ehrlich reagent (14). One unit of activity is defined as that amount of enzyme required to catalyze the production of 1 nmol ALA per hour under the conditions of the assay. Protein concentrations were determined by a modification of the Fluorescamine method (15).
Km Determinations
Each assay contained 1–2 µg purified recombinant wild-type or mutant ALAS2 enzyme. The glycine concentrations were varied from 2.5 to 50 mmol/L at a succinyl-CoA concentration of 100 µmol/L and the succinyl-CoA concentrations ranged from 10 to 100 µmol/L at a glycine concentration of 100 mmol/L. Michaelis constant (Km)values were calculated from Lineweaver-Burk and Eadie-Hofstee plots. Cooperative kinetics were evaluated by using Hill plots with maximum velocity (Vmax) estimates obtained from Lineweaver-Burk plots. Subsequent Lineweaver-Burk plots using reciprocal substrate concentration raised to the Hill number (n) power (1/Sn) were used to iteratively calculate revised Vmax estimates until the Hill number value did not change further.
Thermostability of the Wild-Type and Mutant ALAS2 Apoenzymes
Homogeneous wild-type and mutant ALAS2 enzymes were incubated in tubes at 45°C for varying lengths of time in 50 mmol/L KHEPES, 10 mmol/L MgCl2 and 1 mmol/L DTT, pH 7.4, followed by rapid cooling in an ice-water bath, and then they were assayed for ALAS2 activity. Half-lives were determined from semilog plots of percent activity remaining versus time.
All supplementary materials are available online at https://doi.org/www.molmed.org .
Results
Construction, Expression and Purification of Wild-Type and Mutant ALAS2 Recombinant Enzymes
After construction of the four ALAS2 mutations causing XLP, and the p.F557X and p.Q581X mutant constructs that were engineered to delete the terminal 31 and six amino acids in ALAS2, respectively, each was prokaryotically overexpressed. The ALAS2 wild-type and mutant enzymes were purified to homogeneity in three steps, with 20–56% yields of 2–8 mg per liter of cells (Table 1). Notably, the purification of the ΔG and p.Q548X enzymes resulted in a higher yield (56 and 48%, respectively) than those of the wild-type and other mutant enzymes (20–27%). After amylose affinity purification of the MBP-ALAS2 fusion proteins, the MBP moiety was cleaved by Factor Xa and the MBP was removed.
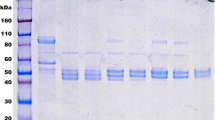
After gel filtration chromatography, the enzyme preparations appeared homogeneous on SDS-PAGE (Figure 2). The purified wild-type recombinant enzyme had two forms of ∼54 and ∼52 kDa, with the lower form due to an ∼2-kb carboxy-terminal cleavage that occurred during expression and/or purification, despite the presence of protease inhibitors. The 52-kDa peptide migrated to a position slightly above that of the engineered p.F557X mutant ALAS2 polypeptide that lacks 31 carboxy-terminal residues (Figures 1 and 2) and similar to that of the purified ΔAT enzyme, which truncated 20 wildtype carboxy-terminal residues and may have had two forms that could not be distinguished on the denaturing gel. The relative amounts of the two enzyme forms were similar for the purified recombinant wild-type and the ΔAGTG mutant enzymes (Figure 2). Of note, the ΔG enzyme had both the 54- and 52-kDa forms; however, the 54-kDa form was the major band (∼80% by densitometry; Figure 2), suggesting altered proteolytic processing of the ΔG enzyme. Similarly, the engineered p.Q581X construct that was truncated immediately after the last wild-type residue of the ΔG enzyme exhibited only the upper ∼54-kDa form on SDS-PAGE.

SDS-PAGE and predicted amino-terminal sequence comparisons. SDS-PAGE of wild-type and mutant ALAS2 proteins after purification to homogeneity by affinity chromatography and gel filtration chromatography. (A) Lanes 1 and 8: protein molecular weight standards; lanes 2 and 7: wild-type ALAS2; lane 3: Δ93 p.F557X carboxy-terminal truncation polypeptide; lane 4: ΔAT polypeptide; lane 5: ΔG polypeptide; lane 6: ΔAGTG polypeptide. (B) Lane 1: protein molecular weight standard; lane 2: wild-type ALAS2; lane 3: p.Q581X truncation polypeptide.
Kinetic and Thermostability Properties of the Wild-Type and Mutant ALAS2 Enzymes
The binding affinity (Km values) of the homogeneous wild-type and mutant enzymes for their substrates, glycine and succinyl-CoA, were similar; however, the specific activities of the purified XLP-causing enzymes were variably increased over that of the wildtype, from 1.6-fold for the p.Q548X mutant to 3.1 for the ΔAGTG mutant enzyme (Table 2). These increases in specific activity appeared to be due to increases in Vmax for both substrates glycine and succinyl-CoA. Notably, Vmax values increased about two-fold for the ΔAT, ΔAGTG, P.F557X and p.Q548X enzymes, while the succinyl-CoA-dependent Hill number values for the ΔAT and ΔAGTG mutation enzymes were increased to 1.8 from the value of 1.5 for the wild-type and ΔG enzymes. This alteration in cooperativity may partially explain the increased in vitro enzyme activities of the ΔAT and ΔAGTG enzymes. The ΔG enzyme, on the other hand, had decreased Vmax values for both substrates (Table 2). There were marked differences in the behavior of the mutant enzymes with the cofactor PLP. The AAT enzyme was completely unstable with no detectable activity after removal of PLP, whereas the ΔG mutant had a much lower affinity constant than wild-type (650 versus 22 nmol/L). With the exception of the ΔG mutation, the thermostabilities of the ALAS2 mutant enzymes were reduced in crude extracts (Table 2), suggesting that the observed gain-of-function for the ΔAT, ΔAGTG and p.Q548X enzymes (1) was primarily due to their altered kinetics, whereas that of the ΔG enzyme was due to increased stability.
Boundaries of the Gain-of-Function Domain Are Between ALAS2 Amino Acids 533 and 580
Because the ΔG XLP mutation produced a protein that had 12 altered amino acids after the end of the wildtype sequence at residue 580, it was of interest to know if these amino acids were responsible for the wild-type activity of this XLP mutation or if it was the truncation alone that determined activity. Therefore, a recombinant p.Q581X mutant was generated, expressed, purified to homogeneity and characterized. This homogeneous mutant enzyme also had nearly wild-type activity specific activity (Figure 1) and a single band on SDS-PAGE that migrated around 54 kDa (Figure 3). Its specific activity was 0.85-fold the wild-type activity compared with 0.95-fold for the ΔG enzyme. Another construct, V533X, was completely inactive (data not shown). Thus, the maximum carboxy-terminal region of gain-of-function in vitro was 57 residues, between amino acids 533 and 580.

SDS-PAGE analysis of succinyl CoA synthetase (SCS) binding to wild-type and Q548X ALAS2. Protein samples from before and after amylose affinity chromatography were visualized by SDS-PAGE. Molecular mass standards were run in lanes 1 and 9. FPLC-purified wild-type MBP-ALAS2, SCS and MBP-Q548X proteins were run in lanes 2, 5 and 8, respectively. Lanes 3 and 7 contained samples of the flow-through of unbound SCS applied to amylose columns containing bound WT-ALAS2 and Q548X MBP fusion proteins, respectively. Lanes 4 and 6 contained samples of the elution of these respective columns with maltose.
The Q548X Mutant ALAS2 Enzyme Did Not Bind to Succinyl-CoA Synthetase
It was recently demonstrated that the carboxy-terminal region of ALAS2 was necessary for binding of succinyl-CoA synthetase, the enzyme that provides ALAS2 with one of its two substrates, succinyl CoA, with the other being glycine. Mutations in this region (p.M567G and p.S568G) that resulted in loss of succinyl-CoA synthetase binding were found in different patients with X-linked sideroblastic anemia, indicating these were loss-of-function mutations in vivo (16). The Q548X mutation was tested for binding to succinyl-CoA synthetase by affinity chromatography. The MBP-wild-type or MBP-Q548 fusion proteins were bound to an amylose affinity column and loaded with succinyl-CoA synthetase. As seen in Figure 3, all the applied succinyl-CoA synthetase was bound to and eluted with the wild-type enzyme, whereas nearly all the applied succinyl-CoA synthetase washed through the mutant enzyme column and only a trace (likely nonspecifically bound) was eluted with maltose, demonstrating that residues in the deleted region were necessary for binding.
Discussion
To investigate the biochemical bases of the ALAS2 exon 11 gain-of-function mutations that cause XLP, the previously reported mutations, ΔAT and ΔAGTG, and two novel mutations, ΔG and p.Q548X, were prokaryotically expressed, were purified to homogeneity and their kinetic and thermostability properties were characterized. Compared to the purified recombinant wild-type enzyme, the purified recombinant ΔAT, ΔAGTG and p.Q548X enzymes had ∼1.6- to 3-fold increased specific activities with increased glycine and succinyl-CoA Vmax values, but similar Km values toward glycine and succinyl-CoA. During heat denaturation, the ΔAT, ΔAGTG, p.Q548X and p.F557X mutant enzymes were all much less stable than the wild-type and ΔG enzymes. Although the ΔAT, ΔAGTG and p.Q548X mutations had reduced thermostability relative to wild-type, apparently the increased turnover rates of these enzymes were dominant over the reduced stability in vivo, since they caused XLP.
The fold-increase in crude extract-specific activities for the recombinant ΔAT, ΔAGTG and p.Q548X enzymes (approximately three-fold over wild-type) were significantly less than the initially estimated 20- to 40-fold increase in crude extract-specific activities (1). Of note, the increased ALAS2 activity and resultant erythroid protoporphyrin accumulation occurred in XLP patients despite the presence of normal FECH activity. That FECH becomes rate-limiting with only modest increases in ALAS2 activity is apparently related to its inability to convert the excess protoporphyrin to heme because of presently ill-defined regulatory mechanisms (17).
Of particular interest, the denaturing gel electrophoretic profiles of the ALAS2 mutant proteins revealed important differences from that of the wild-type enzyme. The wild-type ALAS2 enzyme always had two forms of approximately equal intensity with molecular masses of 54 and 52 kDa, irrespective of the purification method and inclusion of protease inhibitors (11). The human ALAS2 54- and 52-kDa subunits were also seen by immunodetection in human erythroid cell extracts (12). The p.F557X construct was generated because mass spectroscopic analyses indicated that the 52-kDa form was a carboxy-terminal deletion of about 31 amino acids (data not shown). Because this form had 1.3-fold greater specific activity than the wild-type enzyme, it can be concluded that both forms of the enzyme are active. Consistent with this finding, both the p.Q548X and ΔAT mutant enzymes, which prematurely truncated the enzyme polypeptide and had only the ∼52-kDa polypeptide on SDS-PAGE, had increased activity in vitro and apparently in vivo. Whereas the ΔAGTG enzyme had both polypeptide forms in similar amounts, it was notable that the ΔG and p.Q581X enzymes had primarily the 54-kDa form, suggesting that they were more resistant to proteolytic cleavage than the wild-type enzyme and presumably partially explaining their slightly decreased activity in vitro, since the 52-kDa form is more active than the 54-kDa form.
The ΔG mutant enzyme was unique, since in vitro, the homogeneous recombinant enzyme had essentially wild-type specific activity, kinetics and thermostability and reduced affinity for PLP, all of which would argue against a gain-of-function for ALAS2. Nonetheless, the proband with this mutation had elevated erythrocyte protoporphyrin (∼30 times higher than wild-type levels of about 50 µg/ dL), although it was less than the mean increase in erythrocyte protoporphyrin of 10 male probands with the ΔAGTG mutation (110 ± 57 times higher than wild-type) (9). The ΔG mutant enzyme may have significantly greater in vivo stability than the wild-type enzyme, on the basis of its increased yield during expression and purification, possibly due to its increased resistance to proteolysis in vitro and in vivo. The ΔG mutant enzyme had a significantly higher proportion of the large 54-kDa form, which may be more resistant to proteolysis, resulting in a net increase in ALAS2 activity. That the carboxy-terminal ΔG mutation could cause increased stability of ALAS2 is also supported by the recent finding that certain other carboxy-terminal mutations can increase the stability of the enzyme in vivo (16). Finally, sequencing the ALAS2 promoter (1,000 bp) and enhancer elements in intron 8 (18) of affected males and heterozygous females with the ΔG mutation did not identify an alteration that might account for its gain-of-function.
Recently, Ducamp et al. (21) presented results from the expression and characterization of partially purified XLP mutations p.Q548, ΔAT and ΔAGTG as well as a novel del26bp mutation and other engineered constructs. Although these authors demonstrated a similar approximately three-fold increase in activity for the XLP mutant enzymes, the specific activities of their partially purified enzyme were 400- to 1,400-fold lower than those reported here. Their studies narrowed the gain-of-function region to 33 amino acids between residues 544 and 576.
It has been hypothesized that the carboxy-terminal region of ALAS2, which is present in mammals, but not in lower species, is folded such that it limits substrate and/or cofactor entry into the active site of the enzyme (19). The ΔAT enzyme deletes 20 carboxy-terminal wild-type residues, and the ΔAGTG enzyme alters the terminal 18 wild-type residues and introduces six structurally altering proline residues in its elongated sequence. Thus, the deletion or altered elongated sequence may render the active site more open, thereby increasing access to its substrates, resulting in increased enzyme activity. In addition, it has been shown recently that mutations in the carboxy-terminal region of ALAS2 can cause loss of binding to the ATP-using β subunit (SUCLA2) of succinyl-CoA synthetase as well as to the succinyl-CoA synthetase heterodimeric holoenzyme (11). Further, regulation in this region is complicated, since it was shown that two different exon 11 loss-of-function mutations that cause X-linked sideroblastic anemia (M567V and S568G) resulted in elimination of the binding of ALAS2 to SUCLA2. In contrast, the overlapping ΔAT mutation that changes codon S568 to glutamine followed by a stop codon (Figure 1) did not lose the ability to bind SUCLA2, consistent with ALA overproduction by this ALAS2 mutation. In this regard, it is noteworthy that the p.Q548X mutant enzyme does not bind to succinyl-CoA synthetase (Figure 3). Thus, it is possible in vivo that alterations in the carboxy-terminal region of ALAS2 improve the ability of succinyl-CoA synthetase to donate succinyl-CoA to ALAS2 by inducing conformational changes in the ALAS2 protein. This conformational flexibility is highlighted by the recent discovery that ALAS2 enzyme kinetics exhibit positive cooperativity with succinyl-CoA binding (11). Because only the 54-kDa ΔG form binds succinyl-CoA synthase (11) and there is nearly twice the amount of this mutant form compared with wildtype in vitro, the 54-kDa ΔG form could result in gain-of-function relative to wildtype enzyme if this situation also occurs in vivo (11). Proof of this hypothesis will require expression in erythroid cells and further characterization of the processing of the enzyme in situ and its affect on ALA production in vivo.
An alternative hypothesis that could explain these results is that the FECH enzyme activity is affected by the ALAS2 exon 11 mutations. This result could occur if the FECH activity depends on binding to the carboxy-terminal region of ALAS2 that is modified by the ALAS2 gain-of-function mutations. Another mechanism could be that some other protein binds to the carboxy-terminus of ALAS2 and downregulates the wild-type activity in vivo. Loss of this inhibition by deletions of this binding region would then result in increased ALAS2 activity in vivo. Because FECH interacts directly with mitoferrin and/or other iron metabolism factors in the mitochondria to highly regulate the production of erythroid heme (20), this regulatory control prevents the overproduction of heme. Thus, increasing the ALAS2 activity and/or stability does not cause heme excess, but does result in increased protoporphyrin, thereby causing XLP.
Conclusion
These studies provide kinetic data and stability properties for homogeneous recombinant ALAS2 wild-type enzyme and the mutant proteins causing XLP. While in general, they have a modest 2- to 3-fold gain-of-function, the lack of increased activity in vitro for the ΔAmutation highlights the fact that the gain-of-function of ALAS2 in XLP does not necessarily result from the increased catalytic activity of the enzyme, but may be due to alternative causes, such as alterations in protein stability, protein-protein interactions, and/or substrate availability and/or product release in vivo. A fuller understanding of the role of the carboxy-terminal region of ALAS2 awaits in vivo studies in eukaryotic cells.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Whatley SD, et al. (2008) C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am. J. Hum. Genet. 83:408–14.
Anderson KE, Sassa S, Bishop DF, Desnick RJ. (2001) Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver CR, et al. (eds.) McGraw-Hill, New York, pp. 2991–3062.
Magnus IA, Jarrett A, Prankerd TA, Rimington C. (1961) Erythropoietic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinaemia. Lancet 2:448–51.
Bonkowsky HL, Bloomer JR, Ebert PS, Mahoney MJ. (1975) Heme synthetase deficiency in human protoporphyria: demonstration of the defect in liver and cultured skin fibroblasts. J. Clin. Invest. 56:1139–48.
Anderson KE. (2008) The porphyrias. In: Cecil Medicine. Goldman L, Ausiello D (eds.) Saunders, Philadelphia, pp. 1585–93.
Gross U, Frank M, Doss MO. (1998) Hepatic complications of erythropoietic protoporphyria. Photodermatol. Photoimmunol. Photomed. 14:52–7.
Harms J, Lautenschlager S, Minder CE, Minder EI. (2009) An alpha-melanocyte-stimulating hormone analogue in erythropoietic protoporphyria. N. Engl. J. Med. 360:306–7.
Wahlin S, et al. (2010) Combined liver and kidney transplantation in acute intermittent porphyria. Transpl. Int. 23:e18–21.
Balwani M, et al. (2013) Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and X-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol. Med. 19:26–35.
Cotter PD, Rucknagel DL, Bishop DF. (1994) X-linked sideroblastic anemia: identification of the mutation in the erythroid-specific delta-aminolevulinate synthase gene (ALAS2) in the original family described by Cooley. Blood. 84:3915–24.
Bishop DF, Tchaikovskii V, Hoffbrand AV, Fraser ME, Margolis S. (2012) X-linked sideroblastic anemia due to carboxy-terminal ALAS2 mutations that cause loss of binding to the beta-subunit of succinyl-CoA synthetase (SUCLA2). J. Biol. Chem. 287:28943–55.
Furuyama K, et al. (1997) Pyridoxine refractory X-linked sideroblastic anemia caused by a point mutation in the erythroid 5-aminolevulinate synthase gene. Blood. 90:822–30.
Laemmli UK, Favre M. (1973) Maturation of the head of bacteriophage T4. I. DNA packaging events. J. Mol. Biol. 80:575–99.
Urata G, Granick S. (1963) Biosynthesis of alphaaminoketones and the metabolism of aminoacetone. J. Biol. Chem. 238:811–20.
Bishop DF, et al. (1978) Pilot scale purification of alpha-galactosidase A from Cohn fraction IV-1 of human plasma. Biochim. Biophys. Acta. 524:109–20.
Kadirvel S, et al. (2012) The carboxyl-terminal region of erythroid-specific 5-aminolevulinate synthase acts as an intrinsic modifier for its catalytic activity and protein stability. Exp. Hematol. 40:477–86.e1.
Shah DI, et al. (2012) Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature. 491:608–12.
Surinya KH, Cox TC, Masy B. (1997) Transcriptional regulation of the human erythroid 5-aminolevulinase synthase gene. J. Biol. Chem. 272:26585–94.
Lendrihas T, Hunter GA, Ferreira GC. (2010) Targeting the active site gate to yield hyperactive variants of 5-aminolevulinate synthase. J. Biol. Chem. 285:13704–11.
Chen W, Dailey HA, Paw BH. (2010) Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood. 116:628–30.
Ducamp S, et al. (2013) Molecular and functional analysis of the C-terminal region of human erythroid-specific 5-aminolevulinic synthase associated with X-linked dominant protoporphyria (XLDPP). Hum. Mol. Genet. 2013, Jan 3 [Epub ahead of print].
Acknowledgments
This research was supported in part by grants from the National Institutes of Health (NIH), including a research grant (5 R01 DK026824) and a grant (1 U54 DK083909) for the Porphyria Consortium of the NIH Rare Diseases Clinical Research Network as well as a research grant (C024404) from the New York State Department of Health. Funding and/or programmatic support for this project was provided by the NIH Office of Rare Disease Clinical Research Network. The views expressed in written materials or publications do not necessarily reflect the official policies of the Department of Health and Human Services.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Bishop, D.F., Tchaikovskii, V., Nazarenko, I. et al. Molecular Expression and Characterization of Erythroid-Specific 5-Aminolevulinate Synthase Gain-of-Function Mutations Causing X-Linked Protoporphyria. Mol Med 19, 18–25 (2013). https://doi.org/10.2119/molmed.2013.00003
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2013.00003