
Abstract
High mobility group box 1 (HMGB1), the prototypic damage-associated molecular pattern molecule, is released at sites of inflammation and/or tissue damage. There, it promotes cytokine production and chemokine production/cell migration. New work shows that the redox status of HMGB1 distinguishes its cytokine-inducing and chemokine activity. Reduced all-thiol-HMGB1 has sole chemokine activity, whereas disulfide-HMGB1 has only cytokine activity, and oxidized, denatured HMGB1 has neither. Autophagy (programmed cell survival) and apoptosis (programmed cell death) have been implicated in controlling both innate and adaptive immune functions. Reduced HMGB1 protein promotes autophagy, whereas oxidized HMGB1 promotes apoptosis. Thus, the differential activity of HMGB1 in immunity, inflammation and cell death depends on the cellular redox status within tissues.
Similar content being viewed by others

High mobility group box 1 (HMGB1), a nonhistone nuclear factor, acts extracellularly as a damage-associated molecular pattern (DAMP) molecule to modulate inflammation, promoting autophagy and innate immune responses (1–5). HMGB1 has compartment-specific functions: nuclear, intracellular (but extranuclear) and extracellular. Its extracellular functions can now be divided further into cytokine-like or cytokine-inducing, chemokinelike and proangiogenic. Signaling pathways that induce variations on the posttranslational modification, such as phosphorylation and acetylation, have been implicated in the regulation of HMGB1 release. Importantly, HMGB1 contains three cysteines, each of which is susceptible to redox modification (6,7). The redox state of these cysteines is important for the proinflammatory cytokine-stimulating and proautophagic activity of HMGB1 (8–10). Autophagy (literally “self-eating”), a lysosome-mediated catabolic process, contributes to maintenance of intracellular homeostasis and promotes cell survival in response to environmental stress (11–13).
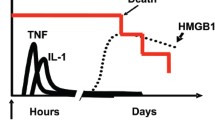
Treatment with reduced but not oxidized HMGB1 protein increases autophagy in cancer cells (9). In contrast, oxidized HMGB1 protein activates the caspase-dependent apoptotic cell death pathway (9). Venereau et al. (14) described a new role for redox control of both the cytokine-inducing and chemokine activity of HMGB1 in the setting of sterile inflammation, regulating leukocyte recruitment and their ability to secrete inflammatory cytokines (Figure 1).

Redox control of HMGB1 activity. To act as a DAMP/danger signal and inflammatory mediator, HMGB1 is transported extracellularly by two principal means: active secretion from living inflammatory cells (for example, macrophages) or passive release from necrotic cells. The activities of extracellular HMGB1 are redox dependent. All-thiol-HMGB1 promotes chemokine production and leukocyte recruitment. Disulfide-HMGB1, originating from infiltrating leukocytes, promotes release of proinflammatory cytokines and thus participates in the inflammatory response. Reactive oxygen species produced by leukocytes induces the terminal oxidation of HMGB1, which is inactivated during resolution of inflammation.
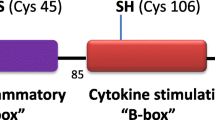
Structurally, HMGB1 is composed of three domains: two positively charged proximal DNA-binding domains (A box and B box) and a negatively charged carboxyl terminus. Three cysteines are encoded within the molecule: two vicinal cysteines in box A (C23 and C45) and a single one in box B (C106). C23 and C45 can form an intermolecular disulfide bond, whereas C106 is unpaired. Therefore, three different redox forms HMGB1 (all-thiol-HMGB1, disulfide-HMGB1 and oxidized HMGB1) were derived from bacterial expression systems (14). In addition, by using tryptic digests and liquid chromatography tandem mass spectrometric analysis, Venereau et al. observed that recombinant HMGB1 can be reversibly oxidized and reduced in the presence of electron donors (for example, dithiothreitol) or acceptors (oxygen) (14).
Next, Venereau et al. assessed whether individual redox forms of HMGB1 have a differential role in cytokine-stimulating and chemoattractant activities (14). They found that disulfide-HMGB1 induced activation of the nuclear factor (NF)-κB pathway and production of proinflammatory cytokines (for example, tumor necrosis factors-α, interleukin [IL]-6 and IL-8) in fibroblasts and macrophages. Interestingly, all-thiol-HMGB1 failed to induce a proinflammatory response. In contrast, all-thiol-HMGB1, but not disulfide-HMGBl, had chemoattractant activity in fibroblasts. These findings prompted them to determine whether HMGB1 inhibitors, such as box A and monoclonal antibody PDH1.1, block the chemoattractant and/or cytokine-inducing activities of HMGB1. Unexpectedly, these inhibitors prevented cell migration but not cytokine production, although they are widely used as HMGB1-targeting agents in experimental inflammatory diseases.
Reactive oxygen species oxidize the HMGB1 released from dying cells, thereby neutralizing its stimulatory activity and promoting tolerance in immune cells (15,16). In addition, oxidation of C106 or lack of a disulfide bridge between C23 and C45 then causes HMGB1 to lose its proinflammatory effects in macrophages (8). Venereau et al. found that terminal oxidation by hydrogen peroxide results in the loss of both the cytokine-stimulating and chemoattractant activities of HMBG1. Moreover, the authors found that the three HMGB1 cysteine residues were required for the cytokine-stimulating activity but not for the chemoattractant activity of HMGB1. Cysteine mutant HMGB1 promotes fibroblast migration, but not cytokine expression in macrophages (14). Collectively, these findings establish a crucial role for redox in the regulation of HMGB1 activity in inflammation and migration.
What is the redox state of HMGB1 in the pathogenesis of individual diseases? The redox state of HMGB1 from the human acute monocytic leukemia cell line THP-1 was measured in the presence or absence of lipopolysaccharide (LPS) and necrotic medium in vitro. Intracellular HMGB1 was all-thiol-HMGB1, whereas secreted HMGB1 contained both all-thiol- and disulfide-HMGB1 (14). Furthermore, disulfide-HMGB1 was present later and time-dependently increased in cardiotoxin-injured muscles in vivo, confirming that the redox state of HMGB1 is altered during tissue damage and inflammation. HMGB1 protein with all three cysteines mutated to serine are resistant to oxidation and induce leukocyte recruitment without inducing cytokine production (14). The activities of HMGB1 are thus redox-dependent and can be modified within the injured tissues after HMGB1 release. Therefore, release of dynamic redox-regulated HMGB1 contributes to the orderly orchestrated recruitment of leukocytes, activation of cytokine release and subsequent resolution of inflammation.
Several issues remain unresolved regarding the redox control of HMGB1 activity. First, HMGB1 is specifically recognized by several cell surface receptors (2), including Toll-like receptor (TLR)-4 and the receptor for advanced glycation end products (RAGE), but most recently was joined by T-cell immunoglobulin and mucin domain 3 (TIM-3) (17). Initial studies suggest that reduced C106 is necessary for the binding of HMGB1 to one of its receptors, TLR4, to stimulate cytokine release (8). HMGB1-induced recruitment of inflammatory cells depends on forming a complex with CXCL12 and signaling via CXCR4 (18). Moreover, RAGE is required for reduced HMGB1-mediated autophagy, but not oxidized HMGB1-induced apoptosis (9). All-thiol-HMGB1, but not disulfide-HMGB1, binds CXCL12 (14). The influence of HMGB1 receptors (for example, RAGE, TLR4, TLR2, CD24, TIM-3 and triggering receptor expressed on myeloid cells 1 [TREM1]) on biological activities of individual redox forms of HMGB1 remains to be carefully investigated. Second, HMGB1 forms highly inflammatory complexes with DNA, lipoteichoic acid, LPS, IL-1ß, chemokine (C-X-C motif) lig-and 12 (CXCL12)/stromal cell-derived factor-1 (SDF-1) and nucleosomes (19). There is great interest in determining whether the individual redox forms of HMGB1 have varying affinity profiles active in inflammation and immunity. Third, HMGB1 has multiple intracellular and extracellular functions in health and disease, including cancer (1,2,6,20). Additional studies will be needed to determine whether redox is required for other functions of HMGB1, such as regeneration and cellular differentiation as well as the complex interactions between autophagy and immunity (5). One additional unanswered question is where and how the formation of the disulfide takes place and whether there is an enzyme specific for regulating this. This is important, knowing that the nuclear form is mostly all thiol. Finally, the development and performance of a simple, sensitive method for the detection of individual HMGB1 redox state iso-forms in clinical specimens remains to be accomplished.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Lotze MT, Tracey KJ. (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5:331–42.
Harris HE, Andersson U, Pisetsky DS. (2012) HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 8:195–202.
Lotze MT, et al. (2007) The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol. Rev. 220:60–81.
Andersson U, Tracey KJ. (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29:139–62.
Tang D, Kang R, Coyne CB, Zeh HJ, Lotze MT. (2012) PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol. Rev. 249:158–75.
Tang D, Kang R, Zeh HJ 3rd, Lotze MT. (2011) High-mobility group box 1, oxidative stress, and disease. Antioxid. Redox. Signal. 14:1315–35.
Rubartelli A, Lotze MT. (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 28:429–36.
Yang H, et al. (2010) A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. U. S. A. 107:11942–7.
Tang D, et al. (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 29:5299–310.
Yang H, et al. (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol. Med. 18:250–9.
Vernon PJ, Tang D. (2012) Eat-me: autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox. Signal. 2012, Sep 18. [Epub ahead of print].
Yang Z, Klionsky DJ. (2010) Eaten alive: a history of macroautophagy. Nat. Cell. Biol. 12:814–22.
Kroemer G, Marino G, Levine B. (2010) Autophagy and the integrated stress response. Mol. Cell. 40:280–93.
Venereau E, et al. (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J. Exp. Med. 209:1519–28.
Kazama H, et al. (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity. 29:21–32.
Lotfi R, et al. (2009) Eosinophils oxidize damage-associated molecular pattern molecules derived from stressed cells. J. Immunol. 183:5023–31.
Chiba S, et al. (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13:832–42.
Schiraldi M, et al. (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med. 209:551–63.
Bianchi ME. (2009) HMGB1 loves company. J. Leukoc. Biol. 86:573–6.
Tang D, Kang R, Zeh HJ 3rd, Lotze MT. (2010) High-mobility group box 1 and cancer. Biochim. Biophys. Acta. 1799:131–40.
Acknowledgments
This work was supported by the National Institutes of Health (R01CA160417 to D Tang) and a grant from the University of Pittsburgh (to D Tang).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Tang, D., Billiar, T.R. & Lotze, M.T. A Janus Tale of Two Active High Mobility Group Box 1 (HMGB1) Redox States. Mol Med 18, 1360–1362 (2012). https://doi.org/10.2119/molmed.2012.00314
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00314