
Abstract
Maraviroc (MVC) is the first licensed antiretroviral therapeutic agent to target a host cell surface molecule, and successful HIV-1 entry blockade by this C-C chemokine receptor type 5 (CCR5)-antagonist potentiates immunomodulation. We hypothesized that MVC intensification impacts immunization responses, T-cell phenotype, function and delayed type hypersensitivity (DTH) in HIV-1+ subjects. A 24-wk, double-blinded, placebo-controlled study of the addition of MVC to suppressive antiretroviral therapy in HIV-1+ persons was performed. Subjects received DTH tests, intramuscular tetanus, meningococcal and oral cholera immunizations. Antibody titers, T-cell function and phenotype were assessed. Of 157 patients referred, 47 were randomized 1:1; MVC:placebo. MVC enhanced meningococcal neo-immunization, blunted cholera response and expedited lymphoproliferation to tetanus boost, without affecting recall humoral response. Anti-HIV-1 group-specific antigen (Gag) and tetanus toxoid (TTox) function improved significantly, HIV-1-associated CD8 T-cell skewing normalized, and the percentage of late-stage and major histocompatibility complex (MHC) class II expressing CD4 T-cells increased. Activated CD4+ CD38+ human leukocyte antigen (HLA)-DR+ T-cells declined, and costimulation shifted to coinhibition. DTH was unchanged. Maraviroc intensification, through antagonism of the cell surface molecule CCR5, favorably influences immune profiles of HIV-1+ patients, supporting its immunomodulatory use in HIV-1 infection and potentially in other immunologically relevant settings.
Similar content being viewed by others

Introduction
The CCR5 antagonist Maraviroc (MVC) which prevents HIV-1 entry into target CD4+ C-C chemokine receptor type 5+ (CD4+ CCR5+)cells, is well tolerated and approved as a combination antiretroviral therapy (cART) component (1,2). CCR5 plays a role in immune activation and lymphocyte recruitment (3). Individuals homozygous for the CCR5Δ32 mutation show reduced immune activation, lower production of interleukin-2 (IL-2) and resistance to HIV-1 infection (4–6), furthermore CCR5−/− mice display significantly lower delayed type hypersensitivity (DTH) than those expressing CCR5 (7). Immune hyperactivation, skewed T-cell differentiation, senescence, exhaustion, anergy and loss of functionality are hallmarks of progressive HIV-1 infection (8–13). MVC may have immunomodulatory properties, potentially facilitating immunological improvement toward normality, when added to successfully suppressive cART (1,13).
Use of MVC in treatment experienced settings (MOTIVATE) associated with delayed category C clinical events (14), and MVC-containing cART increased both memory and naïve CD4+ CCR5+ T-cells (15), and decreased CD8 T-cell activation (1). Stimulation of peripheral blood mononuclear cells (PBMC) with recall antigen leads to CCR5 ligand release, protecting CD4+ memory T-cell targets of R5-tropic HIV-1, and increased levels of CCR5 ligand also occurs in the plasma of MVC-treated patients (16,17). Owing to essential immune orchestration by CD4 T-cells, and chemokine network complexity, it is likely that MVC intensification impacts cell-mediated and humoral immune responses. Increased CCR5 expression on gut and blood CD4 and CD8 T-cells (17), and downregulation of leukocyte trafficking in PBMC of MVC-treated patients (18), may affect peripheral blood and gut-associated lymphoid tissue (GALT) compartments differentially.
We conducted a phase IV, double-blind, placebo-controlled trial to explore the hypothesis that cART intensification using a CCR5 antagonist has a favorable immunological impact on T-cell phenotype, humoral and cell-mediated responses to previously encountered antigen, DTH, and the response to immunization with neoantigens.
Materials and Methods
Study Design
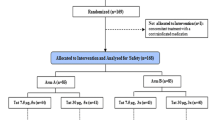
A double-blind placebo-controlled trial in HIV-1+ subjects investigating the impact of the addition of MVC 150 mg twice daily (b.i.d. [bis in die]) to ongoing successful ritonavir-boosted protease inhibitor (PI/r)-based cART, on immune function (Figure 1). Detailed immunology investigated activation (CD38/human leukocyte antigen (HLA)-DR), differentiation (CD27/CD28), senescence (CD57), exhaustion (PD-1) and coinhibition (CTLA-4) immunophenotypes using flow cytometry (12,19). Immune function to HIV-1 and recall antigens (group-specific antigen [Gag] major histocompatibility complex [MHC] class I-restricted peptides [GagMHCI] and overlapping 20 amino acid Gag peptides [Gag20], tetanus toxoid [TTox], and cytomegalovirus [CMV] lysate), was assessed by IFN-γ, IL-2 and perforin ELISpot, lymphoproliferation assays (13), and DTH by Mantoux test. All participants were immunized as detailed (Figure 1). Anti-tetanus and anti-meningococcal subtype C (anti-MenC) immunoglobulin (Ig) G responses were assessed (20,21), with anti-cholera IgA titers assayed by anti-cholera toxin B-subunit (anti-CTB) enzyme-linked immunosorbent assay (ELISA) (Crucell Holland BV, Leiden, the Netherlands).

Study design. Patients enrolled in the study attended for a total of eight clinical visits. Assessments and interventions performed at each visit are detailed. Concomitant medication was continued and adverse events monitored throughout. Blinded 1:1 randomization of participants into MVC and placebo arms occurred at baseline, with drug packs lasting 24 wks. Detailed immunology and HIV clinical care was performed at screen, baseline, wks 4, 12, 16 and 24. Ab, antibody.
Study Population
Patients provided written informed consent prior to study entry, after which eligibility was assessed according to inclusion and exclusion criteria (Figure 2). The study was conducted according to Good Clinical Practice guidelines (22), in accordance with the Declaration of Helsinki (23), approved by UK ethics committee (Riverside Research Ethics Committee, NHS Health Research Authority, Bristol, UK), funded and sponsored by St Stephen’s AIDS Trust (SSAT), with an unrestricted grant from Pfizer Inc. (New York, NY, USA).

Referral, screening, randomization and completion of treatment. Thirty percent of 157 patients referred were randomized into the two treatment arms at baseline. Twenty-four participants were assigned to receive placebo and 23 assigned to receive 150 mg MVC b.i.d.. Seventeen participants in the placebo arm completed the study to wk 24, compared with 20 in the MVC arm. DNA, did not attend; DO, dropout; ITT, intent to treat; LTFU, lost to follow up.
Study Medication
MVC was used at the European Union/United States of America (EU/USA)-approved dose for subjects on PI/r (150 mg b.i.d.). Vaccines were used per manufacturer’s instructions at standard doses, well tolerated in HIV-1+ subjects (24,25). MVC and matching placebo were provided as 24-wk double-blind labeled clinical trial drug packs, randomized by Pfizer Inc. To explore humoral immunity induction in both peripheral blood and gut-associated lymphoid tissue (GALT), neoantigens were selected with different administration modes; namely MenC (Menjugate, Farillon Ltd., Bedford, UK) through intramuscular route, and cholera (Dukoral, Crucell Holland BV) through oral immunization. Tetanus Merieux (Godecke GmbH, Freiberg, Germany) was administered intramuscularly. Resulting plasma antibody titers were assessed. Mantoux test (Farillon Ltd.) involved administration of 0.1 mL tuberculin PPD RT 23 SSI (Statens Serum Insitut, Copenhagen, Denmark) intradermally in the forearm with d-2 reading.
Efficacy Analysis
Changes were measured from baseline for each patient at each visit. Primary end-point was change from baseline in antitetanus IgG titers following immunization. Secondary endpoints were changes from baseline in: i) lymphoproliferative and cytokine response; ii) T-cell phenotype; iii) HIV-1 RNA; iv) humoral response to oral and intramuscular neoantigens; and v) clinical and laboratory safety.
Safety Analysis
Safety monitoring was continuous, with investigator-determined grading and causality assessment, according to the “Division of AIDS Table for Grading the Severity of Adult and Pediatric Adverse Events,” version 1.0, December 2004 (clarification August 2009) (26).
Statistical Analysis
All participants who received at least one dose of study medication formed the statistical analysis. The study was powered to 80% requiring 24 patients to detect a difference of 45% in primary end-point between treatment arms. Between group comparisons of qualitative data were tested using χ2 test with Yates correction where appropriate. Quantitative data were analyzed using the Mann-Whitney U test or by unpaired t test. A linear mixed model (SAS v9.1.3), compared between group data collected over time to derive time-weighted differences from baseline to each time point with values of all study time points as dependent variables, assuming missing data were missing at random. Independent variables included fixed effects of MVC and placebo intervention, study visit time points and interaction of intervention by study time point. Point estimates and 95% CI of changes from baseline were obtained from intervention by study time point interaction. Following analyses of all data, protocol deviators with potential to impact data were considered. Analyses where protocol deviators confounded results are not reported. Owing to the large number of parameters investigated, only analyses resulting in changes significantly different from baseline in the MVC arm, with no such change in placebo, are presented. AE are reported by study arm.
Results
Study Population
A substantial proportion of individuals referred to the study declined consent after detailed information of the trial protocol was provided (Figure 2), most likely reasons for not enrolling appear to have been the number of immunizations and clinic visits required. Written informed consent was obtained prior to screen, with twenty individuals subsequently excluded, as they did not fulfill all inclusion criteria. Baseline patient demographics matched between arms (Table 1). Of 47 patients randomized, 37 attended wk 24 (Figure 2). Three patients in the MVC and seven in the placebo arm did not attend wk 24. Discontinuation was attributed to noncompliance (1 subject; MVC), and personal reasons. No subjects discontinued due to AE.
Efficacy
Humoral immunity. The primary end-point was change in anti-tetanus antibody titer from baseline. Following tetanus immunization, both groups had significantly increased anti-tetanus antibody titers at wk 16, remaining significant at wk 24 (Figure 3A). There was no significant difference between treatment arms.

Humoral and T-cell-mediated responses showing significant changes from baseline over the 24-wk study period, in the context of MVC intensification and immunization. Changes from baseline in: (A) anti-tetanus antibody titers; (B) anti-MenC antibody titers; and (C) anti-CTB antibody titers. Change from baseline in IFN-γ production to stimulation with: (D) HIV-1 Gag20; and (E) TTox. (F) Change from baseline in proliferative response to stimulation with TTox. Mean changes from baseline estimated by mixed model analysis are plotted with bars above and below representing 95% confidence interval. Significance from baseline is shown by error bars that do not bisect the x axis, and is marked with *. Relevant immunizations are indicated by vertical dashed lines. All immunizations were administered at the time point indicated, after blood was drawn for immunological assessments. Ab, antibody.
Antibody response to intramuscular neo-immunization was increased by MVC-intensification, with anti-MenC antibody titers in the MVC group significantly higher than baseline at wks 16 and 24, with no change in the placebo group (Figure 3B). In contrast, MVC appeared to hinder the humoral response to oral immunization (Figure 3C). Although an upward trend was seen at wk 16, anti-CTB antibodies did not increase significantly from baseline in the MVC group, and, compared with wk-16 levels, declined at wk 24 after second dose. However, anti-CTB titers increased significantly from baseline by wk 16 in the placebo arm, increasing further after second dose. No correlation was observed between nadir CD4 count and humoral response to immunization.
Cell-mediated immune function. There was no significant change from baseline in DTH in MVC (κ = 100%; 95% CI: 78% to 100%), or placebo (κ = 100%; 95% CI: 74% to 100%) arms.
Twenty-four weeks of MVC intensification increased the IFN-γ T-cell response to HIV-1 Gag20 and TTox following the wk-12 tetanus booster immunization (Figures 3D, E). Despite changes in IFN-γ production, no differences from baseline were observed in IL-2 release for any antigen tested (data not shown), and lymphocyte proliferation in response to stimulation with GagMHCI, Gag20 or CMV remained unchanged (data not shown). The increase in proliferative response to TTox stimulation following tetanus immunization occurred earlier in the MVC group than in placebo (Figure 3F).
Immunophenotype. No significant changes in absolute numbers or percentage of CD4 or CD8 T-cells were observed in either arm (data not shown). Changes in T-cell subsets are shown in Figure 4. Upon MVC treatment, CD8 T-cells exhibited a shift toward earlier differentiation, with a significant increase in early CD8+ CD28+ CD27+ (Figure 4A) and intermediate CD8+ CD28− CD27+ subsets (Figure 4B), and a contrasting decline in late CD8+ CD28− CD27− T-cells (Figure 4C). CD27 expression on CD8 T-cells increased significantly from baseline by wk 24 (Figure 4D). MVC intensification also resulted in increased density of co-inhibitory CTLA-4 on CD8 T-cells (mean fluorescence intensity, [MFI]) (Figure 4E).

Changes from baseline in phenotypic markers expressed on CD8 and CD4 T-cells over the 24-wk study period. Changes from baseline in percentage of CD8 T-cells expressing (A) CD28+CD27+ (early), (B) CD28−CD27+ (intermediate), (C) CD28−CD27− (late) and (D) CD27+. (E) Changes from baseline in mean fluorescence intensity (MFI) of CTLA−4 expressed on CD8 T-cells. Changes from baseline in the percentage of CD4 T-cells expressing (F) CD28−CD27− (late) and (G) CD28+, and MFI of (H) CD28 and (I) CTLA-4 expressed on CD4+ T-cells. Changes from baseline in the percentage of CD4 T-cells expressing (J) CD38+HLA-DR+ and (K) CD38−HLA-DR+. Mean changes from baseline estimated by mixed model analysis are plotted with bars above and below representing 95% confidence intervals. Significance from baseline is shown by error bars that do not bisect the x axis, and is marked with *. When significant differences from baseline were lost upon removal of protocol deviators from the analysis, data from all patients is plotted with * shown. Immunizations are indicated by vertical dashed lines. All immunizations were administered at the time point indicated, after blood was drawn for immunological assessments.
No changes in early or intermediate CD4 T-cell subsets were observed (data not shown), however, in MVC-treated patients, an increase from baseline in late-stage CD4+ CD28−CD27− T-cells was seen at every time-point (Figure 4F). A corresponding significant decrease from baseline in percentage of CD4+ CD28+ T-cells, and cell-surface density of CD28 was observed in the MVC arm (Figures 4G, H). The opposite was seen for CTLA-4 cell-surface density on CD4 T-cells (Figure 4I).
Activated CD4+ CD38+ HLA-DR+ T-cells were reduced significantly by wk 24 of MVC treatment (Figure 4J), however an increase in CD4 + CD38-HLA-DR + T-cells was observed after 4 wks of MVC, and was sustained at all other time points (Figure 4K). An increase in this subset also occurred transiently at wk 16 in the placebo group (Figure 4K). No significant, sustained changes in senescence (CD57) or exhaustion (PD-1) markers were observed.
Safety. No clinically relevant changes occurred in hematology, biochemistry, lymphocyte subset or virological parameters investigated (HIV-1 clinical care) (Figure 1). No serious AE were reported. Forty-nine AE were reported in the MVC (11 grade 2, 38 grade 1) and 37 in the placebo arm (1 grade 3, 4 grade 2 and 32 grade 1); most considered unrelated to study drug.
Discussion
This is the first study to assess MVC-intensification using vaccine response endpoints. Although cART successfully controls HIV-1 replication to levels below standard detection limits, immune senescence, increased activation, skewed differentiation and suboptimal immunization response (8–13,24), remain unmet needs to be addressed through novel immunotherapeutic strategies. Our data reveal, for the first time, no detrimental effect of CCR5 antagonism on humoral recall response, and divergent impact on oral versus intramuscular neo-responses. Furthermore, we show that antiviral cytokine production to HIV-1 significantly increased upon administration of MVC, as did the cell-mediated response to TTox, coupled with expedited proliferation following tetanus boost. MVC favorably alters the HIV-1-associated skewing of CD8 T-cell differentiation; increases coinhibition of T-cells, the proportion of late-stage CD4 T-cells and those expressing cell-surface MHC class II (possibly via protection from residual R5-tropic virus); and decreases costimulation and HIV-1-associated activation of CD4 T-cells. MVC intensification in treatment-experienced individuals with reduced CD4 counts increases reconstitution (14,27); however, in our study, where all patients were receiving successful cART, CD4 count was unchanged. MVC was well tolerated with no excess of AE, laboratory or clinical changes, concurring with previous reports (1,28).
T-cell phenotype is indicative of function, and the HIV-1-associated skewing of CD8 T-cell differentiation toward enrichment of later stage T-cells, is coupled with a shift from an IL-2 producing, replicatively competent T-cell pool, toward the IFN-γ production, IL-2 deficiency and inadequate antiviral activity characteristic of chronic HIV-1 disease (9,13,19). Increased proportions of highly differentiated CD8 T-cells also have been shown to correlate with disease progression (29), and so the reversal of this skewing toward normalization in the CD8 T-cell compartment after 24 wks of MVC, suggests favorable impact on clinical outcome. It is of note that subsets are expressed as a proportion of the entire T-cell pool, and so compartment percentages are relative to one another. The increase in proportion of early and intermediate CD8 T-cells, and decrease of those at late stage after MVC intensification may result from reduced plasma lipopolysaccharide (17) and/or prevention of macrophage and dendritic cell recirculation (18), decreasing consequent signature chronic inflammation (8), thereby preventing HIV-1-induced skewing. CCR5 antagonism also may revert late phenotype toward intermediate, as T-cell subsets exhibit differential expression of CCR5 (30). In contrast, late-stage CD4 T-cells increased as early as 4 wks into MVC intensification, possibly due to reduced early and intermediate subsets and/or a CD4 T-cell protective effect of MVC CCR5 antagonism (1,30). Central and transitional memory CD27+ CD4+ Tcells, at earlier stages of differentiation, have been shown to harbor the majority of the proviral reservoir (31), with more terminally differentiated CD4 T cells exhibiting a lower level of HIV-1 infection (32). The increased proportion of late-stage CD4 T cells in the MVC-treated arm may, therefore, represent a desirable reduction in the HIV-1 reservoir (28,33).
Ligand binding to costimulatory CD28 reduces CCR5 expression, and coinhibitory CTLA-4 binding leads to CCR5 upregulation and inhibition of T-cell activation (30). The decline in CD28 expression and increase in CTLA-4 density in the T-cell compartment reported here may, therefore, be due to MVC antagonism downregulating CCR5. It is thought that T-cell differentiation, costimulation and activation are inter-linked processes, so this shift from costimulatory to coinhibitory molecule expression is likely to have ramifications with other phenotypic markers and corresponding T-cell functionality.
Activation marker expression on CD4 T-cells correlates with HIV-1 disease progression and higher risk of mortality (34). Here we confirm previous observations that MVC-intensification reduces activation (1,28), which may occur through direct CCR5 antagonism and/or a decline in chronic immune activation (17,18). CCR5 and CD38 density on CD4 T-cells correlate (35), supporting the observation that CCR5 antagonism reduced the proportion of CD4 T-cells expressing CD38. Other studies have shown that CCR5 antagonism causes T-cells to associate with lymph node macrophages instead of dendritic cells, diminishing T-cell activation (36), and may decrease monocyte trafficking in vivo, lessening chronic inflammation (18). A combination of such mechanisms may contribute to reduced CD4 T-cell activation, considered beneficial to disease prognosis (34).
CD4+ CD38−HLA-DR+ T-cells are hypothesized to be a substrate for CCR5-tropic viral replication (35), and the sustained increase in this subset from wk 4 until study completion may be explained by MVC preventing HIV-1 from infecting these memory CD4 T-cells. HLA-DR is an MHC class II molecule involved in presentation of exogenous antigen and, so, the increase in this subset, also transiently seen in the placebo group at wk 16, may be, in part, attributable to the immunization schedule. These HLA-DR+ T-cells provide help to B-cells during formation of humoral immunity, so an increase may be beneficial to antibody production.
It would be of interest to investigate the T-cell profiles and immunization responses of HIV-1 negative CCR5Δ32 individuals who have a lower occurrence of graft rejection (37), and less inflammation-associated mortality in dialysis (38), to determine whether the phenotype induced by MVC-mediated CCR5 antagonism resembles that of Δ32 hetero- and homozygotes.
Dysfunctional IL-2 production is characteristic of chronic HIV-1 infection (13), and, although surface expression of CCR5 correlates with IL-2 mRNA and protein levels and promotes T-cell proliferation (3), MVC-intensification did not affect IL-2 production in response to any of the antigens tested. Nevertheless IFN-γ production to HIV-1 Gag peptides is an important immune correlate, associated with lower viremia and delayed disease progression (13,39), and the increase in IFN-γ production to Gag20 upon MVC-intensification reported here may convey beneficial effects.
Responses to immunization in cART-treated HIV-1+ individuals, although observed, are often deficient either in quality or longevity (21,24). The immunization schedule and routes of administration for each individual vaccine were in accordance with the HIV immunization guidelines (24), however this particular combination is not commonly used in the clinical setting. The resulting immunomonitoring took into account kinetics of immune responses following antigenic stimulation. The neo-response to MenC immunization was increased in patients receiving MVC, suggesting that CCR5 antagonism improves responses to this novel antigen when encountered intramuscularly. This may be due to protection of circulating CCR5+ CD4+ T-cells from HIV-1 infection, and increased availability of MHC class II-presenting CD4 T-cells. MenC vaccine consists of Neisseria meningitidis group C oligosaccharide conjugated to Corynebacterium diphtheriae protein. Although not measured in this study, if the T-cell response to the protein component increased similarly to the anti-TTox response, then the resulting larger pool of vaccine-specific T helper cells may account for increased antibody production.
In contrast to this, the formation of neo-responses to enterally encountered antigen was impaired by MVC. Oral cholera vaccine contains killed whole Vibrio cholerae and nontoxic recombinant CTB. MVC accumulation in the GALT exceeds that of peripheral blood (40), suggesting a potentially greater effect in this compartment. HIV-1 infection selectively depletes CCR5+ CD4+ T-cells of the GALT (41), and MVC treatment has been shown to result in lower microbial translocation (8,17), suggesting possible restoration of GALT integrity. This may lead to less orally administered immunogen crossing the gut-blood barrier, affording explanation for the lower humoral response reported in the MVC arm. However, of note is the significant increase from baseline in anti-CTB IgA titers observed in the plasma of HIV-1 seronegative individuals receiving a single dose of Dukoral, which then significantly increased further upon second dose administration (42).
Antigen localization within the tissue, vaccine components and differences in dose and bioavailability also may explain the divergence observed between neo-immunogens. The effect of CCR5 antagonism on CCR5 ligands (43), and chemokine network trafficking (18), is not necessarily apparent from humoral response to immunization, which may not predict challenge outcome owing to pathogen tropism and infection compartmentalization. CCR5 density has been shown to govern T-cell migration into rheumatoid joints (44), and West Nile virus infection is more likely to result in severe symptoms in CCR5Δ32 individuals as it is thought that CCR5 acts to recruit leukocytes into the infected central nervous system (6). Blockade of CCR5 signaling reduces leukocyte trafficking and chemotaxis of monocytes (18), and interferes with T-cell activation at the draining lymph node (36), all of which could have a differential impact on immunization responses at specific anatomical sites. Alternatively, regulatory T-cells, if protected through CCR5 antagonism (45), may induce antigen-specific tolerance to orally administered immunogen (46).
Importantly, in concurrence with the reported lack of effect of MVC on influenza immunization (47), no difference was observed in humoral response to tetanus boost between the two groups, demonstrating no detrimental effect of in vivo CCR5 antagonism on recall immunity. However MVC administration did increase IFN-γ production to TTox, and expedited the expected peak in T-cell proliferation following immunization (48), indicating that the cell-mediated immune response to the protein vaccine component occurred more rapidly.
Conclusion
MVC intensification of cART favorably adjusts HIV-1-associated activation and differentiation profiles, increases certain anti-HIV-1 immune function, expedites cell-mediated response to boost immunization, and displays route-specific divergence of neo-immunization response. This data supports the use of MVC as an immunomodulator in HIV-1+ patients, and, potentially, in other immunologically relevant settings.
Disclosure
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Wilkin TJ, Gulick RM. (2012) CCR5 Antagonism in HIV infection: current concepts and future opportunities. Annu. Rev. Med. 63:81–93.
Gulick RM, et al. (2008) Maraviroc for previously treated patients with R5 HIV-1 infection. N. Engl. J. Med. 359:1429–41.
Camargo JF, et al. (2009) CCR5 expression levels influence NFAT translocation, IL-2 production, and subsequent signaling events during T lymphocyte activation. J. Immunol. 182:171–82.
Samson M, et al. (1996) Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 382:722–5.
Hutter G, et al. (2011) The effect of the CCR5-delta32 deletion on global gene expression considering immune response and inflammation. J. Inflamm. (Lond.). 8:29.
Lim JK, et al. (2010) CCR5 deficiency is a risk factor for early clinical manifestations of West Nile virus infection but not for viral transmission. J. Infect. Dis. 201:178–85.
Dolan MJ, et al. (2007) CCL3L1 and CCR5 influence cell-mediated immunity and affect HIV-AIDS pathogenesis via viral entry-independent mechanisms. Nat. Immunol. 8:1324–36.
Brenchley JM, et al. (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 12:1365–71.
Champagne P, et al. (2001) Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature. 410:106–11.
Brenchley JM, et al. (2003) Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood. 101:2711–20.
Wherry EJ, et al. (2007) Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 27:670–84.
Rosignoli G, et al. (2007) Expression of PD-L1, a marker of disease status, is not reduced by HAART in aviraemic patients. AIDS. 21:1379–81.
Westrop SJ, et al. (2009) Transient nature of long-term nonprogression and broad virus-specific proliferative T-cell responses with sustained thymic output in HIV-1 controllers. PLoS ONE. 4:e5474.
Asmuth DM, et al. (2010) CD4+ T-cell restoration after 48 weeks in the maraviroc treatment-experienced trials MOTIVATE 1 and 2. J. Acquir. Immune Defic. Syndr. 54:394–7.
Cossarini F, et al. (2011) Increased levels of CD4+ T cells expressing CCR5 during effective treatment with MRC. In: 18th Conference on Retroviruses and Opportunistic Infection; 2011 Feb 27–Mar 2; Boston, MA. Paper no. 573; poster no. L-202. Available from: https://doi.org/retroconference.org/AbstractSearch/Default.aspx?Conf=20
Sun L, Abdelwahab SF, Lewis GK, Garzino-Demo A. (2004) Recall antigen activation induces prompt release of CCR5 ligands from PBMC: implication in memory responses and immunization. Int. Immunol. 16:1623–31.
Hunt P, et al. (2011) Immunomodulatory effects of MVC intensification in HIV-infected individuals with incomplete CD4+ T cell recovery during suppressive ART. In: 18th Conference on Retroviruses and Opportunistic Infection; 2011 Feb 27–Mar 2; Boston, MA. Paper no. 153LB. Available from: https://doi.org/retroconference.org/AbstractSearch/Default.aspx?Conf=20
Rossi R, et al. (2011) In vitro effect of anti-human immunodeficiency virus CCR5 antagonist maraviroc on chemotactic activity of monocytes, macrophages and dendritic cells. Clin. Exp. Immunol. 166:184–90.
Appay V, van Lier RA, Sallusto F, Roederer M. (2008) Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 73:975–83.
Gheesling LL, et al. (1994) Multicenter comparison of Neisseria meningitidis serogroup C anti-capsular polysaccharide antibody levels measured by a standardized enzyme-linked immunosorbent assay. J. Clin. Microbiol. 32:1475–82.
Hart M, et al. (2007) Loss of discrete memory B cell subsets is associated with impaired immunization responses in HIV-1 infection and may be a risk factor for invasive pneumococcal disease. J. Immunol. 178:8212–20.
The European Parliament and the Council of the European Union. (2001) Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Off. J. Eur. Communities. L121/34–44.
World Medical Association [WMA]. (1964) WMA Declaration of Helsinki — ethical principles for medical research involving humans. Last amended 2008 Oct. [cited 2012 Oct 15]. Available from: https://doi.org/www.wma.net/en/30publications/10policies/b3/index.html
Geretti AM, et al. (2008) British HIV Association guidelines for immunization of HIV-infected adults 2008. HIV Med. 9:795–848.
Lewis DJ, et al. (1994) Immune response following oral administration of cholera toxin B subunit to HIV-1-infected UK and Kenyan subjects. AIDS 8:779–85.
Divisio of AIDS (DAIDS). (2004, 2009). Division of AIDS table for grading the severity of adult and pediatric adverse events. Version 1.0 (2004); clarification (2009). [cited 2012 Oct 23]. Available from: https://doi.org/rsc.tech-res.com/safetyandpharmacovigilance/gradingtables.aspx
Espiau M, et al. (2011) Maraviroc intensification for suboptimal CD4 T cell response in a perinatally HIV-infected adolescent. AIDS. 25:1243–4.
Gutierrez C, et al. (2011) Intensification of antiretroviral therapy with a CCR5 antagonist in patients with chronic HIV-1 infection: effect on T cells latently infected. PLoS One. 6:e27864.
Papagno L, et al. (2004) Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. 2:E20.
Westrop SJ, Moyle GJ, Imami N. (2010) Nature and function of chemokine receptor 5 in immune activation, HIV-1 entry and targeted therapeutics. J. Viral Entry. 4:1–8.
Chomont N, et al. (2009) HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15:893–900.
Brenchley JM, et al. (2004) T-cell subsets that harbor human immunodeficiency virus (HIV) in vivo: implications for HIV pathogenesis. J. Virol. 78:1160–8.
Descours B, et al. (2012) Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clin. Infect. Dis. 54:1495–503.
Giorgi JV, et al. (1999) Shorter survival in advanced human immunodeficiency virus type 1 infection is more closely associated with T lymphocyte activation than with plasma virus burden or virus chemokine coreceptor usage. J. Infect. Dis. 179:859–70.
Nicholson JK, et al. (2001) CCR5 and CXCR4 expression on memory and naive T cells in HIV-1 infection and response to highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 27:105–15.
Hickman HD, et al. (2011) Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J. Exp. Med. 208:2511–24.
Fischereder M, et al. (2001) CC chemokine receptor 5 and renal-transplant survival. Lancet. 357:1758–61.
Muntinghe FL, et al. (2009) CCR5 deletion protects against inflammation-associated mortality in dialysis patients. J. Am. Soc. Nephrol. 20:1641–9.
Kiepiela P, et al. (2007) CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53.
Walker DK, et al. (2008) Preclinical assessment of the distribution of maraviroc to potential human immunodeficiency virus (HIV) sanctuary sites in the central nervous system (CNS) and gut-associated lymphoid tissue (GALT). Xenobiotica. 38:1330–9.
Gordon SN, et al. (2010) Disruption of intestinal CD4+ T cell homeostasis is a key marker of systemic CD4+ T cell activation in HIV-infected individuals. J. Immunol. 185:5169–79.
Alam MM, et al. (2011) Antigen-specific memory B-cell responses in Bangladeshi adults after one- or two-dose oral killed cholera vaccination and comparison with responses in patients with naturally acquired cholera. Clin. Vaccine Immunol. 18:844–50.
Lin YL, et al. (2008) The chemokine CCL5 regulates the in vivo cell surface expression of its receptor, CCR5. AIDS. 22:430–2.
Desmetz C, et al. (2007) Cell surface CCR5 density determines the intensity of T cell migration towards rheumatoid arthritis synoviocytes. Clin. Immunol. 123:148–54.
Moreno-Fernandez ME, Zapata W, Blackard JT, Franchini G, Chougnet CA. (2009) Human regulatory T cells are targets for human immunodeficiency Virus (HIV) infection, and their susceptibility differs depending on the HIV type 1 strain. J. Virol. 83:12925–33.
Mucida D, et al. (2005) Oral tolerance in the absence of naturally occurring Tregs. J. Clin. Invest. 115:1923–33.
Canestri A, et al. (2010) Maraviroc does not affect humoral response to the pandemic influenza A-H1N1v 2009 adjuvated vaccine in HIV-1-infected patients. AIDS. 24:2887–9.
Burton CT, et al. (2008) Restoration of anti-tetanus toxoid responses in patients initiating highly active antiretroviral therapy with or without a boost immunization: an INITIO substudy. Clin. Exp. Immunol. 152:252–7.
Acknowledgments
We wish to thank the patients and staff of the Saint Stephen’s Center, without whom this study would not have been possible. This study was funded and sponsored by SSAT, with an unrestricted grant from Pfizer Inc. awarded to GMoyle and N Imami. N Imami also was supported by funding from the Medical Research Council (MRC) (grant number G0501957). All authors have no relevant financial interests to disclose. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Westrop, S.J., Moyle, G., Jackson, A. et al. CCR5 Antagonism Impacts Vaccination Response and Immune Profile in HIV-1 Infection. Mol Med 18, 1240–1248 (2012). https://doi.org/10.2119/molmed.2012.00206
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00206