
Abstract
Flurbiprofen acts as a nonselective inhibitor for cyclooxygenases (COX-1 and COX-2), but its impact on hepatic ischemia/reperfusion (I/R) injury remains unclear. Mice were randomized into sham, I/R and flurbiprofen (Flurb) groups. The hepatic artery and portal vein to the left and median liver lobes were occluded for 90 min and unclamped for reperfusion to establish a model of segmental (70%) warm hepatic ischemia. Pretreatment of animals with flurbiprofen prior to I/R insult significantly decreased serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH), and prevented hepatocytes from I/R-induced apoptosis/necrosis. Moreover, flurbiprofen dramatically inhibited mitochondrial permeability transition (MPT) pore opening, and thus prevented mitochondrial-related cell death and apoptosis. Mechanistic studies revealed that flurbiprofen markedly inhibited glycogen synthase kinase (GSK)-3β activity and increased phosphorylation of GSK-3β at Ser9, which, consequently, could modulate the adenine nucleotide translocase (ANT)-cyclophilin D (CyP-D) complex and the susceptibility to MPT induction. Therefore, administration of flurbiprofen prior to hepatic I/R ameliorates mitochondrial and hepatocellular damage through inhibition of MPT and inactivation of GSK-3β, and provides experimental evidence for clinical use of flurbiprofen to protect liver function in surgical settings in addition to its conventional use for pain relief.
Similar content being viewed by others

Introduction
Hepatic ischemia/reperfusion (I/R) injury is a series of complicated cellular events that occur upon restoration of hepatic blood flow after a period of ischemia (1). The injury could be severe enough to cause a significant morbidity and mortality in two main settings. Firstly, it is associated with liver transplantation and hepatic resections where temporary clamping and unclamping of the vascular inflow takes place (2,3). Secondly, it may occur as a consequence of systemic hypoxia or in conditions including hemorrhagic, cardiogenic or septic shock (4).
Although the topic of hepatic I/R injury has been studied extensively in recent decades, the molecular mechanisms underlying this effect largely remain poorly understood. Hepatic injuries caused by I/R are the result of complex interactions between various inflammatory mediators (5), among which, cyclooxygenase (COX)-derived prostanoids such as prostacyclin (PGI) and prostaglandin E (PGE) have been shown to play a pivotal role in I/R injury (6,7). There are at least two cyclooxygenase isoenzymes, COX-1 and COX-2 (8). COX-1 is usually expressed constitutively, while COX-2 is undetectable in most tissues in the physiological condition but is rapidly induced upon pathological insults (9). Studies have shown that both COX-1 and COX-2 are involved in skeletal muscle and gastric I/R injuries (9,10). Furthermore, inhibition of COX-2 by selective inhibitors or gene knock-out leads to a significant reduction in I/R-induced hepatic damage (5,11,12). However, the mechanisms underlying this protective effect are yet to be investigated.
It was interestingly noted that pharmaceutical inhibition of glycogen synthase kinase (GSK)-3β protects the liver from I/R-induced injury by regulating mitochondrial permeability transition (MPT), a phenomenon relevant to the formation and opening of the MPT pores in the inner mitochondrial membrane (13,14). Given the fact that COX-2 regulates the phosphorylation of GSK-3β (15), it is conceivable that COX-2 may play a role in GSK-3β signaling and in the regulation of mitochondrial function during hepatic I/R injury. Thus, we used flurbiprofen, a nonselective COX-1/COX-2 inhibitor, to further gain insight into the role of COX in mitochondrial function relevant to hepatic I/R injury. Although flurbiprofen is considered conventionally for treatment of acute and chronic pain (16), it has been suggested recently to have a neuroprotective effect in cerebral ischemia (17,18). Therefore, we employed liver I/R injury as a model to dissect the role of flurbiprofen in protecting I/R-induced hepatic injury. Our results demonstrate that flurbiprofen pretreatment protects the liver from I/R-induced injury by increasing GSK-3β phosphorylation and inhibiting MPT induction.
Materials and Methods
Materials
Flurbiprofen was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Antibodies for GSK-3β phospho-Ser9 subunit, GSK-3β, COX-1, COX-2, caspase-9 and cytochrome c were purchased from Cell Signaling Technology (CST) (Boston, MA, USA). Antibody for adenine nucleotide translocase (ANT) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cyclophilin D (anti-CyP-D) was obtained from MitoSciences (Eugene, OR, USA). Anti-caspase-3 was from Millipore (Temecula, CA, USA). Calcium Green-5N probe was from Invitrogen (Carlsbad, CA, USA). All other chemical reagents are in analytic pure grade.
Animals and Surgery
Male 8-wk-old C57BL/6 mice (∼22 to ∼25 g) obtained from Sino-British Sippr/BK Lab Animal Ltd. (Shanghai, China) were housed in the SPF condition according to the institutional guidelines. The mice were divided randomly into three groups: (i) sham-operation (Sham) group; (ii) hepatic ischemia/reperfusion (I/R) group; and (iii) flurbiprofenpretreated I/R (Flurb) group. All animals were fasted for 12 h prior to surgery, and anesthetized with pentobarbital (1%, 40 mg/kg) intraperitoneally. A model of segmental (70%) hepatic ischemia was used with minor modifications (19). Briefly, after a midline laparotomy, all structures in the portal triad (hepatic artery, portal vein and bile duct) to the left and median liver lobes were occluded for 90 min. This method of partial hepatic ischemia prevented mesenteric venous congestion by permitting portal decompression through the right and caudate lobes. Reperfusion was initiated by removal of the clamp. Animals were again anesthetized at different periods after reperfusion and liver biopsies were taken for further evaluation. In the Sham group, mice, in which all structures in the portal triad (hepatic artery, portal vein, and bile duct) to the left and median liver lobes were occluded for 3 to 5 s, followed by placing them on the platform for 90 min to resemble the conditions of experimental mice, were used for controls. In the Flurb group, flurbiprofen (10 mg/kg) was administered in the caudal vein 20 min before ischemia; while in the I/R group, an appropriate volume (3 mL/kg) of vehicle (ethanol: saline, 5:95) was used. Sham mice were injected with same volume of vehicle before laparotomy (the interruption of portal venous blood supply only lasted for 3 to 5 s). Following 2 h or 6 h of reperfusion, the mice were euthanized, and liver and blood samples were collected.
Mitochondria Isolation
Mitochondria isolation was performed by gradient centrifugation as described previously (14,20). Briefly, the fresh liver tissues (1 g) were homogenized with 8 mL of isolation buffer (ISA) containing 220 mmol/L d-mannitol, 70 mmol/L sucrose, 10 mmol/L Tris-HCL, 1 mmol/L EGTA and 0.4% bovine serum albumin (pH 7.4). The homogenates were centrifuged at 850g for 10 min to collect supernatants, followed by centrifuging at 10,000g for an additional 10 min. The mitochondrial pellet next was resuspended in a final washing buffer (ISB) containing 220 mmol/L d-mannitol, 70 mmol/L sucrose and 10 mmol/L Tris-HCL (pH 7.4). Protein concentration was determined by biuret method (21) and calibrated with bovine serum albumin.
Western Blot Analysis
The levels for COX-1, COX-2, GSK-3β, phospho-Ser9-GSK-3β, caspase-3 and caspase-9 were determined in liver lysates. Cytochrome c was determined in mitochondrial and cytoplasm extracts. The levels of prohibitin and β-tubulin were evaluated as loading controls for mitochondrial extracts and cytosol extracts, respectively. For protein extraction, liver tissues were homogenized in lysis buffer (Promega, Madison, WI, USA). The homogenates were centrifuged at 850g for 10 min to collect supernatants, followed by centrifuging at 10,000g for additional 10 min. The supernatant (cytoplasmic extract) and pellet (mitochondrial extract) were applied for Western blot analysis. Protein concentration of the extracts was determined by BCA protein assay (Pierce, Rockford, IL, USA). Equal amount of protein extracts was separated on a SDS polyacrylamide gel, and then transferred onto a nitrocellulose membrane (Millipore, Temecula, CA, USA). After incubation with indicated primary antibodies, the blots were probed with a goat anti-rabbit or an anti-mouse secondary horseradish peroxidase (HRP) antibody (Santa Cruz), and developed by the enhanced chemiluminescence (Pierce). The relative amount of each targeted protein was normalized by the ratio with β-actin and analyzed densitometrically using a Gel Pro Analyzer (Media Cybernetics, Silver Spring, MD, USA).
Plasma Biochemical Assays
Mice were euthanized 6 h after reperfusion by decapitation. Blood samples were collected immediately from the heart. Serum levels for alanine aminotransferase (ALT), aspartate aminotransferase (AST) and lactate dehydrogenase (LDH) were determined by a serum autoanalyzer (H-7600; Hitachi Ltd., Tokyo, Japan).
Immunohistochemical Analysis
Livers were fixed with formalin and embedded with paraffin. Hepatocellular necrosis was determined in hematoxylin and eosin (H&E)-stained tissue using a semiquantitative scale by a point counting method as previously described. Briefly, a total of 30 random fields were counted for each H&E-stained tissue sample under high power magnification (400×) in a blinded fashion to determine the percentage of necrotic cells. In this study, only grade 3 injury with disintegration of hepatic cords was counted as necrosis (19). The terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) test was performed using a commercial kit from Roche (Rotkreuz, Switzerland) as instructed. In each section, areas from 10 visual fields (400×) were analyzed for TUNEL-positive cells. The TUNEL index was determined using the following formula: (number of stained cells/number of stained and unstained cells) × 100 (22).
Electronic Microscopy
Electronic microscopy was performed on isolated mitochondria at the end of the preincubation period (that is, before Ca2+ loading). Briefly, mitochondrial samples were fixed for 6 h in 2.5% glutaraldehyde and 100 mmol/L phosphate buffer (pH 7.4), and postfixed in 1% osmium tetroxide. Dehydration was performed in a series of ethanol and propylene oxide extractions before sample embedding in Epon (Miller-Stephenson Chemical Company, Danbury, CT, USA). Ultrathin sections were examined with a transmission electron microscope (H-7650; Hitachi Ltd.) at the Second Military Medical University.
Calcium Retention Capacity
The assay was adapted from the description by Ichas et al. (23). Briefly, calcium retention capacity (CRC) was defined as the amount of Ca2+ required to trigger a massive Ca2+ release by isolated liver mitochondria. It was used as an indicator for the resistance of the MPT pore to open after matrix Ca2+ accumulation and expressed as nmol CaCl2 per mg mitochondrial proteins. Extramitochondrial Ca2+ concentration was recorded by a Fluorescence Microplate Reader controlled by the SOFTmax PR software (Molecular Devices, Sunnyvale, CA, USA) in the presence of 1 µmol/L Calcium Green-5N molecular probe, with the excitation and emission wavelengths set at 500 and 530 nm, respectively. The fluorescence scan interval was set at 12 s with the system. Isolated mitochondria (2 mg proteins) were suspended in 2 mL of incubation buffer (220 mmol/L d-mannitol, 70 mmol/L sucrose, 1 mmol/L Pi-Tris, 10 mmol/L Tris-MOPS, 5 mmol/L glutamate-Tris, 2.5 mmol/L malate-Tris, pH 7.4, containing 0.01% [w/v] bovine serum albumin and 1 µmol/L of the Ca2+ indicator Calcium Green-5N) in a clear 24-well plate. At the end of the preincubation period (120 s), 20 nmol CaCl2 pulses were performed every 60 s for calculating the CRC. After sufficient calcium loading, extramitochondrial calcium concentration abruptly increased, indicating a massive release of calcium by mitochondria as a result of MPT pore opening as described previously (24).
Evaluation of GSK-3β Activity
GSK-3β activity was evaluated by promoting the in vitro reaction of phosphorylation of exogenous tau by GSK-3β present in the liver homogenates. To promote the reaction (100 µL), 250 µg of liver homogenates were incubated with 10 ng/mL exogenous tau and 200 µmol/L ATP for 1 h at 30°C. Phosphorylated tau was detected using a Tau (pS199) phosphoELISA kit (Invitrogen) according to the manufacturer’s instructions. Lithium chloride (10 mmol/L) was used as an inhibitor of GSK-3β. Samples and standard dilutions were read in a Multiskan FC plate reader (Thermo Scientific, part of Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm.
Immunoprecipitation
Immunoprecipitation (IP) was performed as reported previously (25). In brief, 1,000 µg of mitochondrial fractions were solubilized by 500 µL of IP buffer (20 mmol/L Tris-HCl [pH 7.4], 1 mmol/L EGTA, 5 mmol/L NaN3, 50 mmol/L NaCl, 1 mmol/L PMSF, 50 mmol/L Na3VO4, 1% Triton X-100, 0.5% NP-40 and a protease inhibitor cocktail) and preincubated with 50 µL protein G magnetic beads (New England BioLabs, Beverly, MA, USA) for 1 h to remove proteins that can bind nonspecifically to the beads. The supernatant was taken and incubated with antibodies against ANT (Santa Cruz) for 1 h, and the mixture incubated with 50 µL of fresh beads for 1 h. A magnetic field was applied to this IP mixture, and the supernatant was removed. The beads were washed two times using 500 µL of IP buffer, resuspended in 30 µL of SDS sample loading buffer (125 mmol/L Tris-HCl [pH 6.8], 4.3% SDS, 30% glycerol, 10% β-mercaptoethanol, and 0.01% bromophenol blue), and incubated at 70°C for 5 min. Finally, 20 µL of the supernatant was taken after applying a magnetic field to the mixture and was used for immunoblotting with the use of antibodies against ANT and CyP-D. The blots were visualized and quantified as described above.
Statistical Analysis
All data are expressed as mean ± standard deviation (SD). Statistical analysis was carried out with a two-way analysis of variance (ANOVA), and comparisons between tested groups were carried out using the LSD tests. SPSS 10.0 (SPSS Inc, Chicago, IL, USA) was used for the statistical analysis. In all cases, a p value <0.05 was considered to be statistically significant.
Results
Flurbiprofen Pretreatment Reduces I/R-Induced Hepatic Injury
We first sought to determine the effect of flurbiprofen pretreatment on I/R-induced hepatic injury. To this end, mice were euthanized 6 h after I/R insult. It was noted that I/R insult significantly induced apoptosis (40.6% in I/R versus 11.2% in Sham, p < 0.01) in the liver. Given that late stage of apoptotic cells would undergo necrosis, we therefore further examined necrosis. Indeed, necrosis was noted in 79.0% of hepatocytes in the sections of I/R-insulted mice, while only around 14.8% of cells undergoing necrosis in the sections originated from Sham mice (p < 0.01). Importantly, administration of flurbiprofen prior to (20 min) ischemic insult markedly reduced the percentage of apoptotic (26.2% in Flurb versus 40.6% in I/R, p = 0.01) and necrotic cells (60.0% in Flurb versus 79.0% in I/R, p = 0.01) (Figures 1A–D). Next, we examined serum ALT and AST in all animals. As shown in Figure 1E, flurbiprofen pretreatment drastically reduced the levels for ALT (U/L, 2752 versus 6785, p < 0.01) and AST (U/L, 2064 versus 5399, p < 0.01) as compared with that of I/R group.

The effects of flurbiprofen pretreatment prior to ischemia on liver damage. (A) H&E staining results of liver sections 6 h after reperfusion (100× magnification). (B) TUNEL assay results of liver sections 6 h after reperfusion (100× magnification). (C) Bar graphs showing the percentage of necrotic cells in the tissue sections. (D) Bar graphs showing the percentages of apoptotic cells in the tissue sections. (E) Serum levels for ALT and AST. (F) Serum levels for LDH. Six mice were included in each study group. **p < 0.05 versus I/R.
To further confirm the above results, we examined LDH, a soluble cytosolic enzyme passively released by necrotic cells. Consistent with the above results, I/R insult significantly increased LDH release (p < 0.01) in the liver as compared with that of Sham controls; however, flurbiprofen pretreatment markedly reduced I/R-mediated LDH release (p < 0.01) (Figure 1F). Together, these results suggest that flurbiprofen pretreatment protects mice from I/R-induced hepatic injury.
Flurbiprofen Regulates the Opening of MPT Pores
An important mechanism leading to cell death in hepatic I/R injury is the onset of MPT (26). Several factors such as GSK-3β, protein kinase A (PKA) and c-Jun N-terminal kinase 2 (JNK2) are known factors to regulate the MPT pore after hepatic I/R insult (14,26,27). However, the role of COX in MPT onset remains unclear. We therefore evaluated the effect of flurbiprofen on MPT pore susceptibility after I/R insult.
We first examined mitochondria morphology by electron microscopy. It was found that mitochondria isolated from I/R animals revealed greater contortion and increased vacuoles than that from mice with flurbiprofen pretreatment, suggesting that mitochondria in Flurb group suffered less damage (Figure 2). We next measured MPT pore opening in isolated mitochondria by CRC detection. As shown in Figure 3A, flurbiprofen pretreatment significantly restored the ability of mitochondria to tolerate calcium induction, as compared with that of I/R group. We further applied cyclosporine A (CsA), a well-known inhibitor for CyP-D and a blockade for the opening of MPT pore, to the isolated mitochondria derived from I/R livers. Our data showed that flurbiprofen strongly improved CRC over 2 h and 6 h of reperfusion, and this effect could be enhanced by CsA treatment (Figures 3B–C). Since MPT pore opening is an important factor in I/R-induced cell death, our findings strongly suggest that flurbiprofen may protect the hepatocytes from I/R injury by inhibiting MPT pore opening.

Flurbiprofen protects mitochondria from morphological changes after ischemic insult. The mitochondria isolated from I/R animals revealed greater contortion and increased vacuoles than that from mice with flurbiprofen pretreatment.

The effects of flurbiprofen treatment on mitochondrial calcium tolerance. Mitochondria were isolated from animals euthanized after 90 min of hepatic ischemia plus 2 h or 6 h of reperfusion in each group. Calcium pulse were fluorometrically monitored using the probe Ca2+-Green 5N. (A) Determination of extramitochondrial Ca2+ after subsequent addition of 10 nmol/L CaCl2 pulses in mitochondria isolated from 6 h of reperfusion. (B) and (C) Calcium retention capacity after 2 h and 6 h of reperfusion in each groups (n = 6 per group), the CRC was measured without (−CsA) or with (+CsA) addition of 0.8 mmol/L of CsA in the cuvette to fully inhibit the interaction of CyP-D with the MPT pore. **p < 0.05 versus CRC in the I/R group (same strain).
Flurbiprofen Suppresses Cytochrome c Release and Caspase Activation
Given that the opening of MPT pore causes cytochrome c release and caspase activation (28), we then investigated the effect of flurbiprofen on cytochrome c release and caspase activation in I/R injury. It was noted that animals in the I/R group displayed decreased levels for mitochondrial cytochrome c and increased levels for cytosol cytochrome c as compared with that of Sham animals. On the contrary, administration of flurbiprofen prior to I/R insult greatly prevented cytochrome c release (Figures 4A–C). Since the release of cytochrome c is associated with caspase-3 and caspase-9 activation (29), we next analyzed caspase-3 and caspase-9 cleavage by Western blot analysis. As expected, flurbiprofen pretreatment markedly reduced the cleavage of caspase 3 (Figures 4D–E) and caspase 9 (Figures 4D, F). Taken together, these data suggest that flurbiprofen may play a role to prevent cytochrome c release and caspase activation during I/R injury.

The effect of flurbiprofen on cytochrome c release and caspase-3/9 activation. (A) A representative results for Western blot analysis of mitochondrial and cytoplasmic cytochrome c. (B) Relative amount of mitochondrial cytochrome c. (C) Relative amount of cytoplasmic cytochrome c. (D) Representative Western blot results for the cleaved caspase 3 and caspase 9. (E) Relative amount for the cleaved caspase 3. (F) Relative amount for the cleaved caspase 9. The studies were carried out with three replications. **p < 0.05 versus I/R.
The Effects of Flurbiprofen on GSK-3β and the Interaction of ANT with CyP-D
COX-2 has been shown to regulate the phosphorylation of GSK-3β (15), a pivotal enzyme implicated in MPT regulation (14). Thus, we assessed the effect of Flurb pretreatment on GSK-3β activity at 2 h and 6 h after I/R injury. I/R livers exhibited increased GSK-3β activity as compared with both Sham and Flurb animals determined by a tau (pS199) phosphoELISA kit (Figure 5). Since phosphorylation of GSK-3β at Ser9 suppresses its enzymatic activity, we thus examined the impact of flurbiprofen on GSK-3β activity. For this purpose, we determined the relative amount of Ser9 phosphorylated GSK-3β by Western blot analysis at 2 h and 6 h after I/R injury (Figure 6A). We noticed that I/R insult induced a significant decrease for Ser9 phosphorylated GSK-3β at both time points, while administration of flurbiprofen significantly enhanced Ser9 phosphorylation of GSK-3β (Figure 6B, p < 0.05). All together, our results suggest that flurbiprofen treatment can inhibit the activity of GSK-3β strongly by suppressing the phosphorylation of GSK-3β at Ser9.

The effects of flurbiprofen administered before ischemia on GSK-3β activity on liver homogenates. Tau phosphorylation by GSK-3β was detected using a Tau (pS199) phosphoELISA kit. Animals were euthanized after 90 min of hepatic ischemia plus 2 h or 6 h of reperfusion, with (Flurb) or without (I/R) flurbiprofen (10 mg/kg) in the caudal vein 20 min before ischemia. I/R livers exhibited increased GSK-3β activity, when compared with both Sham and Flurb animals. **p < 0.05 versus I/R, n = 6.

The effects of flurbiprofen administered before ischemia on GSK-3β and phosphorylated GSK-3β at Ser9 (pSer9-GSK-3β) content in liver homogenates. (A) Western blot results for the total GSK-3β and pSer9-GSK-3β. (B) Relative amount for the pSer9-GSK-3β. **p < 0.05 versus I/R, n = 6. I/R animals exhibited decreased content of phosphorylated GSK-3β at Ser9, when compared with both Sham and Flurb animals.
CyP-D is known as a component responsible for triggering MPT opening by its binding to ANT, a major subunit of ANT-CyP-D complex. It has been reported that phospho-GSK-3β-ANT interaction and associated ANT-CyP-D interaction are responsible for suppression of MPT pore opening and myocardial protection after I/R injury (25). Therefore, we assumed that phospho-GSK-3β-ANT-CyP-D may play a role in flurbiprofen-mediated protective effect on hepatic I/R injury. As shown in Figure 7, flurbiprofen pretreatment markedly reduced the levels for ANT-CyP-D complex as determined by IP, which could be caused by increased binding of phospho-GSK-3β to ANT as reported previously (25). Accumulatively, our findings indicate that flurbiprofen may reduce hepatic I/R injury by induction of GSK-3β phosphorylation and suppression of ANT-CyP-D complex formation.

The effects of flurbiprofen on interaction of ANT with CyP-D in mitochondria. The effects of flurbiprofen administered before ischemia on interaction of ANT with CyP-D in mitochondria isolated from animals euthanized after 90 min of hepatic ischemia plus 2 h or 6 h of reperfusion in each group. (A) A representative immunoblot results for the ANT and CyP-D. (B) Relative amount for the CyP-D compared with ANT. **p < 0.05 versus I/R, n = 6. I/R animals exhibited increased content of CyP-D-ANT complex, when compared with both Sham and Flurb animals. IP: immunoprecipitation; IB: immunoblotting. the formation of ANT-CyP-D complex (Figure 7), which could be caused by the increased binding of phospho-GSK-3β to ANT (25). Thus, our findings indicate that flurbiprofen reduces hepatic I/R injury by suppressing GSK-3β activity and the formation of ANT-CyP-D MPT pore complex. Interestingly, flurbiprofen seems to be more potent in suppressing the expression of COX-2 versus COX-1 upon hepatic I/R insult (Figure 8). This result supports the assumption that COX-2 plays a predominant role in the regulation of mitochondrial function in liver I/R injury. Of important note, the previously reported COX inhibitors (sc-560, NS-398, celecoxib and FK3301) have not been reported to be involved in the protection of mitochondrial function (32,36). Our data in the current study also demonstrate that mechanisms underlying COX1 or COX2 inhibitors protection of liver I/R injury
The Effect of Flurbiprofen on COX-1 and COX-2 Expression
Next, we examined the effect of flurbiprofen on COX-1 and COX-2 expressions by Western blot analysis (Figure 8). Interestingly, we failed to detect a dramatic change for COX-1 levels between animals in I/R and Flurb group (Figure 8B). However, COX-2 was induced significantly upon I/R insult, while pretreatment of animals with flurbiprofen suppressed COX-2 expression significantly (Figure 8C). These results suggest that flurbiprofen protection of hepatic I/R injury is likely through inhibition of COX-2 expression. Therefore, our results provide evidence supporting that the reduced MPT pore opening in animals of the Flurb group could be a consequence of inhibited COX-2 expression.

The impact of flurbiprofen on COX expressions. (A) Representative Western blot results for COX-1 and COX-2, (B) Relative expression levels for COX1. (C) Relative expression levels for COX2. The relative expression levels for each target were assessed by densitometry and normalized by β-actin. Three replicates were included for the analyses. **p < 0.05 versus I/R.
Discussion
COX, also known as prostaglandin endoperoxide synthases, is an enzyme that catalyzes the oxygenation of arachidonic acid to prostaglandin endoperoxides, which are subsequently converted into prostaglandins. COX-1 is constitutively expressed in many mammalian cells, whereas COX-2 is usually expressed transiently upon induction. COX has been implicated in modulating angiogenesis, vascular function, chronic inflammation and cancer development (30,31).
I/R is a common pathophysiological process, and I/R insult induces altered COX-1/COX-2 expression in various organs or tissues such as in the brain, stomach, kidney and skeleton muscle (9). Studies also have shown that in hepatic I/R injury, both COX-1 and COX-2 contribute to I/R-induced hepatic microvascular and hepatocellular injury (32). The effect of selective COX-1 inhibitor, SC-560, and selective COX-2 inhibitors, NS-398, celecoxib and FK 3311, as well as COX-2 deficiency have been examined extensively in hepatic I/R injury (5,11, 12,32). However, the effect of flurbiprofen, a nonselective COX-1/COX-2 inhibitor, in hepatic I/R injury is yet to be elucidated.
In the present report, we evaluated the potential role of flurbiprofen in a model of 70% warm hepatic I/R. Our data revealed that administration of flurbiprofen 20 min before ischemia significantly protected mice against I/R-induced hepatic injury as manifested by the reduced cell death (apoptosis and necrosis), decreased release of ALT and AST (Figure 1). Our data further suggest that flurbiprofen protects hepatocytes undergoing apoptosis/necrosis by inhibiting MPT pore opening (Figures 2, 3). Opening of MPT pores initiates the onset of MPT, which is a causative event leading to apoptosis (transient pore opening) and necrosis (extensive pore opening) in hepatocytes after I/R insult (33). Our mechanistic studies further support that flurbiprofen pretreatment drastically prevented mitochondrial swelling, improved CRC and thus ameliorated mitochondria damage caused by hepatic I/R insult (Figures 2, 3). Consistent with these results, the subsequent release of cytochrome c and activation of caspase-3 and caspase-9 were strongly attenuated along with flurbiprofen treatment (Figure 4). These findings provided strong evidence indicating a role for flurbiprofen in the regulation of hepatic I/R injury.
To dissect how flurbiprofen modulates MPT pore opening and mitochondrial function, we examined the phosphorylation of GSK-3β, a possible signaling molecule of COX-2 signaling pathway. It was noted that administration of flurbiprofen prior to I/R insult significantly decreased GSK-3β activity and enhanced phosphorylation of GSK-3β at Ser9 (Figures 5, 6). Previous studies demonstrated that Ser9 phosphorylated GSK-3β interacts with regulators of MPT, and limits MPT induction (34,35). Therefore, inhibition of GSK-3β phosphorylation can modulate the susceptibility to MPT induction and preserve mitochondrial function after I/R (14). More importantly, we found that flurbiprofen pretreatment also decreased could be different from that of flurbiprofen, in which sc-560 (a selective COX-1 inhibitor) has been revealed to ameliorate mouse hepatic I/R injury by improving microcirculation and decreasing inflammatory factors such as interleukin family and TNF-α, while FK3311 (a selective COX-2 inhibitor) can protect liver from I/R injury by inhibiting TXA2 and improving microcirculation. Given that I/R-induced hepatocyte death is a very complicated process, the studies in the report could only address some major mechanistic pathways relevant to this process, additional mechanisms could be involved and are yet to be addressed.
Conclusion
In conclusion, flurbiprofen, as a nonsteroidal antiinflammatory drug (NSAID), has been used widely to relieve perioperative pain (16). Our data in the present report, however, demonstrate a novel function for flurbiprofen, in which it restores mitochondrial function and protects hepatic cells from I/R-induced injury. This discovery possesses important clinical and scientific significance, suggesting that flurbiprofen could be a useful drug to protect liver function in surgical settings such as liver transplantation or tumor resections other than its conventional role in pain relief.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Siriussawakul A, Zaky A, Lang JD. (2010) Role of nitric oxide in hepatic ischemia-reperfusion injury. World J. Gastroenterol. 16:6079–86.
Caldwell-Kenkel JC, Currin RT, Tanaka Y, Thurman RG, Lemasters JJ. (1991) Kupffer cell activation and endothelial cell damage after storage of rat livers: effects of reperfusion. Hepatology. 13:83–95.
Liu DL, Jeppsson B, Hakansson CH, Odselius R. (1996) Multiple-system organ damage resulting from prolonged hepatic inflow interruption. Arch. Surg. 131:442–7.
Glantzounis GK, Salacinski HJ, Yang W, Davidson BR, Seifalian AM. (2005) The contemporary role of antioxidant therapy in attenuating liver ischemia-reperfusion injury: a review. Liver Transpl. 11:1031–47.
Hamada T, et al. (2008) Cyclooxygenase-2 deficiency enhances Th2 immune responses and impairs neutrophil recruitment in hepatic ischemia/reperfusion injury. J. Immunol. 180:1843–53.
Kawai M, Harada N, Takeyama H, Okajima K. (2011) Neutrophil elastase contributes to the development of ischemia/reperfusion-induced liver injury by decreasing the production of insulin-like growth factor-I in rats. Transl. Res. 155:294–304.
Hafez T, et al. (2007) The effect of intraportal prostaglandin E1 on adhesion molecule expression, inflammatory modulator function, and histology in canine hepatic ischemia/reperfusion injury. J. Surg. Res. 138:88–99.
Yokoyama C, Tanabe T. (1989) Cloning of human gene encoding prostaglandin endoperoxide synthase and primary structure of the enzyme. Biochem. Biophys. Res. Commun. 165:888–94.
Dupouy VM, Ferre PJ, Uro-Coste E, Lefebvre HP. (2006) Time course of COX-1 and COX-2 expression during ischemia-reperfusion in rat skeletal muscle. J. Appl. Physiol. 100:233–9.
Hiratsuka T, et al. (2005) COX-1 and COX-2 conversely promote and suppress ischemia-reperfusion gastric injury in mice. Scand. J. Gastroenterol. 40:903–13.
Takeyoshi I, et al. (2001) The effect of a selective cyclooxygenase-2 inhibitor in extended liver resection with ischemia in dogs. J. Surg. Res. 100:25–31.
Ozturk H, Gezici A, Ozturk H. (2006) The effect of celecoxib, a selective COX-2 inhibitor, on liver ischemia/reperfusion-induced oxidative stress in rats. Hepatol. Res. 34:76–83.
Moreno AJ, et al. (2007) Unaltered hepatic oxidative phosphorylation and mitochondrial permeability transition in wistar rats treated with nimesulide: Relevance for nimesulide toxicity characterization. J. Biochem. Mol. Toxicol. 21:53–61.
Varela AT, et al. (2010) Indirubin-3′-oxime prevents hepatic I/R damage by inhibiting GSK-3beta and mitochondrial permeability transition. Mitochondrion. 10:456–63.
Fan X, et al. (2011) Celecoxib and 2,5-dimethylcelecoxib prevent cardiac remodeling inhibiting Akt-mediated signal transduction in an inherited DCM mouse model. J. Pharmacol. Exp. Ther. 338:2–11.
Sultan A, McQuay HJ, Moore RA, Derry S. (2009) Single dose oral flurbiprofen for acute postoperative pain in adults. Cochrane Database Syst. Rev. (3):CD007358.
Hewett SJ, Uliasz TF, Vidwans AS, Hewett JA. (2000) Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture. J. Pharmacol. Exp. Ther. 293:417–25.
Wang C, et al. (2008) Therapeutic time window of flurbiprofen axetil’s neuroprotective effect in a rat model of transient focal cerebral ischemia. Chin. Med. J. (Engl). 121:2572–7.
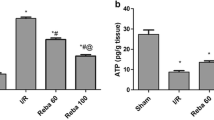
Selzner N, Selzner M, Jochum W, Clavien PA. (2003) Ischemic preconditioning protects the steatotic mouse liver against reperfusion injury: an ATP dependent mechanism. J. Hepatol. 39:55–61.
Selzner M, Selzner N, Jochum W, Graf R, Clavien PA. (2007) Increased ischemic injury in old mouse liver: an ATP-dependent mechanism. Liver Transpl. 13:382–90.
Gornall AG, Bardawill CJ, David MM. (1949) Determination of serum proteins by means of the biuret reaction. J. Biol. Chem. 177:751–66.
Yamada F, et al. (2007) Ischemic preconditioning enhances regenerative capacity of hepatocytes in long-term ischemically damaged rat livers. J. Gastroenterol. Hepatol. 22:1971–7.
Ichas F, Jouaville LS, Sidash SS, Mazat JP, Holmuhamedov EL. (1994) Mitochondrial calcium spiking: a transduction mechanism based on calcium-induced permeability transition involved in cell calcium signalling. FEBS Lett. 348:211–5.
Basso E, Petronilli V, Forte MA, Bernardi P. (2008) Phosphate is essential for inhibition of the mitochondrial permeability transition pore by cyclosporin A and by cyclophilin D ablation. J. Biol. Chem. 283:26307–11.
Nishihara M, et al. (2007) Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J. Mol. Cell Cardiol. 43:564–70.
Pediaditakis P, et al. (2010) Inhibition of the mitochondrial permeability transition by protein kinase A in rat liver mitochondria and hepatocytes. Biochem. J. 431:411–21.
Theruvath TP, Snoddy MC, Zhong Z, Lemasters JJ. (2008) Mitochondrial permeability transition in liver ischemia and reperfusion: role of c-Jun N-terminal kinase 2. Transplantation. 85:1500–4.
Argaud L, et al. (2004) Preconditioning delays Ca2+-induced mitochondrial permeability transition. Cardiovasc. Res. 61:115–22.
Lee HJ, et al. (2008) Mitochondria-cytochrome C-caspase-9 cascade mediates isorhamnetin-induced apoptosis. Cancer Lett. 270:342–53.
Mbonye UR, Song I. (2009) Posttranscriptional and posttranslational determinants of cyclooxygenase expression. BMB Rep. 42:552–60.
Namkoong S, et al. (2005) Prostaglandin E2 stimulates angiogenesis by activating the nitric oxide/cGMP pathway in human umbilical vein endothelial cells. Exp. Mol. Med. 37:588–600.
Ito Y, et al. (2003) Effects of selective cyclooxygenase inhibitors on ischemia/reperfusion-induced hepatic microcirculatory dysfunction in mice. Eur. Surg. Res. 35:408–16.
Kim JS, He L, Lemasters JJ. (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem. Biophys. Res. Commun. 304:463–70.
Das S, Wong R, Rajapakse N, Murphy E, Steenbergen C. (2008) Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 103:983–91.
Xi J, Wang H, Mueller RA, Norfleet EA, Xu Z. (2009) Mechanism for resveratrol-induced cardioprotection against reperfusion injury involves glycogen synthase kinase 3beta and mitochondrial permeability transition pore. Eur. J. Pharmacol. 604:111–6.
Kobayashi M, Takeyoshi I, Kurabayashi M, Matsumoto K, Morishita Y. (2007) The effects of a cyclooxygenase-2 inhibitor, FK3311, on total hepatic ischemia-reperfusion injury of the rat. Hepatogastroenterology. 54:522–6.
Acknowledgements
This work was partially supported by grants from the National Key Basic Research Program of China (2009CB522402), the Key Basic Research Projects from Science and Technology Commission of Shanghai Municipality (STCSM) (09JC1405600), the Key Medical Research Projects from the Division of Biomedicine, STCSM (10411951300), projects from Shanghai Municipal Health Bureau (SMHB) (2011227) and Foundation of Changzheng Hospital (200910). The funders did not play any role in study design, data collection and analysis, decision for data release or manuscript preparation.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Fu, H., Chen, H., Wang, C. et al. Flurbiprofen, a Cyclooxygenase Inhibitor, Protects Mice from Hepatic Ischemia/Reperfusion Injury by Inhibiting GSK-3β Signaling and Mitochondrial Permeability Transition. Mol Med 18, 1128–1135 (2012). https://doi.org/10.2119/molmed.2012.00088
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00088