
Abstract
Sustained inflammatory reactions are common pathological events associated with neuron loss in neurodegenerative diseases. Reported evidence suggests that Toll-like receptor 4 (TLR4) is a key player of neuroinflammation in several neurodegenerative diseases. However, the mechanisms by which TLR4 mediates neurotoxic signals remain poorly understood. We investigated the role of TLR4 in in vitro and in vivosettings of motor neuron degeneration. Using primary cultures from mouse spinal cords, we characterized both the proinflammatory and neurotoxic effects of TLR4 activation with lipopolysaccharide (activation of microglial cells, release of proinflammatory cytokines and motor neuron death) and the protective effects of a cyanobacteriaderived TLR4 antagonist (VB3323). With the use of TLR4-deficient cells, a critical role of the microglial component with functionally active TLR4 emerged in this setting. The in vivo experiments were carried out in a mouse model of spontaneous motor neuron degeneration, the wobbler mouse, where we preliminarily confirmed a protective effect of TLR4 antagonism. Compared with vehicle- and riluzole-treated mice, those chronically treated with VB3323 showed a decrease in microglial activation and morphological alterations of spinal cord neurons and a better performance in the paw abnormality and grip-strength tests. Taken together, our data add new understanding of the role of TLR4 in mediating neurotoxicity in the spinal cord and suggest that TLR4 antagonists could be considered in future studies as candidate protective agents for motor neurons in degenerative diseases.
Similar content being viewed by others


Introduction
In pathological conditions, persistent immune-stimulating signals may induce aberrant microglial activation with elevated cytokine release, eventually leading to neuronal injury. Toll-like receptors (TLRs) recognize highly conserved structural motifs from either pathogens or damaged and stressed tissues and are involved in microglial response to physiological and pathological signals. TLR4, in particular, reportedly mediates neuroprotective (1,2) and neurotoxic (3–5) effects, suggesting that TLR4 activation needs to be tightly controlled, to regulate the switch between its different roles in immune surveillance or neuroinflammatory propagation.
A well-known natural ligand of TLR4 is endotoxin/lipopolysaccharide (LPS), one of the major cell wall components of Gram-negative bacteria (6). LPS has potent immunostimulatory effects (7), mainly exerted through TLR4/MD2/CD14-bearing cells (8,9). The membrane anchor of LPS is a glucosamine-based phospholipid called lipid A, which represents the endotoxic principle of LPS and is responsible for its pathophysiological effects (10) via TLR4 (11,12). There is convincing evidence that LPS/TLR4 signaling is involved in various human and experimental central nervous system (CNS) diseases. In fact, increased expression of microglial TLR4 was found in animal models of Alzheimer’s disease (13) and Parkinson’s disease (14). Altered levels of the receptor have been reported in brain samples from Alzheimer’s disease (4) and multiple sclerosis (15). Increased CD14 (a TLR4 coreceptor) levels were found in amyotrophic lateral sclerosis (ALS) patients (16). Increased expression of TLR4 was found in reactive glia in human ALS spinal cord (17), suggesting a possible role for the TLR/receptor for advanced glycation end products (RAGE) signaling pathways. The activation of these pathways in the spinal cord may contribute to the progression of inflammation, resulting in motor neuron injury. However, the specific role of TLR4 in the spinal cord cell population, and the mechanism(s) by which TLR4 activation triggers inflammation and neurotoxicity in this region, remain poorly understood.
By using primary spinal cord cultures from mouse embryos as an in vitro model for the study of motor neuron injuries, we investigated the effects induced by TLR4 activation in neurons and glial cells. Cultures from TLR4-deficient mice were also used to understand the cell-specific role of this receptor in mediating motor neuron death. The effects of a novel cyanobacteria-derived TLR4 antagonist (VB3323) were analyzed in this in vitro setting and were compared with those of a well-known commercially available TLR4 antagonist, the LPS from Rhodobacter sphaeroides (RsLPS).
VB3323 was also tested in vivo in a mouse model of spontaneous motor neuron degeneration. The wobbler mouse, carrying a mutation in the vacuolar-vesicular protein sorting S4 (Vps54) gene (18) coding for a protein involved in the retrograde transport of late endosomes from the periphery to the Golgi apparatus (19), shows early-onset selective motor neuron death in the cervical spinal cord (reviewed in [20]). Glial activation (21–23) and upregulation of tumor necrosis factor (TNF)-α (24) have been reported in the cervical spinal cord of presymptomatic or early symptomatic mice. Thus, the wobbler mouse represents a useful model for testing candidate treatments aimed at modifying the neuroinflammatory mechanisms underlying the motor neuron loss.
Materials and Methods
Primary Cell Cultures
Procedures involving animals and their care were conducted in conformity with the institutional guidelines that comply with national (25,26) and international laws and policies (27,28). Primary cultures of motor neurons or neuron/glia cocultures were obtained from the spinal cord of 13-d-old C57 BL/6J (wild-type [WT]) or C57BL/10ScNJ (bearing a TLR4-null mutation, TLR4LPS-del; Charles River Laboratories International, Calco, Italy) mouse embryos, as previously described (29). Briefly, ventral horns were dissected from spinal cords, exposed to DNAse and trypsin (Sigma-Aldrich, Milan, Italy) and centrifuged through a bovine serum albumin (BSA) cushion. Cells obtained at this step were a mixed neuron/glia population and were centrifuged (800g for 15 min) through a 6% iodixanol (OptiPrep™; Sigma-Aldrich) cushion for motor neuron enrichment. A sharp band (motor neuron-enriched fraction) on the top of the iodixanol cushion and a pellet (glial fraction) were obtained. The glial feeder layer was prepared by plating the glial fraction at a density of 25,000 cells/cm2 into 12-well plates or into flasks, both previously precoated with poly-l-lysine (Sigma-Aldrich). Purified microglia were obtained by adapting the protocols of Hamby et al. (30) and Gingras et al. (31) with minor modifications. Flasks containing confluent mixed glial cultures were shaken overnight at 275 rpm in incubators. The supernatants (containing microglial cells) were collected and seeded at a density of 20,000 cells/cm2 into new flasks for Western blot analysis, plates or microslides (8-well µSlides; Ibidi GmbH, Martinsried, Germany) for time-lapse analysis. The purification yield of microglial cells with this protocol was about 98% (not shown). Astrocyte-enriched cultures were obtained by treating the glial cultures, from which microglia had been previously harvested, with 60 mmol/L l-leucine methyl ester (Sigma-Aldrich) for 90 min. To establish neuron/glia cocultures, the motor neuron-enriched fraction (obtained from the iodixanol-based separation) was seeded at a density of 10,000 cells/cm2 onto mature glial layers composed of either mixed glial cells or purified astrocytes. In these cocultures, about 84 ± 5% (n = 6) of the neuronal cells (revealed by neurofilament staining; Supplementary Figure S1A) were SMI32-positive cells with the typical motor neuron morphology (Supplementary Figure S1B; SMI32-positive cells merged with neurofilament staining in Supplementary Figure S1C; cell nuclei in blue). About 96 ± 2% (n = 3) of SMI32-positive neurons were also stained by another antibody specific for motor neurons, Hb9 (Supplementary Figures S1D-F).
For combined mixed cocultures from wild-type and TLR4LPS-del mouse embryos, microglial cells were added (10% of the astrocyte number) to motor neuron/astrocyte cocultures on the third day in vitro.
Motor Neuron Viability
The viability of motor neurons was assayed as follows: only the SMI32-positive cells, with typical morphology (triangular shape, single well defined axon), large bodies (>20 µm) and intact axons and dendrites were counted, at a magnification of 200µ, following the length of the coverslip in four nonoverlapping pathways (29). This number was normalized to the mean of SMI32-positive cells counted in the appropriate control wells. In a typical experiment with cocultures, the number of counted SMI32-positive cells in control (untreated) wells was 70 ± 11 (n = 12).
Culture Treatments
Cocultures were exposed to the different TLR4 ligands (diluted to the appropriate concentrations in culture medium) on the sixth day in vitro. Cultures maintained with normal medium served as the control condition. Motor neuron death was determined after 24-h incubation with 1 µg/mL LPS (from Escherichia coli 0111:B4) or lipid A (from E. coli F583; Sigma-Aldrich). The protective effects of 20 µg/mL VB3323 (Bluegreen Biotech, Milan, Italy) or 20 µg/mL RsLPS (Sigma-Aldrich) were evaluated after simultaneous treatment with the TLR4 agonists (LPS or lipid A). For the other investigations (microglial activation, cytokine release, TLR4 expression), we tested several exposure times to the TLR4 ligands (from 0.5 to 24 h). Motor neuron viability was assessed by counting SMI32-positive cells in each treatment condition, as previously reported (29). Treatment effects were compared by one-way analysis of variance (ANOVA) and the Tukey or Dunnett test, or two-way ANOVA and the Bonferroni posttest, using GraphPad v5.01 (GraphPad Software).
VB3323
VB3323 is a highly (95%) purified form of cyanobacterial LPS-like molecule (CyP), extracted from the freshwater cyanobacterium Oscillatoria planktothrix sp. by TRI reagent (Sigma-Aldrich), as described previously (32,33). The product was treated with DNase (20 µg/mL) and RNase (10 µg/ mL) in 50 mmol/L Tris, 10 mmol/L MgCl2, pH 7.5, for 2 h at room temperature before addition of proteinase K (100 µg/mL) and incubation overnight at 37°C. The sample was reextracted with TRI reagent and then purified by ion exchange chromatography.
Immunocytochemical and Immunofluorescent Assays
Cells were fixed with 4% paraformaldehyde or methanol (for glial fibrillary acidic protein [GFAP] staining). When indicated, cells were permeabilized by 0.2% Triton X-100 (Sigma-Aldrich). Staining was carried out by overnight incubation with the primary antibody, followed by incubation with an appropriate fluorescent secondary antibody for immunofluorescence (Dy-light; Rockland Immuno-chemicals, Gilbertsville, PA, USA). Cell nuclei were labeled with Hoechst 33258 by incubation with a 250 ng/mL solution. Double or triple staining was done by overnight incubation of the cultures separately with each primary antibody. In each experiment, some wells were processed without the primary antibody to verify the specificity of the staining.
Primary antibodies were as follows: SMI32 (anti-nonphosphorylated neurofilament H antibody, mouse, 1:6,000; Covance, Princeton, NJ, USA), anti-neurofilament 200 (1:500; Sigma-Aldrich), Hb9 (1:200; Abcam, Cambridge, MA, USA), GFAP (rabbit, 1:500; Santa Cruz Biotechnology, Santa Cruz, CA, USA), TLR4 (goat, 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and CD11b (rat, 1:1,000; eBioscience, San Diego, CA, USA). Appropriate fluorescent secondary antibodies conjugated to different fluorochromes were used at 1:1,000 dilution. Biotinylated anti-mouse secondary antibody (1:200; Vector Laboratories, Burlingame, CA, USA) was used to analyze motor neuron viability.
Microscopy and Live Imaging Analysis
For the immunocytochemistry experiments, pictures of stained cells were obtained with a laser scanning microscope (Olympus Fluoview BX61 microscope with a FV500 confocal system; Olympus, Milan, Italy), and images were analyzed with ImageJ v1.43 (National Institutes of Health). An optical microscope (Olympus BX51) was used for the neurotoxicity analysis. For time-lapse analysis, images were collected and analyzed with a CellR imaging station (Olympus) coupled to an inverted microscope (Olympus IX 81) equipped with an incubator to maintain constant temperature (37°C) and CO2 (5%) in at least three wells for each condition. Images were acquired at 30-min intervals for 18 h. For this analysis, purified microglial cultures were seeded at a density of 15,000 cells/cm2 into eight-well microslides and maintained in culture for 1 wk. They were then infected with lentiviral vectors (2.0 × 109 titer unit (T.U.)/well) expressing green fluorescent protein (GFP) reporter, to allow cell tracking in live-cell imaging experiments.
Viral Vector Production
High-titer viral vectors were produced by transfecting 293T cells with pRSV-Rev and pCMVdR8.74 plasmids (Trono Laboratories, Lausanne, Switzerland) encoding gag/pol/rev proteins, pVSV-G (BD Biosciences, Franklin Lakes, NJ, USA), for expression of VSV-G protein and pWPXLd transfer vector (Addgene Plasmid 12257) encoding GFP reporter gene under the EF1a ubiquitous promoter. Briefly, 293T cells were plated at 70% confluence in Dulbecco’s modified Eagle’s medium, 10% fetal calf serum, 1% glutamine, 1% penicillin and 1% streptomycin and maintained at 37°C in a humidified 5% CO2 incubator. Four plasmids were cotransfected with an Effectene kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions, and the medium was replaced after 16 h. Supernatants containing the viral particles were harvested 24 and 48 h later, filtered through a 0.45-µm filter and concentrated by ultracentrifugation at 50,000g at 4°C for 2 h. Viral pellets were resuspended in sterile phosphate-buffered saline and stored at −80°C until used. The titer of the viral stocks was in the range of 1.0−2.0 × 109 T.U./mL, assessed by an endpoint dilution on HEK293 cells (34).
Quantitative Enzyme-Linked Immunosorbent Assays
TNF-α, interleukin (IL)-6 and IL-1β concentrations in cell culture supernatants were quantified by solid-phase sandwich enzyme-linked immunosorbent assay (ELISA) (eBioscience). Samples from each experiment were tested in triplicate, according to the manufacturer’s instructions. The sensitivity of the kits was 8 pg/mL for TNF-α and IL-1β and 4 pg/mL for IL-6.
Immunoblotting
Microglial cultures were treated with ice-cold lysis buffer, and the protein concentration from supernatants was determined by the Bradford method (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were separated by 10% sodium do-decyl sulfate-polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. Blots were blocked and probed with the primary antibody anti-CD68 (rat, 1:500 dilution; AbD Serotec, Kidlington, UK). The membranes were then washed and incubated with horseradish peroxidase-conjugated anti-rat secondary antibodies (1:5,000 dilution; Sigma-Aldrich). Blots were developed, and the densitometric analysis of autoradiographic bands was done using image analysis software (ImageJ v1.43; National Institutes of Health [NIH], Bethesda, MD, USA). The integrated optical density values (normalized for the total protein content of each sample) were used as individual data for the statistical analysis.
In Vivo Treatment
Wobbler mice were originally obtained from NIH genetics and then bred at Charles River (Calco, Italy). Because heterozygous mice do not show any phenotypic difference compared with homozygous healthy littermates, heterozygous founders were designed by genotyping (23).
Thirty wobbler mice (fourth week of life) and the same number of age-matched healthy littermates were randomly recruited in the following experimental groups: (a) vehicle-treated mice; (b) riluzole-treated mice; and (c) VB3323-treated mice. The group of riluzoletreated mice followed the treatment schedule already reported by our group (35). Riluzole was provided by Sanofi-Aventis (Sanofi-Aventis Centre de Récherche de Paris, Vitry Sur Seine, Cedex, France). VB3323 was intraperitoneally injected three times a week at a dose of 5 mg/kg/d and at a final concentration of 0.5 mg/mL. All experimental groups included the same numbers of male and female mice, since no sex-related differences were detected during the symptom progression.
Behavioral Evaluations and Tissue Preparation
Mice were weighed twice a week. Behavioral trials for wobbler mice were done twice a week by the same operator blinded to the treatment. Semiquantitative score evaluation (paw abnormality) and quantitative measurements (grip strength) were done, as for previous protocols (35).
Immunohistochemistry and Histology
To ensure optimal quality for cryostat sections, animals were transcardial perfused as previously described (36). For Nissl staining, cryostatic sections of cervical spinal cord (C2–C6) were serially cut (30 µm thick) and processed by the method reported by our group (37). The mean number of neurons was calculated for each animal and used for statistical analysis. Free-floating sections (30 µm thick) were incubated with GFAP (mouse monoclonal antibody, Immunological Science, Rome, Italy; dilution 1:1,000) or CD11b (mouse monoclonal antibody OX 42; AbD Serotec; dilution 1:300), and double-staining experiments were carried out by using an antibody against TNF-α (rat monoclonal antibody; HyCult Biotech, Uden, the Netherlands; dilution 1:200). To exclude antibody cross-reactions, each mouse monoclonal antibody was tested with non-species-specific secondary antibody, and no signal was revealed in this condition.
All supplementary materials are available online at www.molmed.org .
Results
Distribution of TLR4 in Spinal Cord Cells
In a first set of experiments, TLR4 expression was analyzed in the resident cell types of the spinal cord. Motor neurons, in coculture with glia, were exposed to 1 µg/mL LPS, with or without 20 µg/mL VB3323, for different times, to check whether these agents altered the expression of TLR4. Immunocytochemistry for SMI32 and TLR4 was then done. As shown in Figure 1, immunopositivity for TLR4 (red) was widespread in control conditions (Figure 1B). SMI32-positive motor neurons (Figure 1A, green) were stained with the specific anti-TLR4 antibody (Figure 1C, merge). Treatment with the TLR4 ligands did not change the fluorescent signal (not shown). In cocultures from TLR4LPS-del embryos, used to verify the specificity of immunostaining with the TLR4 antibody, no signal was obtained (Figure 1D–F).

TLR4 expression on motor neurons from WT or TLR4LPS-del mice. Motor neuron/glia cocultures from WT (A, B) or TLR4LPS-del (D, E) mouse embryos underwent immunocyto-chemistry for SMI32 (A, D, green) and TLR4 (B, E, red). Images were acquired by a confocal microscope. WT motor neurons showed a widespread TLR4 distribution colocalizing with SMI32-positive dendrites and axons (C, merge). TLR4LPS-del motor neurons gave only a very weak, aspecific red fluorescence (E). Scale bar, 20 µm,
We then analyzed the expression of TLR4 on different glial cells, using purified glial cultures. In purified microglia cultures, all cells were double-stained by CD11b-specific (microglial marker, Figure 2A) and TLR4-specific (Figure 2B) antibodies. The intensity and distribution of TLR4 staining was not affected by treatments with LPS, alone (Figures 2D–F, J–L) or in combination with VB3323 (Figures 2G–I, M–O). TLR4 was not detected in purified astrocyte cultures, in either control conditions or after LPS treatment (not shown).

TLR4 distribution and TLR4-mediated morphological alterations in purified microglia cultures. Purified microglial cultures were treated with 1 µg/mL LPS alone or with 20 µg/mL VB3323, for 6 or 18 h. Cells were then double-stained by CD11b and TLR4. Nuclei were revealed by Hoechst 33258 dye (blue). TLR4 was expressed in basal conditions (B) and appeared mostly unaltered during LPS treatment with (H, N) or without (E, K) TLR4 antagonist. After 18 h, LPS induced morphological alterations in CD11b-positive cells (J) that were prevented by cotreatment with VB3323 (M). (P) Morphometric parameters of CD11b-positive microglia were measured with Olympus DPSoft software. The LPS-induced increase (***p < 0.001; one-way ANOVA and Tukey test) in the mean microglial cell area and perimeter was counteracted by VB3323 cotreatment. At least 120 cells for each condition were analyzed from three independent experiments. (Q) Purified microglial cultures were harvested after treatment, and cell lysates were processed by Western blot. In the autoradiograms, the anti-CD68 antibody recognizes two protein bands at different molecular weights (87–115 kDa), which were used for quantification by optical densitometry. Expression data were normalized relative to actin and are expressed as the percentage of the mean CD68 levels in untreated cells (control). Data from three independent experiments were analyzed by one-way ANOVA and Tukey test. LPS doubled the CD68 level (***p < 0.001 versus control), and cotreatment with VB3323 prevented this. There was no stimulation by LPS in TLR4LPS-del cultures. Scale bar, 20 µm.
Inhibition of LPS-Induced Microglial Activation by the TLR4 Antagonist VB3323
We initially characterized the effects of LPS on the morphology of microglia and astrocytes in purified cultures. Cotreatment with VB3323 was also investigated, to verify whether TLR4 activation was involved in LPS-induced alterations.
Morphological and Biochemical Alterations in Glial Cultures
Morphological changes in microglia were investigated by immunocytochemistry or time-lapse analysis. In a first set of experiments, purified microglial cells were processed by immunocytochemistry for CD11b, and perimeters and areas of CD11b-positive cells were measured by an imaging software tool. The exposure of cultures to 1 µg/mL LPS for 18 h induced the activation of microglia, which acquired a reactive phenotype (Figures 2J–L). At this time, increases in the mean area and perimeter (Figures 2J, P) were evident in treated cells, compared with both basal (Figure 2A) and 6-h exposure (Figure 2D) conditions. VB3323 counteracted these effects (Figures 2M, P).
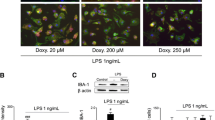
To investigate whether these morphological changes were accompanied by quantitative alterations in specific microglial biomarkers associated with phagocytosis, immunoblot analysis for CD68 was done on cell lysates from purified microglial cultures. Western blot experiments confirmed that VB3323 prevented the activation of microglia induced by LPS. Optical densitometry analysis of the CD68-specific bands showed a significant increase of CD68 in the LPS-treated cultures compared with control (p < 0.001) or VB3323 treatment (p < 0.01; Figure 2Q). As expected, LPS did not affect the basal CD68 levels in TLR4LPS-del microglia (Figure 2Q).
For easy cell tracking in time-lapse experiments, microglial cells were infected with GFP-expressing lentiviral vectors. This step led to highly efficient transduction of microglia, as demonstrated by the widespread GFP expression in the cultures. Cultures were then treated with 1 µg/mL LPS, alone or in combination with 20 µ g/mL VB3323, and time-lapse recording was done. In representative video frames (Figure 3) after 10–15 h of LPS exposure, striking morphological changes were clearly visible in microglia, toward an activated phenotype (increased cell area). Phagocytic events were also observed in LPS-treated cultures (for example, cell debris processed by phagocytosis indicated by the arrows in Figure 3), but not in the control condition or when VB3323 was present (not shown).

Live imaging analysis of microglial activation induced by LPS. Purified microglial cultures were incubated with a GFP-lentiviral construct for 72 h. The success of the infection was first confirmed by optical microscopy, and the cultures under the different experimental conditions were then analyzed by a time-lapse recording. Pictures at different time points from the recording of a LPS-treated (1 µg/mL) culture are reported, showing cell debris phagocytosis by an activated microglial cell (white arrows). Scale bar, 40 nm.
Reduction of LPS-Dependent Cytokine Release by VB3323
The proinflammatory effect of LPS, and the counteracting action of VB3323, were further investigated in cocultures of motor neurons and glia by analyzing the release of cytokines in the culture medium. Cultures were exposed to 1 µg/mL LPS. Conditioned medium was collected at different times after treatment (3, 6, 18 and 24 h), and the concentrations of TNF-α, IL-1β and IL-6 were measured by ELISA. The time course of cytokine release showed that the highest concentrations were reached 18–24 h after treatment (Figure 4A). Cocultures were treated with LPS and one TLR4 antagonist (20 µg/mL VB3323 or RsLPS), and cytokine release was measured after 18 h of exposure. Both TLR4 antagonists significantly reduced the cytokine release triggered by LPS (Figure 4B). VB3323 reduced the release of the three cytokines as follows: TNF-α by 47.5 ± 16.1% (p < 0.001 versus LPS), IL-1β by 59.7 ± 20.4% (p < 0.001) and IL-6 by 29.3 ± 16.3% (p < 0.05). RsLPS had similar effects for TNF-α and IL-6, but the reduction of IL-1β release was significantly greater (87.2 ± 14.1%, p < 0.05; see Figure 4B).

TLR4 ligands regulated cytokine release. Motor neuron/glia cocultures were treated with 1 µg/mL LPS, alone or with 20 µg/mL VB3323 or RsLPS. Culture media were collected at different times, and cytokine concentrations were measured by ELISA. (A) Time course of TNF-α, IL-6 and IL-1 β release induced by LPS. Concentrations of cytokines in the culture media peaked between 18 and 24 h after exposure to LPS. (B) VB3323 or RsLPS significantly reduced the release of TNF-α, IL-6 or IL-1 β induced by LPS after 24 h. ***p < 0.001, **p < 0.01, *p < 0.05; one-way ANOVA and Tukey test.
When the cytokine release was measured in culture medium from cocultures of motor neurons and astrocytes (without microglia), slightly but not statistically different levels were revealed.
Prevention of Motor Neuron Degeneration by VB3323
We investigated whether, in addition to the proinflammatory effects, TLR4 activation also affected motor neuron viability. Motor neuron/glia cocultures were exposed to 1 µg/mL LPS, with or without 20 µg/mL VB3323 or 20 µg/mL RsLPS, for 24 h. Neurotoxicity was evaluated by comparing motor neuron viability in treated and untreated cultures. LPS reduced cell viability by 30.8 ± 11.9% (p < 0.001 versus control; Figure 5A and representative pictures of LPS-treated motor neurons in Figures 5E and H). This toxic effect was significantly counteracted (Figure 5 Finteraction < 0.001) by VB3323 (representative pictures in Figures 5F and I) or RsLPS, which both almost completely restored motor neuron viability (91.3 ± 9.9% with p < 0.001 versus LPS, or 93.4 ± 7.9% with p < 0.001 versus LPS, respectively). We also tested the effect of the toxic residue of LPS, lipid A, in the same experimental setting. Like the full-length endotoxin, 1 µg/mL lipid A significantly reduced motor neuron survival by 30% (Figure 5B), without any dose-dependent effect at higher concentrations (not shown). VB3323 (20 µg/mL) also counteracted lipid A-induced motor neuron death (see Figure 5B).

TLR4 modulation affected motor neuron survival. Viability of motor neurons directly (A and B) or indirectly (C) exposed to TLR4 agonists (1 µg/mL LPS or lipid A) with or without an antagonist (20 µg/mL VB3323 or RsLPS), under different coculture conditions, is shown. (A) Motor neuron/glia cocultures were exposed to LPS ± TLR4 antagonists for 24 h. ***p < 0.001; two-way ANOVA and Bonferroni posttest. Finteraction < 0.001. (B) Motor neuron/glia cocultures were exposed to lipid A ± VB3323 for 24 h. **p < 0.01: one-way ANOVA and Tukey test. C: Mixed glial layers were exposed to LPS ± VB3323 for 24 h. After removing the TLR4 ligands, purified motor neurons were added to this conditioned glial layer. These co-cultures were allowed to grow for 1 wk, and motor neuron viability was assessed. ***p < 0.001, *p < 0.05; two-way ANOVA and Bonferroni posttest. Data are mean number ± standard deviation (SD) of viable motor neurons normalized against control. At least nine wells for each condition were analyzed from three independent experiments. (D-I) Representative pictures of SMI32-positive motor neurons maintained in control conditions (D, G) or treated with LPS alone (E, H) or in combination with VB3323 (F, I). Scale bar (D-F), 40 µm; scale bar (G-I), 20 µm.
To evaluate whether LPS-induced activation of glial cells was enough to induce neurotoxicity, purified motor neurons were cultured with a LPS-conditioned glial layer pretreated with 1 ng/mL LPS for 24 h before cocultures were established. Motor neuron survival was significantly reduced by 35.1 ± 17.4% (p < 0.001 versus control), and cotreatment with 20 µg/mL VB3323 completely counteracted this effect (Figure 5C). To investigate which of the main glial populations mediated this process, cocultures of motor neurons on purified astrocytes were established. When microglia were absent, LPS did not affect motor neuron viability (not shown).
Absence of TLR4 on Microglia Prevents LPS Neurotoxicity
To verify the cell-specific role of TLR4 in LPS neurotoxic effects, we took motor neurons or glial cells from TLR4LPS-del mice. This step enabled us to establish cocultures of motor neurons, astrocytes and microglia with different combinations of cells lacking TLR4, which could be assembled with a controlled cell composition. When TLR4LPS-del motor neuron/ glia cocultures were treated with LPS (1 µg/mL), motor neuron viability was not affected (Figure 6A). When cocultures of TLR4LPS-del motor neurons over WT glia were used, LPS reduced viability by 20% (Figure 6B). This result suggests that functionally active TLR4 on glial cells are required for LPS to induce neurotoxicity. We further distinguished the cell-specific role of TLR4 in the different glial cell types by establishing cocultures of purified microglia from TLR4LPS-del or WT mice, plus WT motor neurons and astrocytes. When microglia were from TLR4LPS-del mice, LPS (1 µg/mL) did not affect motor neuron viability (Figure 6C). Thus, TLR4-expressing microglia are required by LPS to exert neurotoxicity on motor neurons.

LPS reduced motor neuron viability through functionally active TLR4 of microglia. Motor neuron/glia cocultures were established by mixing the different neural populations from the spinal cords of WT or TLR4LPS-del mouse embryos. Cocultures were then exposed to 1 µg/mL LPS, alone or together with 20 µg/mL VB3323, for 24 h. (A) LPS did not affect motor neuron viability when both motor neuron and glial cells came from TLR4LPS-del mice. (B) LPS significantly reduced viability of TLR4LPS-del motor neurons in coculture with WT glia. VB3323 prevented the neurotoxic effects of LPS. (C) LPS did not affect the viability of WT motor neurons in cocultures with WT astrocytes and TLR4LPS-del microglia. Data are mean number ± SD of viable motor neurons normalized against control. Nine wells for each condition were analyzed from three independent experiments. *p < 0.05, one-way ANOVA and Tukey test.
Muscular Decay in Wobbler Mice Is Reduced by VB3323
There were no significant differences in the growth rate and in the body weight progression among the three different experimental groups (not shown). Four-week-old vehicle-treated wobbler mice showed a rapid symptoms progression, as scored by the paw abnormality degree and the forelegs grip-strength measurements. Riluzole significantly improved both behavioral trials until the end of the clinical observation (10th wk of life). VB3323-treated mice showed improvement in both behavioral scores with earlier and stronger effects compared with the riluzole-treated group (Figures 7A, B).
Glial Activation and TNF-α Expression in the Cervical Spinal Cord of Wobbler Mice Are Reduced by VB3323
Compared to control mice (Figure 7C), the morphology of Nissl-stained large neurons from vehicle-treated wobbler mice reveals a marked morphological alteration (swollen cell bodies in Figure 7D). Such alterations seem to be reduced after treatment with riluzole (Figure 7E) or VB3323 (Figure 7F). In addition, the relevant process of chromatolysis observed in motor neurons of untreated wobbler mice, which is characterized by an almost overall loss of cresyl-violet positive staining (see Figure 7D and representative neuronal cell bodies in high-magnified pictures in the panel below), is notably reduced in riluzole-treated wobbler mice (Figure 7E). Interestingly, quite intense staining for cresyl-violet is also observed in VB3323-treated wobbler mice (Figure 7F). In the cervical spinal cord of 10-wk-old healthy mice, the immunoreactivity for the astrocyte marker GFAP (red) was very low and no immunoreactivity for TNF-α (green signal) was revealed (Figure 7G). As previously reported (35,38), an increase in GFAP and TNF-α staining was observed in vehicle-treated wobbler mice (Figure 7H). Treatment with riluzole reduced both GFAP and TNF-α immunoreactivity (Figure 7I). VB3323 induced a greater reduction of GFAP immunoreactivity, and TNF-α expression was reduced by a similar extent as after riluzole treatment (Figure 7J).

VB3323 treatment significantly improved the behavior performance in wobbler mice and reduced reactive astroglyosis. (A, B) Behavioral tests in wobbler mice treated with vehicle (solid line), riluzole (dotted line) or VB3323 (broken line). Riluzole was given in drinking water at the dose of 40 mg/kg/d; VB3323 was administered intraperitoneally three times a week at the dose of 5 mg/kg/d. The treatments were carried out from the 4th to the 10th week of age. Each point represents the mean ± standard error of the mean of 10 mice. Statistical analysis for VB3323-treated wobbler mice: **p < 0.01, ***p < 0.001, different from vehicle-treated wobbler mice; °p < 0.05, °°°p < 0.001, different from riluzole-treated wobbler mice. Riluzole-treated wobbler mice: ∧p < 0.05, ∧∧p < 0.01, ∧∧∧p < 0.001 different from vehicle-treated wobbler mice (Bonferroni posttest). (C-F) Representative images showing the Nissl-positive stained large neurons from the ventral horn of cervical spinal cord of 10-wk-old healthy mice (C), and age-matched vehicle-treated wobbler mice (D), riluzole-treated wobbler mice (E) and VB3323-treated wobbler mice (F); high magnified (1,000×) neuronal cell bodies are reported in the lower panel for each treatment. (G-J) Colocalization experiments showing the profile of immunoreactivity for GFAP (red), TNF-α (green) and their merge (yellow) from the ventral horn of cervical spinal cord of 10-wk-old healthy mice (G) and age-matched vehicle-treated wobbler mice (H), in riluzole-treated wobbler mice (I) and in VB3323-treated wobbler mice (J). (K-N) Colocalization experiments showing the profile of immunoreactivity for CD11b (blue), TNF-α (green) and their merge (light green) from the ventral horn of cervical spinal cord of 10-wk-old healthy mice (K), and age-matched vehicle-treated wobbler mice (L), in riluzole-treated wobbler mice (M) and in VB3323-treated wobbler mice (N). Scale bar (C-F, upper panels), 45 nm; scale bar (C-F, lower panels), 10 µm; scale bar (G-N), 60 µm.
Control mice did not show either CD11b (blue) or TNF-α (green) positive staining (Figure 7K). Both markers were markedly increased in vehicle-treated wobbler mice (Figure 7L) with a relevant colocalization (light-blue staining). Both riluzole (Figure 7M) and VB3323 (Figure 7N) clearly reduced the immunoreactivity for CD11b and TNF-α in wobbler mice. In VB3323-treated wobbler mice, a reduced colocalization of the two markers was also shown (light-blue in Figure 7N).
Discussion
In this work, we investigated the role of TLR4 in in vitro and in vivo settings of motor neuron degeneration. Primary cell cultures from the mouse spinal cord were used to investigate (a) the effects of TLR4 activation in specific cell types, (b) the different contribution of glial populations in motor neuron toxicity and (c) the protective effects of a specific TLR4 antagonist. LPS was used to activate the TLR4-dependent pathways. We showed, by multiple approaches (immunocytochemistry, immunoblotting and live-cell imaging analyses), that in vitro LPS had a potent proinflammatory effect on microglia, which acquired a reactive and phagocytic phenotype, accompanied by functional activation, as demonstrated by the massive increase in TNF-α, IL-1β and IL-6 release in the culture medium. No effects were seen in astrocyte cultures, in accordance with the lack of receptor expression we found for this population (15,39). Such microglia-specific proinflammatory changes may represent the leading cause of neurotoxicity on motor neurons. In fact, only in the presence of microglial cells was LPS able to reduce the viability of motor neurons in cocultures. Proinflammatory cytokines including TNF-α (40), IL-1β (41) and IL-6 (42) are known to contribute to the process of motor neuron degeneration. Thus, a primary role for these cytokines in LPS-induced neurotoxicity could be hypothesized also in our model. By combining cell types from spinal cords of control or TLR4-deficient mice, we also specifically demonstrated the primary role of microglial TLR4 in mediating LPS neurotoxicity. When TLR4 was lacking in microglia, in fact, LPS did not affect motor neuron viability.
Although widely used to induce neuroinflammation in experimental models, stimulation of neural TLR4 by LPS in mammalian CNS diseases probably occurs only with bacterial infections. LPS is a high-molecular-weight hydrophilic molecule that cannot cross the blood-brain barrier (BBB) (43). We therefore also tested the effects of the hydrophobic component of LPS, lipid A, which is responsible for most of the bioactivity of endotoxin and might, because of its lipophilic properties, cross the BBB. Interestingly, lipid A induced motor neuron death to an extent similar to the full-length endotoxin. Lipid A acts through TLR4 (11,12), and, in accordance, cotreatment with TLR4 antagonists counteracted its toxicity on motor neurons.
An LPS-like molecule extracted from the cyanobacterium Oscillatoria Planktothrix FP1 (CyP) dose-dependently inhibited TLR4-mediated inflammatory effects in vitro (32,33) and in vivo in a model of lethal endotoxin shock (32). TLR4 inhibition by CyP had an anticonvulsant effect on kainate-induced seizures in mice (44). We investigated the effect of a highly purified (95%) derivative of CyP, VB3323. It showed potent antiinflammatory and neuroprotective effects in spinal cord cultures, significantly counteracting the effects of TLR4 stimulation by LPS (that is, microglial morphological changes and release of proinflammatory cytokines and motor neuron toxicity). The effects of VB3323 were comparable to those of a well-known, commercially available TLR4 antagonist, the LPS from RsLPS, which is commonly used to inhibit the TLR4-dependent inflammatory mechanisms (45–47).
Considering the antiinflammatory and neuroprotective effects shown by VB3323 in vitro, it was of interest to test the compound also in an in vivo setting of motor neuron degeneration.
Inflammation is associated with motor neuron death in ALS and has been well characterized in animal models of motor neuron degeneration. Marked activation of microglia and astrocytes, and cytokine overexpression (that is, TNF-α), were commonly detected in the SOD1 transgenic mouse (48–50) and in the wobbler mouse. The wobbler mouse shows early-onset selective motor neuron death in the cervical spinal cord (37) accompanied by glial activation (21–23) and upregulation of TNF-α (24) and the activation of both jun N-terminal kinase (JNK) and p38 mitogen-activated stress kinase (p38MAPK) in glial cells and neurons at the early phases of symptom progression (38). The beneficial effect shown by the chronic treatment with a TNF-α binding protein (rhTBP-1; 38) further strengthens the primary role of inflammation in motor neuron degeneration in this model. Thus, we considered the wobbler mouse as a valid and useful model to preliminarily test the effects of chronic treatment by VB3323 on symptoms progression, motor neuron protection and glial activation. Because, similarly to ALS patients (51–53), the wobbler mouse shows symptom amelioration after riluzole administration (35), the effect of VB3323 was compared not only to vehicle-treated mice, but also to riluzole, used as a reference drug. Chronic treatments with the TLR4 antagonist induced a remarkable reduction of gliosis and TNF-α production in the cervical region of the wobbler spinal cord. Notably, VB3323 also induced significant improvements in motor functional tests if compared with either vehicle or riluzole. Whether or not VB3323 has a direct effect in the CNS by passing the BBB remains unknown, but, as suggested also for other treatments (riluzole, rhTBP-1), peripheral administration of antiinflammatory compounds might represents an effective pharmacological approach aimed at modifying the pathological outcome of neurodegenerative diseases.
Conclusion
These combined in vitro/in vivo investigations add new understanding on the role of TLR4 in mediating neurotoxicity in the spinal cord and suggest that TLR4 antagonists could be considered in future studies as candidate protective agents for motor neurons in degenerative diseases.
Disclosure
M Molteni was employed by Bluegreen Biotech.
References
Marsh BJ, Stevens SL, Hunter B, Stenzel-Poore MP. (2009) Inflammation and the emerging role of the toll-like receptor system in acute brain ischemia. Stroke. 40:S34–7.
Tahara K, et al. (2006) Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 129:3006–19.
Chakravarty S, Herkenham M. (2005) Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. J. Neurosci. 25:1788–96.
Tang SC, et al. (2008) Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp. Neurol. 213:114–21.
Abate W, Alghaithy AA, Parton J, Jones KP, Jackson SK. (2009) Surfactant lipids regulate LPS-induced interleukin-8 production in A549 lung epithelial cells by inhibiting translocation of TLR4 into lipid raft domains. J. Lipid Res. 51:334–44.
Hoshino K, et al. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–52.
Takeda K, Akira S. (2001) Roles of Toll-like receptors in innate immune responses. Genes Cells. 6:733–42.
Flo TH, et al. (2000) Involvement of CD14 and beta2-integrins in activating cells with soluble and particulate lipopolysaccharides and mannuronic acid polymers. Infect. Immun. 68:6770–6.
Tobias PS, Tapping RI, Gegner JA. (1999) Endotoxin interactions with lipopolysaccharideresponsive cells. Clin. Infect. Dis. 28:476–81.
Muller-Loennies S, et al. (1998) What we know and don’t know about the chemical and physical structure of lipopolysaccharide in relation to biological activity. Prog. Clin. Biol. Res. 397:51–72.
Lien E, et al. (2000) Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J. Clin. Invest. 105:497–504.
Shimazu R, et al. (1999) MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 189:1777–82.
Walter S, et al. (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 20:947–56.
Panaro MA, et al. (2008) Expression of TLR4 and CD14 in the central nervous system (CNS) in a MPTP mouse model of Parkinson’s-like disease. Immunopharmacol. Immunotoxicol. 30:729–40.
Bsibsi M, Ravid R, Gveric D, van Noort JM. (2002) Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61:1013–21.
Letiembre M, et al. (2009) Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol. Aging. 30:759–68.
Casula M, et al. (2011) Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience. 179:233–43.
Schmitt-John T, et al. (2005) Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 37:1213–5.
Liewen H, et al. (2005) Characterization of the human GARP (Golgi associated retrograde protein) complex. Exp. Cell Res. 306:24–34.
Beghi E, Mennini T. (2004) Basic and clinical research on amyotrophic lateral sclerosis and other motor neuron disorders in Italy: recent findings and achievements from a network of laboratories. Neurol. Sci. 25 (Suppl. 2):S41–60.
Bigini P, Bastone A, Mennini T. (2001) Glutamate transporters in the spinal cord of the wobbler mouse. Neuroreport. 12:1815–20.
Boillee S, Viala L, Peschanski M, Dreyfus PA. (2001) Differential microglial response to progressive neurodegeneration in the murine mutant Wobbler. Glia. 33:277–87.
Rathke-Hartlieb S, Schmidt VC, Jockusch H, Schmitt-John T, Bartsch JW. (1999) Spatiotemporal progression of neurodegeneration and glia activation in the wobbler neuropathy of the mouse. Neuroreport. 10:3411–6.
Schlomann U, Rathke-Hartlieb S, Yamamoto S, Jockusch H, Bartsch JW. (2000) Tumor necrosis factor alpha induces a metalloproteasedisintegrin, ADAM8 (CD 156): implications for neuron-glia interactions during neurodegeneration. J. Neurosci. 20:7964–71.
D.L. no. 116, G.U. suppl. 40, 18 February 1992.
Circolare no. 8, G.U., 14 Luglio 1994.
EEC Council Directive 86/609, OJ L 358, 1, Dec. 12, 1987.
Institute of Laboratory Animal Resources; Commission on Life Sciences; National Research Council. (1996) Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academy Press. [cited 2012 Jul 30]. Available from: https://doi.org/www.nap.edu/openbook.php?record_id=5140
De Paola M, Diana V, Bigini P, Mennini T. (2008) Morphological features and responses to AMPA receptor-mediated excitotoxicity of mouse motor neurons: comparison in purified, mixed anterior horn or motor neuron/glia cocultures. J. Neurosci. Methods. 170:85–95.
Hamby ME, Uliasz TF, Hewett SJ, Hewett JA. (2006) Characterization of an improved procedure for the removal of microglia from confluent monolayers of primary astrocytes. J. Neurosci. Methods. 150:128–37.
Gingras M, Gagnon V, Minotti S, Durham HD, Berthod F. (2007) Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J. Neurosci. Methods. 163:111–18.
Macagno A, et al. (2006) A cyanobacterial LPS antagonist prevents endotoxin shock and blocks sustained TLR4 stimulation required for cytokine expression. J. Exp. Med. 203:1481–92.
Jemmett K, et al. (2008) A cyanobacterial lipopolysaccharide antagonist inhibits cytokine production induced by Neisseria meningitidis in a human whole-blood model of septicemia. Infect. Immun. 76:3156–63.
Blesch A. (2004) Lentiviral and MLV based retroviral vectors for ex vivo and in vivo gene transfer. Methods. 33:164–72.
Fumagalli E, Bigini P, Barbera S, De Paola M, Mennini T. (2006) Riluzole, unlike the AMPA antagonist RPR119990, reduces motor impairment and partially prevents motoneuron death in the wobbler mouse, a model of neurodegenerative disease. Exp. Neurol. 198:114–28.
Bigini P, Mennini T. (2004) Immunohistochemical localization of TNFalpha and its receptors in the rodent central nervous system. Methods Mol. Med. 98:73–80.
Bigini P, et al. (2007) Lack of caspase-dependent apoptosis in spinal motor neurons of the wobbler mouse. Neurosci. Lett. 426:106–10.
Bigini P, et al. (2008) Recombinant human TNF-binding protein-1 (rhTBP-1) treatment delays both symptoms progression and motor neuron loss in the wobbler mouse. Neurobiol. Dis. 29:465–76.
Lehnardt S, et al. (2002) The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J. Neurosci. 22:2478–86.
Wood PL. (1995) Microglia as a unique cellular target in the treatment of stroke: potential neurotoxic mediators produced by activated microglia. Neurol. Res. 17:242–8.
Madrigal JL, Feinstein DL, Dello Russo C. (2005) Norepinephrine protects cortical neurons against microglial-induced cell death. J. Neurosci. Res. 81:390–6.
Heneka MT, et al. (2002) Noradrenergic depletion potentiates beta-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. J. Neurosci. 22:2434–42.
Banks WA, Erickson MA. (2010) The blood-brain barrier and immune function and dysfunction. Neurobiol. Dis. 37:26–32.
Maroso M, et al. (2010) Toll-like receptor 4 and high-mobility group box-1 are involved in ictogenesis and can be targeted to reduce seizures. Nat. Med. 16:413–9.
Christianson CA, et al. (2011) Spinal TLR4 mediates the transition to a persistent mechanical hypersensitivity after the resolution of inflammation in serum-transferred arthritis. Pain. 152:2881–91.
Mazarati A, Maroso M, Iori V, Vezzani A, Carli M. (2011) High-mobility group box-1 impairs memory in mice through both toll-like receptor 4 and receptor for advanced glycation end products. Exp. Neurol. 232:143–8.
Netea MG, van Deuren M, Kullberg BJ, Cavaillon JM, Van der Meer JW. (2002) Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? Trends Immunol. 23:135–9.
Alexianu ME, Kozovska M, Appel SH. (2001) Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 57:1282–9.
Tortarolo M, et al. (2003) Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol. Cell. Neurosci. 23:180–92.
Yoshihara T, et al. (2002) Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 80:158–67.
Bensimon G, Lacomblez L, Meininger V. (1994) A controlled trial of riluzole in amyotrophic lateral sclerosis: ALS/Riluzole Study Group. N. Engl. J. Med. 330:585–91.
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. (1996) Dose-ranging study of riluzole in amyotrophic lateral sclerosis: Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 347:1425–31.
Miller RG, Mitchell JD, Lyon M, Moore DH. (2003) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4:191–206.
Acknowledgments
This work was partially supported by the Lombardy Region Found for the Promotion of Institutional Agreements (NEPENTE [Network lombardo di eccellenza per lo sviluppo di farmaci di origine naturale diretti alla modulazione del microambiente tissutale per la prevenzione e terapia dei tumori e delle malattie neurodegenerative] project ID 14501). A fellowship to A Mariani was funded by the Cariplo Foundation (grant 2009–2426) (Milan, Italy). The authors acknowledge Alessandro Soave for artwork and Judy Baggott for the English revision.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
10020_2012_1806971_MOESM1_ESM.pdf
Neuroprotective Effects of Toll-Like Receptor 4 Antagonism in Spinal Cord Cultures and in a Mouse Model of Motor Neuron Degeneration
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
De Paola, M., Mariani, A., Bigini, P. et al. Neuroprotective Effects of Toll-Like Receptor 4 Antagonism in Spinal Cord Cultures and in a Mouse Model of Motor Neuron Degeneration. Mol Med 18, 971–981 (2012). https://doi.org/10.2119/molmed.2012.00020
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00020