
Abstract
Fabry disease is a panethnic, X-linked, inborn error of glycosphingolipid metabolism resulting from mutations in the α-galactosidase A gene (GLA) that lead to the deficient activity of the lysosomal enzyme, α-galactosidase A (α-Gal A). Affected males with no α-Gal A activity have the early-onset classic phenotype, whereas those with residual activity present with the later-onset subtype. Recently, we reported that newborn enzyme-based screening using dried blood spots (DBS) in Taiwan revealed a high incidence of newborn males who had the GLA c.936+919G→A (IVS4+919G→A) mutation. This lesion causes cryptic splicing, markedly reducing the amount of wild-type GLA mRNA, and has been found in males with the later-onset Fabry phenotype, manifesting as cardiac, renal and/or cerebrovascular disease. To more accurately determine the incidence of the IVS4+919G→A mutation, 20,063 consecutive newborns were screened by a deoxyribonucleic acid (DNA)-based assay. Of the 10,499 males, 12 (1/875) and 24 of the 9,564 females (1/399) had the mutation. On the basis of these frequencies, the previous newborn enzyme-based DBS screening (cutoff: <30% of the normal mean) only identified 67% and 17% of mutation-positive males and females, respectively. The mean DBS α-Gal A activities in the mutation-positive males and females were 23% (1.54 U) and 55% (3.63 U) of normal mean male/female values, respectively. These studies confirm the high incidence of the IVS4+919G→A mutation in the Taiwanese population and indicate that its detectability by enzyme-based DBS screening is unreliable, especially in females. These studies emphasize the superiority of DNA-based newborn screening for common mutations, particularly for X-linked diseases.
Similar content being viewed by others

Introduction
Fabry disease (MIM 301500) is an X-linked lysosomal storage disorder caused by the markedly deficient activity of the lysosomal glycohydrolase, α-galactosidase A (α-Gal A) (1). This enzymatic defect leads to the progressive accumulation of globotriaosylceramide (GL-3) and related glycosphingolipids, primarily in the vascular endothelium of the heart, kidney, skin and brain. Affected males with the classic phenotype have little, if any, α-Gal A enzyme activity (<1% of mean normal), whereas males with the later-onset phenotype have residual enzymatic activity, typically >1% of normal. In classically affected males, the microvascular pathology leads to the onset of angioker-atomas, acroparesthesias, hypohidrosis, gastrointestinal abnormalities and a characteristic corneal opacity early in childhood or adolescence. With advancing age, manifestations include renal insufficiency and failure, cardiac involvement and cerebrovascular disease. Affected males with the later-onset phe-notype present in adulthood usually lack the classic early manifestations and typically develop renal and/or cardiac disease in the third to sixth decades of life. Clinical manifestations in heterozygous females from Fabry families with the classic phenotype range from asymptomatic to as severe as affected males, whereas heterozygous females from later-onset families may have milder symptoms later in life, including cardiac and renal manifestations (1).
Previous studies revealed that enzyme replacement therapy with recombinant α-Gal A can improve the outcome of patients with Fabry disease, particularly when initiated before significant renal (2,3) or cardiac involvement (4). Therefore, we and others have initiated pilot newborn screening programs for Fabry disease on the basis of dried blood spot (DBS) α-Gal A activity (5–7). These studies revealed a surprisingly high incidence of the disease-causing α-Gal A (GLA) gene mutations, particularly those causing the later-onset phenotype. In Italy, enzyme-based screening of over 37,000 consecutive newborn males identified 1 in ~3,100 who were subsequently confirmed to have GLA mutations; the ratio of classic to later-onset GLA mutations was 11 to 1 (7). In Taiwan, enzyme-based screening of over 90,000 males by our group identified 1 in ~1,250 as having confirmed GLA mutations. Of these, 81.9% (or 1 in ~1,530 males) had the c.936+919G→A (IVS4+919G→A) later-onset cryptic splice mutation, whereas the frequency causing the classic phenotype was 1 in ~22,570; the ratio of classic to later-onset GLA mutations was 16 to 1 (5). Lin et al. (6) also screened over 57,000 Taiwanese males, of which 1 in ~1,370 males had confirmed GLA mutations (6). Of these, 83% (or 1 in ~1,640) had the IVS4+919G→A mutation. Of the over 600 known mutations causing Fabry disease, the IV4+919G→A is the first common disease-causing mutation in any racial, ethnic or demographic group (https://doi.org/www.hgmd.cf.ac.uk).
In the Taiwanese studies, the detection of newborn females by enzyme screening was dramatically inefficient, since only five enzyme-positive and mutation-confirmed females of the 134,265 screened (1 in →26,850) were detected in the two reported studies (5,6). Thus, newborn screening by DBS α-Gal A activity underestimated the incidence of this mutation in females, since most female heterozygotes had >30% of normal mean α-Gal A activity (5) due to random X-inactivation (1,8–11). Therefore, an investigation was undertaken to determine the incidence of the IVS4+919G→A mutation by direct mutation analysis in over 20,000 consecutive newborn males and females in Taiwan.
Materials and Methods
Newborn screening for Fabry disease was carried out by the Taiwanese Newborn Screening Program, and over 92% of newborns were from mothers of Southern Chinese ancestry, while ~4% each were from other parts of China or from Southeastern Asia. The study was approved by the Ethics Committee of the National Taiwan University Hospital.
Genomic deoxyribonucleic acid (DNA) was extracted from DBS as previously described (12). Genotyping for the GLA IVS4+919G→A mutation was performed by a TaqMan® assay designed and manufactured by Applied Biosystem’s Custom TaqMan Assays Service using the StepOnePlus™ Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). DBS α-Gal A activity was measured as previously described, and neutral α-glucosidase (NAG) served as a control enzyme activity (5). One unit (U) of enzymatic activity was defined as 1 μmol/h of substrate cleaved per liter whole blood.
The following studies documented the sensitivity and specificity of the TaqMan assay for the IVS4+919G→A mutation. The first 200 DNA samples were also Sanger sequenced to confirm the TaqMan assay results (see the supplementary material for primer sequences). All IVS4+919G→A-positive DNA samples were confirmed by Sanger sequencing as previously described (13). In addition, all newborn DBS with low α-Gal A levels (<30% of normal) were sequenced to detect →-Gal A mutations, including IVS4+919G→A.
All supplementary materials are available online at https://doi.org/www.molmed.org .
Results
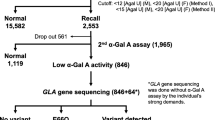
Among 20,063 consecutive newborn samples analyzed specifically for the GLA IVS4+919G→A mutation, 12 of the 10,499 males (0.114% or 1 in ~875) and 24 of the 9,564 (0.25% or 1 in ~399) female newborns had the IVS4+919G→A mutation. The combined frequency of this mutation was 1 in 823 X-chromosomes. There were no distribution anomalies in either the sequence of newborns or their geographical distribution.
The mean and median DBS α-Gal A activities of 12 male newborns with the mutation were 1.54 and 1.30 U (standard deviation [SD] 0.612, range 0.91–2.97), which were 23.4% and 19.8%, respectively, of the population mean (6.58 U, SD 3.13, range 0.26–60.6, n = 20,027; mean of normal male 6.45 U and normal female 6.73 U). The mean and median α-Gal A activities of the 24 female new-borns with the mutation were 3.63 and 3.41 U (SD 1.52, range 1.77–7.66), which were 55.2% and 51.8% of the normal mean (Figure 1A). For DBS newborn screening, the initial cutoff level of α-Gal A activity was <30% of the normal mean for both males and females. Using that cutoff, DBS screening would have detected 67% of the mutation-positive male newborns and only 17% of the mutationpositive female newborns.

Distribution of α-Gal A activity and different GLA genotypes. (A) Distribution of α-Gal A activity and IVS4+919 G (wild type) or A (mutant) genotype. Note that the α-Gal A activity in the different genotypes of males and females overlapped. (B) Distribution of NAG/α-Gal A activity ratio and IVS4+919 genotypes. By use of this ratio cutoff, male patients, but not female herterozygotes, were more accurately detected. Male-G, males with IVS4+919G; Male-A, males with IVS4+919A; Female-GG, females with IVS4+919 GG; Female-AG, females with IVS4+919 AG; AGAL, α-Gal A.
Because the α-Gal A activities of affected Fabry newborns and normal newborns overlapped, we used the NAG/ α-Gal A ratio as the cutoff for Fabry disease screening (Figure 1B). With this activity ratio, a cutoff ratio of 20 would have identified 83% of the mutation-positive male newborns, but none of the mutation-positive females. Of particular note, by raising the cutoff activity ratio to 40 (avoiding detection for newborns with the later-onset IVS4+919G→A), only two IVS4+919G→A males would be detected in this series of over 10,000 male newborns.
Discussion
Previously, our group (5) and an independent Taiwanese group (6) performed α-Gal A DBS enzyme-based screening, and both reported that the GLA IVS4+919G→A mutation was the most common detected. When combined, the two groups screened by the enzyme-based assay had >147,700 and >134,260 newborn males and females, respectively. Sequencing the enzyme-positive DBS DNA samples revealed a combined incidence of the IVS4+919G→A later-onset mutation of 1 in ~1,570 Taiwanese males and 1 in ~41,420 females. Since the IVS4+919G→A mutation encoded a mutant enzyme with residual activity, enzyme-based detection in males might be decreased, whereas detection of females was clearly decreased from that expected (that is, twice the male frequency) by the presence of one normal allele and random X-inactivation (1,8–11). Thus, to accurately determine the incidence of the IVS4+919G→A mutation in the Taiwanese newborns and to compare the detection rates of the enzyme- and molecular-based assays, an additional 20,063 consecutive newborn male and female DBS samples were screened by both assays.
Molecular detection of the IVS4+919G→A mutation revealed a higher incidence than that detected using our previously described α-Gal A DBS assay (5). Among the >20,000 newborns tested by the molecular-based assay, 1 in ~875 males and 1 in ~399 females had the IVS4+919G→A mutation (a ratio of 2.2:1, close to the expected 2:1 male-to-female ratio). On the basis of these findings, the IVS4+919G→A is clearly the most common mutation identified among over 600 mutations causing Fabry disease. When compared with the molecular assay, the DBS enzyme assay detected only 67% of the males and 17% of the females with the later-onset mutation. The difference was because the relatively common IVS4+919G→A mutation causes a splicing defect that generates both mutant and normal α-Gal A mRNAs (13). Therefore, detection by α-Gal A enzymatic activity in DBS is not sensitive or specific enough to detect all males and most females with this mutation. The reduced detection of female IVS4+919G→A heterozygotes by enzyme assay was due to the inherent 50% of normal activity of the wild-type allele and the skewing of X-chromosomal inactivation. Those with >50% inactivation of the X-chromosome carrying the wildtype GLA gene were more likely to be detected. The level of α-Gal A activity, therefore, could range from that seen in IVS4+919G→A-positive newborn males to normal levels. Thus, most mutationpositive female newborns will have more DBS α-Gal A activity than the cutoff value of 30% of mean normal activity.
The finding that enzyme-based DBS newborn screening did not identify most of the mutation-positive females and missed one-third of the IVS4+919G→A-positive males has direct implications for detecting patients with common later-onset α-Gal A mutations by DBS enzyme-based screening of newborns or of adults in hemodialysis, cardiac or stroke clinics. For newborn screening, it would be recommended that all enzyme-positive males and females first be tested for the common later-onset mutation(s) in their respective populations using rapid, relatively inexpensive and reliable mutation-specific techniques such as the TaqMan assay used to detect the IVS4+919G→A mutation. Second, newborn screening programs should consider at least a pilot screening of all males for selected, relatively common, later-onset mutations in their population. If these mutations are known to cause disease manifestations in heterozygous females, then pilot screening should include female newborns. Mutation-based detection of later-onset α-Gal A mutations in new-borns should more reliably determine the penetrance and natural history of these mutations in their maternal grandparents, thereby providing more accurate genetic counseling for the parents of mutation-positive newborns. In addition, it will permit identification of older at-risk relatives who may benefit from more frequent medical monitoring and treatment by enzyme replacement therapy.
The IVS4+919G→A mutation has been identified in Japan (13) and Taiwan (5). We also encountered individuals with the mutation from Vietnam, Indonesia and the United States who had Chinese ancestors. Because this mutation has not been identified in Northern China, Korea and Japan (except in the Kagoshima area), the observation of a north-south differentiation of East Asian populations suggests a “southern origin” of the ancient mutation (14,15), although its distribution due to more recent migrations in the last centuries may be quite substantial. Subsequent studies in various Asian communities will further establish the distribution of this mutation. To date, the mutation has not been reported in other ethnic, racial or demographic groups.
The IVS4+919G→A mutation was first reported in a Japanese male who had later-onset Fabry disease, as manifested by left ventricular hypertrophy (LVH), which lead to hypertrophic cardiomyopathy (HCM) (13). Affected males with later-onset mutations such as IVS4+919G→A typically lack the angiokeratomas, acroparesthesias, hypohidrosis, gastrointestinal abnormalities and corneal opacities that are characteristic of the classic, early-onset, more severe phenotype and may manifest renal and/or cardiac disease with LVH leading to HCM in the third to sixth decades of life (1,16–18). Most later-onset males have been identified by α-Gal A enzyme screening or GLA mutation analyses in hemodialysis, cardiac and stroke clinics (for example, 17–22). Screening of male patients with LVH or HCM revealed that up to 3.0% had previously unrecognized Fabry disease, all having been confirmed by GLA mutation analyses (17,22–27). A recent European collaborative study of over 1,380 European patients (64% males) with unexplained LVH and/or HCM who were ≥40 years of age identified 0.5% (1 in ~200) who had previously unrecognized Fabry disease. Of these, 0.34% (3/887 or 1 in 296) were males and 0.8% (4/499 or 1 in 112) were females, all with known later-onset GLA mutations including A143T, N215S, R118C, D244N and T410A (21). Thus, the mutation-based incidence of the IVS4+919G→A mutation in Taiwanese males (1 in ~875) was high, but was reasonably consistent with the prevalence (0.5%) of GLA mutations causing the later-onset Fabry cardiac phenotype in European males with unexplained LVH or HCM (21).
Although the prevalence of Fabry disease among LVH and HCM patients in Taiwan is unknown, the finding of the IVS4+919G→A mutation in 1 in ~875 newborn males and 1 in ~400 newborn females should contribute to the prevalence of cardiac and renal disease in Taiwan. However, no systematic studies have evaluated the frequency of this mutation in older men and women with LVH, HCM or renal disease in Taiwan. The only relevant information, reported by Lin et al. (6), identified the IVS4+919G→A mutation in 9 maternal grandfathers and 11 maternal grandmothers of mutation-positive new-borns, of which 33% of the grandfathers and none of the grandmothers had HCM (6). They also evaluated 16 unrelated Taiwanese adult males with HCM and found that 25% had the IVS4+919G→A mutation. Subsequently, echocardiography of mutation-positive men and women >40 years of age revealed that 67% (14/21) of the men and 20% (5/25) of the women had LVH (28). They also documented the presence of microalbuminuria in 20% of 86 mutation-positive adults. On the basis of these findings, it is estimated that the prevalence of LVH due to the IVS4+919G→A mutation among the ~10 million adults ≥40 years of age in Taiwan would be about 63 per 100,000 (77/100,000 for males and 50/100,000 for females). Although these estimates may not accurately reflect the number of Taiwanese patients with cardiac disease, it is likely that the IVS4+919G≥A lesion contributes to the occurrence of unexplained LVH, HCM and renal and cerebrovascular disease in Taiwan. Future targeted screening of this mutation among Taiwanese males and females in cardiac, renal and stroke clinics would provide information on the prevalence and natural history of this later-onset mutation. In conclusion, we confirmed a high incidence of the IVS4+919G≥A mutation in the Taiwanese population. However, the natural history of individuals carrying this mutation remains to be clarified.
Conclusion
This study comparing the detectability of enzyme versus molecular assays for the Fabry IVS4+919G≥A mutation is relevant to screening for patients with other diseases resulting from common mutations, especially for X-linked diseases. Clearly, for detecting common mutations, screening by a more sensitive and specific molecular-based assay is strongly recommended.
Disclosure
The Mount Sinai School of Medicine, the Department of Genetics and Genomic Sciences and some faculty members in the department (including the chair emeritus and dean for Genetic and Genomic Medicine, RJ Desnick) receive financial benefit from the Genzyme Corporation for the sale of Fabrazyme® and from Shire HGT for the sale of Replagal, enzyme replacement drugs for the treatment of Fabry disease. RJ Desnick is a consultant for the Genzyme Corporation and Synageva Biopharmaceuticals, receives grants from the Genzyme Corporation, and has founders’ stock in Amicus Therapeutics.
References
Desnick RJ, Ioannou YA, Eng CM. (2001) α-Galactosidase A deficiency: Fabry disease. In: The Metabolic and Molecular Bases of Inherited Disease. Vol. III. 8th ed. Scriver CR, Beaudet AL, Sly WS, Valle D, Kinzler KE, Vogelstein B (eds.). New York: McGraw-Hill, pp. 3733–74.
Banikazemi M, et al. (2007) Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann. Intern. Med. 146:77–86.
Wilcox WR, et al. (2004) Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am. J. Hum. Genet. 75:65–74.
Weidemann F, et al. (2003) Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation. 108:1299–301.
Hwu WL, et al. (2009) Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G<A (IVS4+919G<A). Hum. Mutat. 30:1397–405.
Lin HY, et al. (2009) High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ. Cardiovasc. Genet. 2:450–6.
Spada M, et al. (2006) High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 79:31–40.
Morrone A, et al. (2003) Fabry disease: molecular studies in Italian patients with X-inactivation analysis in manifesting carriers. J. Med. Genet. 40:e103–9.
Dobrovolny R, et al. (2005) Relationship between X-inactivation and clinical involvement in Fabry heterozygotes: eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J. Mol. Med. 83:647–54.
Maier EM, et al. (2006) Disease manifestations and X-inactivation in heterozygous females with Fabry disease. Acta. Paediatr. Suppl. 95:30–8.
Linthorst GE, Poorthuis BJ, Hollak CE. (2008) Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J. Am. Coll. Cardiol. 51:2082.
Baker MW, et al. (2009) Development of a routine newborn screening protocol for severe combined immunodeficiency. J. Allergy Clin. Immunol. 124:522–7.
Ishii S, et al. (2002) Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am. J. Hum. Genet. 70:994–1002.
Chu JY, et al. (1998) Genetic relationship of popu lations in China. Proc. Natl. Acad. Sci. U. S. A. 95:11763–8.
Su B, et al. (1999) Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am. J. Hum. Genet. 65:1718–24.
von Scheidt W, et al. (1991) An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N. Engl. J. Med. 324:395–9.
Nakao S, et al. (1995) An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N. Engl. J. Med. 333:288–93.
Nakao S, et al. (2003) Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 64:801–7.
Linthorst GE, et al. (2010) Screening for Fabry disease in high-risk populations: a systematic review. J. Med. Genet. 47:217–22.
Wozniak MA, et al. (2010) Frequency of unrecognized Fabry disease among young European-American and African-American men with first ischemic stroke. Stroke. 41:78–81.
Elliott P, et al. (2011) Prevalence of Anderson-Fabry disease in patients with hypertrophic car-diomyopathy: the European Anderson-Fabry Disease Survey. Heart. 97:1957–60.
Hagege AA, et al. (2011) Screening patients with hypertrophic cardiomyopathy for Fabry disease using a filter-paper test: the FOCUS study. Heart. 97:131–6.
Sachdev B, et al. (2002) Prevalence of Anderson-Fabry disease in male patients with late onset hy-pertrophic cardiomyopathy. Circulation. 105:1407–11.
Ommen SR, Nishimura RA, Edwards WD. (2003) Fabry disease: a mimic for obstructive hyper-trophic cardiomyopathy? Heart. 89:929–30.
Arad M, et al. (2005) Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 352:362–72.
Morita H, et al. (2006) Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 113:2697–705.
Monserrat L, et al. (2007) Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 50:2399–403.
Lin HY, et al. (2010) Enzyme assay and clinical assessment in subjects with a Chinese hotspot late-onset Fabry mutation (IVS4+919G→A). J. Inherit. Metab. Dis. 33:619–24.
Acknowledgments
The authors acknowledge the contributions of the participants in these research endeavors and Nicole Kelly for her excellent administrative expertise. This project was partially supported by the Bureau of Health Promotion, Department of Health, Taiwan (DOH100-HP-1214), and by Shire Pharmaceuticals, neither of which had a role in the study design; the collection, analysis and interpretation of data; the writing of the manuscript; or the decision to submit the paper for publication.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Chien, YH., Lee, NC., Chiang, SC. et al. Fabry Disease: Incidence of the Common Later-Onset α-Galactosidase A IVS4+919G→A Mutation in Taiwanese Newborns—Superiority of DNA-Based to Enzyme-Based Newborn Screening for Common Mutations. Mol Med 18, 780–784 (2012). https://doi.org/10.2119/molmed.2012.00002
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2012.00002