
Abstract
Severe burn injury is associated with induction of the hepatic endoplasmic reticulum (ER) stress response. ER stress leads to activation of c-Jun N-terminal kinase (JNK), suppression of insulin receptor signaling via phosphorylation of insulin receptor substrate 1 and subsequent insulin resistance. Marked and sustained increases in catecholamines are prominent after a burn. Here, we show that administration of propranolol, a nonselective β1/2 adrenergic receptor antagonist, attenuates ER stress and JNK activation. Attenuation of ER stress by propranolol results in increased insulin sensitivity, as determined by activation of hepatic phosphatidylinositol 3-kinase and Akt. We conclude that catecholamine release is responsible for the ER stress response and impaired insulin receptor signaling after burn injury.
Similar content being viewed by others


Introduction
Burn is a severe form of trauma that affects over 2 million people in the United States annually (1), with multiple organ failure after severe burn injury being the most common cause of death (2). The liver, which regulates whole-body energy homeostasis, plays a critical role after a burn, since hepatic function correlates closely to patient morbidity and mortality (3). In an animal burn model, we have shown that burn injury is associated with hepatic ER stress (4) that persists for at least 3 wks after injury (5). Further, we have shown that ER stress after burn injury is accompanied by impairment of the phosphatidylinositol 3-kinase (PI3K)/Akt signaling cascade and insulin resistance (6).
A causative link between ER stress and suppressed insulin signaling in the liver has been well established in models of type 2 diabetes (7). Activation of the inositol requiring enzyme (IRE)-1 branch of the ER stress pathway leads to activation of the cellular stress kinase, c-Jun N-terminal kinase (JNK) (8). Activated JNK has been shown to negatively regulate insulin signaling and downstream activation of PI3K/Akt signaling by serine phosphorylation of the insulin receptor substrate (IRS)-1 (7), a critical regulator of the insulin receptor signaling pathway (9).
Marked and sustained increases in catecholamines are thought to initiate the stress response after severe burn injury (10). We have shown that catecholamine levels in patients remain elevated up to 2 years after severe burn injury (11). Catecholamines induce ER stress in various cell types (12,13). The role of catecholamines in potentiating burn-induced ER stress and PI3K/Akt dysregulation in the liver has yet to be examined. Here, we show that ER stress and JNK-dependent inhibition of the PI3K/Akt pathway after a burn is induced by catecholamines. Further, administration of propranolol, a nonselective β adrenergic receptor antagonist attenuates ER stress and improves PI3K/Akt signaling.
Materials and Methods
Animal Model of Burn Injury
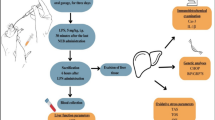
All animal manipulations were approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch at Galveston. The National Institutes of Health Guide for the Care and Use of Laboratory Animals were met (14). A well-established method was used to induce a full-thickness scald burn (15). Male Sprague-Dawley rats (n = 6 each for experimental and control groups, 325–350 g) were allowed to acclimate for 1 wk before conducting experiments. Rats were housed in an institutional animal care facility and received a high-protein diet (Ensure, Abbott Laboatories, IL, USA) and water ad libitum throughout the study. Ensure was administered 7 d before the study to adjust the animals to the liquid diet. Animals were anesthetized with general anesthesia (ketamine, 40 mg/kg body weight, and xylazine, 5 mg/kg body weight, both injected intraperitoneally) and received analgesia (Buprenorphine, 0.05 mg/kg body weight, injected subcutaneously). The dorsum of the trunk and the abdomen was shaved, and a 60% total body surface area burn was administered by placing the animals in a mold that exposed defined areas of the skin on the back and abdomen under general anesthesia and analgesia. The mold was placed in 96–98°C water, scalding the back 10 s and the abdomen 1.5 s. This method delivers a full-thickness cutaneous burn, as confirmed by histological section. Lactated Ringers solution (40 mL/kg body weight) was administered intraperitoneally immediately after the burn for resuscitation. After burn and resuscitation, animals were observed, received oxygen and were placed into cages.
Experimental Design
The treatment groups included sham, burn and burn with propranolol. Sham animals received anesthesia and analgesia but were not burned. Animals in the propranolol treatment group received 5 mg/kg/d orally beginning after burn injury. We have previously shown that burn injury significantly upregulates sensors of hepatic ER stress and associated impairment of the PI3K/Akt signaling cascade at 72 h (6). Therefore, we chose 72 h after burn injury as a time point for analysis in the current study.
Insulin Challenge
To determine the effects of burn injury on hepatic insulin signaling pathways, a laparotomy was performed at 72 h after the burn/sham procedure as described (6). Briefly, the right lobe of the liver was ligated and stored (preinsulin). Insulin (1 IU/kg, Novolin R; Novo Nordisk, Mainz, Germany) was injected into the portal vein, and the remaining liver was harvested 1 min thereafter.
Antibodies and Reagents
Antibodies against murine phosphoJNK, JNK, phospho-IRS-1, IRS-1, phospho-protein kinase R (PKR)-like ER kinase (phospho-PERK), phospho-Akt and Akt were purchased from Cell Signaling (Danvers, MA, USA); 78 kDa glucose-regulated protein/binding immunoglobulin protein (GRP78/BiP), phospho-IRE-1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Abcam (Cambridge, MA, USA); and activating transcription factor 6 (ATF-6) was purchased from Lifespan Biosciences (Seattle, WA, USA). SuperSignal West Pico Chemiluminescent Substrate was purchased from Thermo Scientific (Rockford, IL, USA).
Western Blotting
Approximately 100 mg frozen liver tissue was homogenized in 150 mmol/L NaCl, 50 mmol/L Tris-HCl, pH 7.8, 1% (w/v) Triton X-100, 1 mmol/L EDTA, 0.5 mmol/L phenylmethanesulfonyl fluoride, 1x Complete protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN, USA) and a phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). The homogenate was centrifuged at 20,000g for 30 min at 4°C and the pellet discarded. Western blotting was performed with 50 µg protein. Band intensities were quantified with the Image J software (National Institutes of Health, Bethesda, MD, USA). GAPDH was used as a loading control.
Statistical Analysis
Statistical analysis was performed using one-way analysis of variance. Statistical analysis of pre- and postinsulin administration within each experimental group was performed using a paired Student t test. Data are presented as mean ± standard error of the mean (SEM). Significance was accepted at P < 0.05.
Results
Propranolol Attenuates Burn-Induced Hepatic ER Stress
In agreement with previous studies, we found that burn produced a significant increase in markers associated with ER stress (P < 0.05; Figures 1A, B). Pro-pranolol administration significantly decreased burn-induced activation of type 1 ER transmembrane protein kinase (IRE-1) and PERK, as determined with phospho-specific antibodies. Furthermore, propranolol reduced the amount of the cleaved, active form of ATF-6 (P < 0.05; Figures 1A, B). These results indicate that propranolol attenuates burn-induced hepatic ER stress.

Propranolol attenuates activation of proximal sensors of hepatic burn-induced UPR. (A) Activation of Bip, IRE-1, PERK and ATF-6 were analyzed in sham, burn and burn + propranolol (Prop) groups. Proteins levels are normalized to GAPDH. (B) Data represent mean ± SEM. #P < 0.05, value for sham versus burn; *P < 0.05, value for burn versus burn + prop 72 h after burn injury. Data are representative of three separate experiments.
Propranolol Attenuates JNK-Mediated Phosphorylation of IRS-1
ER stress-induced activation of IRE-1 is known to activate the downstream cellular stress kinase JNK (8). Thermal injury results in marked JNK activation, as determined using phospho-specific antibodies (P < 0.05; Figure 2A). Importantly, propranolol administration significantly decreases hepatic JNK activation, consistent with our finding that propranolol inhibits ER stress (P < 0.05; Figure 2A).

Propranolol reduces JNK activation and JNK associated IRS-1 phosphorylation. Activation of JNK (A) and IRS-1 phosphorylation (B) at serine 307 was analyzed in sham, burn and burn + propranolol (Prop) groups. Protein levels were normalized to total levels of protein. Data represent mean ± SEM. #P < 0.05, value for sham versus burn; *P < 0.05, value for burn versus burn + prop 72 h after burn injury. Data are representative of three separate experiments.
Activation of JNK has been shown to negatively regulate insulin signaling through serine 307 phosphorylation of IRS-1 (16). Burn injury is associated with a significant increase in IRS-1 phosphorylated at serine 307 (P < 0.05; Figure 2B). Propranolol significantly decreases this inhibitory phosphorylation of IRS-1 (P < 0.05; Figure 2B). These results suggest that elevated catecholamines after burn injury provoke insulin resistance by activation of JNK.
Propranolol Improves Postburn Insulin Sensitivity
To determine if propranolol administration improves insulin sensitivity after burn injury, insulin was injected directly into the portal vein of animals in all treatment groups. Phosphorylation of the insulin receptor was analyzed. Insulin administration results in significant phosphorylation of the receptor in sham animals (P < 0.05; Figure 3A). In contrast, phosphorylation of the insulin receptor was not significantly altered in either thermally injured or propranolol treatment groups after insulin administration (Figure 3A). Activation of IRS-1 by the insulin receptor is mediated by phosphorylation of tyrosine residue 612. As expected, insulin administration results in significant phosphorylation of tyrosine 612 in sham animals (P < 0.05; Figure 3B). In contrast, phosphorylation of tyrosine 612 is inhibited in thermally injured animals, indicating postburn insulin resistance (Figure 3B). Propranolol treatment restored IRS-1 tyrosine phosphorylation in thermally injured animals (P < 0.05; Figure 3B). Activation of Akt by the insulin receptor is mediated by phosphorylation of serine residue 473. Insulin administration results in significant phosphorylation of serine 473, which is abolished in thermally injured animals (P < 0.05; Figure 3C). Importantly, phosphorylation of Akt by insulin stimulation is restored with propranolol administration (P < 0.05; Figure 3C). These results indicate that propranolol restores insulin sensitivity in thermally injured animals. Therefore, catecholamines are a significant contributor to postburn ER stress and insulin resistance.

Propranolol improves impaired PI3K/Akt signaling postburn injury after insulin administration. Phosphorylation of insulin receptor β (A), IRS-1 (B) and Akt (C) and associated total protein levels were analyzed in sham, burn and burn + propranolol (Prop) groups 1 min before (−) and 1 min after (+) insulin injection. Phosphorylated protein levels were normalized to respective total protein levels. Data represent mean ± SEM. #P < 0.05, value for before (−) and after (+) insulin 72 h after burn injury. Data are representative of three separate experiments.
Discussion
We demonstrate that blockade of catecholamine signaling by the nonselective β adrenergic receptor antagonist propranolol attenuates ER stress and improves postburn insulin resistance. These findings suggest that catecholamine release after burn injury is responsible for up-regulation of the ER stress response pathway. The actions of the catecholamines appear to be mediated through the β adrenergic receptors, since blockade of these receptors by propranolol attenuates postburn hepatic ER stress and improves PI3K/Akt signaling.
We have shown that propranolol administration after burn injury significantly attenuates all branches of the unfolded protein response. These results suggest that catecholamines play a role in potentiating ER stress. Consistent with this, norepinephrine has been shown to induce the ER stress response in both cardiomyocytes and PC12 cells (12,13). Furthermore, we demonstrated that this result is mediated by JNK activation and suppression of signaling downstream of IRS-1, ultimately resulting in insulin resistance (17). In the liver, however, IRS-2 preferentially mediates insulin sensitivity (18). Indeed, JNK has been shown to negatively phosphorylate IRS-2, thereby inhibiting the downstream insulin signaling pathways (19). The role of IRS-2 in mediating insulin sensitivity postburn injury will be explored in future studies.
Burn injury is associated with impaired PI3K/Akt signaling in both the liver (6) and muscle (20,21). Given that prolonged β adrenergic receptor stimulation in cardiomyocytes inhibited insulin receptor signaling (22), we hypothesized that β adrenergic receptor blockade might improve insulin receptor signaling. As expected, in addition to reducing inhibitory IRS-1 phosphorylation, propranolol administration promoted tyrosine phosphorylation of IRS-1 and phosphorylation of Akt. Administration of the chemical chaperone taurine-conjugated derivative (TUDCA) to diabetic animals was shown to reduce ER stress in addition to improving the phosphorylation of both the insulin receptor and Akt after insulin administration (23). Given that propranolol significantly attenuated the ER stress response, we hypothesized that its administration would improve phosphorylation of the insulin receptor. Interestingly, administration of propranolol significantly improved Akt phosphorylation but not the insulin receptor after insulin injection.
Future studies will explore the mechanisms by which catecholamines mediate these effects. Specifically, it is unclear whether catecholamines directly or indirectly cause ER stress-dependent inhibition of PI3K/Akt signaling transduction machinery after burn injury. Studies conducted in cardiomyocytes and neuronal cells indicate that administration of norepinephrine directly induces the ER stress response (12,13). Epinephrine has not yet been shown to induce the ER stress response.
The metabolic changes induced by catecholamines after burn injury have been shown to increase adipose tissue lipolysis, which increases free fatty acids in the liver (10). Accumulation of fat in the liver has been shown to lead to pathogenesis (24). Saturated fatty acids induced ER stress and apoptosis in hepatocytes (25). Further, saturated fatty acids led to sustained JNK activation and inhibition of Akt signaling transduction machinery (19). Administration of a diet high in saturated fat led to ER stress, apoptosis and hepatic damage and dysfunction in rats (26). Further studies will also determine which β adrenergic receptor mediates these effects. The predominant β adrenergic receptor subtypes in the liver are β1 and β2 (27). In cardiomyocytes, inhibition of the β1 receptor attenuated ER stress (28).
Conclusion
In conclusion, attenuation of ER stress and improved insulin sensitivity after propranolol treatment indicates that catecholamine release is responsible for the ER stress response and impaired insulin receptor signaling after burn injury. These findings improve our understanding of ER stress and impaired insulin signaling after burn injury and provide a new therapeutic approach. When translated to the clinical setting, propranolol treatment has the potential to suppress the hypermetabolic response after severe thermal injury, by preventing hepatic ER stress and improving insulin sensitivity.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Bringham PA, McLoughlin E. (1996) Burn incidence and medical care use in the United States: estimates, trends and data sources. J. Burn Care Rehabil. 17:95–107.
Bloemsma GC, Dokter J, Boxma H, Oen IMMH. (2008) Mortality and causes of death in a burn center. Burns. 34:1103–7.
Price LA, Thombs B, Chen CL, Milner SM. (2007) Liver disease in burn injury: evidence from a national sample of 31,338 adult patients. J. Burns Wounds. 7:e1.
Jeschke MG, et al. (2009) Calcium and ER stress mediate hepatic apoptosis after burn injury. J. Cell Mol. Med. 13:1857–65.
Song J, Finnerty CC, Herndon DN, Boehning D, Jeschke MG. (2009) Severe burn-induced endoplasmic reticulum stress and hepatic damage in mice. Mol. Med. 15:316–20.
Gauglitz GG, et al. (2010) Post-burn hepatic insulin resistance is associated with ER stress. Shock. 33:299–305.
Ozcan U, et al. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 306:457–61.
Urano F, et al. (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 287:664–66.
Herschkovitz A, et al. (2007) Common inhibitory serine sites phosphorylated by IRS-1 kinases, triggered by insulin and inducers of insulin resistance. J. Biol. Chem. 282:18018–27.
Gauglitz GG, Herndon DN, Jeschke MG. (2008) Insulin resistance postburn: underlying mechanisms and current therapeutic strategies. J. Burn Care Res. 29:683–94.
Kulp GA, Herndon DN, Lee JO, Suman OE, Jeschke MG. (2010) Extent and magnitude of catecholamine surge in pediatric burned patients. Shock. 33:269–74.
Mao W, et al. (2004) Extracellular norepinephrine reduces neuronal uptake of norepinephrine by oxidative stress in PC12 cells. Am. J. Physiol. Heart Circ. Physiol. 287:H29–39.
Mao W, et al. (2007) Cardiomyocyte apoptosis in autoimmune cardiomyopathy: mediated via endoplasmic reticulum stress and exaggerated by norepinephrine. Am. J. Physiol. Heart Circ. Physiol. 293:H1636–45.
Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies. (2011) Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press. [cited 2012 Jun 11]. Available from: https://doi.org/oacu.od.nih.gov/regs/
Herndon DN, Wilmore DW, Mason AD Jr. (1978) Development and analysis of a small animal model simulating the human postburn hypermetabolic response. J. Surg. Res. 25:394–403.
Aguirre V, Uchida T, Yenush L, Davis R, White MF. (2000) The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J. Biol. Chem. 275:9047–54.
Hirosumi J, et al. (2002) A central role for JNK in obesity and insulin resistance. Nature. 420:333–36.
Previs SF, Withers DJ, Ren J, White MF, Shulman GI. (2000) Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J. Biol. Chem. 275:3890–94.
Solinas G, Naugler W, Galimi F, Lee MS, Karin M. (2006) Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. U. S. A. 103:16454–59.
Ikezu T, Okamoto T, Yonezawa K, Thompkins RG, Martyn JA. (1997) Analysis of thermal injury-induced insulin resistance in rodents: Implication of postreceptor mechanisms. J. Biol. Chem. 272:25289–95.
Sugita H, et al. (2005) Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 288:E585–91.
Morisco C, et al. (2005) Akt mediates the cross-talk between beta-adrenergic and insulin receptors in neonatal cardiomyocytes. Circ. Res. 96:180–88.
Ozcan U, et al. (2006) Chemical chaperones reduce ER Stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 313:1137–40.
Unger RH, Orci L. (2002) Lipoapoptosis: its mechanism and its diseases. Biochim. Biophys. Acta. 1585:202–12.
Wei Y, Wang D, Topczewski F, Pagliassotti MJ. (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 291:E275–81.
Wang D, Wei Y, Pagliassotti MJ. (2006) Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 147:943–51.
Arner P, et al. (1990) Beta-adrenoreceptor subtype expression in human liver. J. Clin. Endocrinol. Metab. 71:1119–26.
George I, Sabbah HN, Xu K, Wang N, Wang J. (2011) Beta-adrenergic receptor blockade reduces endoplasmic reticulum stress and normalizes calcium handling in coronary embolization model of heart failure in canines. Cardiovasc. Res. 91:447–55.
Acknowledgments
The authors would like to thank Paul McKeever for his technical assistance, in addition to Alexandra Smith and Yaeko Hiyama for critically reading the manuscript. This work was supported by grants from Shriners Hospitals for Children (SHG 8660 and 6840), the National Institutes of Health (R01-GM087285-0182, R01-GM56687 and P50-GM60338), CFI Leader’s Opportunity Fund (Project 25407) and the Physicians’ Services Incorporated Foundation, Health Research Grant Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Brooks, N.C., Song, J., Boehning, D. et al. Propranolol Improves Impaired Hepatic Phosphatidylinositol 3-Kinase/Akt Signaling after Burn Injury. Mol Med 18, 707–711 (2012). https://doi.org/10.2119/molmed.2011.00277
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00277