
Abstract
Many tumors present with increased activation of the phosphatidylinositol 3-kinase (PI3K)-PtdIns(3,4,5)P3-protein kinase B (PKB/Akt) signaling pathway. It has long been thought that the lipid phosphatases SH2 domain-containing inositol-5′-phosphatase 1 (SHIP1) and SHIP2 act as tumor suppressors by counteracting with the survival signal induced by this pathway through hydrolysis or PtdIns(3/4/5)P3 to PtdIns(3,4)P2. However, a growing body of evidence suggests that PtdInd(3,4)P2 is capable of, and essential for, Akt activation, thus suggesting a potential role for SHIP1/2 enzymes as proto-oncogenes. We recently described a novel SHIP1-selective chemical inhibitor (3α-aminocholestane (3AC)) that is capable of killing malignant hematologic cells. In this study, we further investigate the biochemical consequences of 3AC treatment in multiple myeloma (MM) and demonstrate that SHIP1 inhibition arrests MM cell lines in either G0/G1 or G2/M stages of the cell cycle, leading to caspase activation and apoptosis. In addition, we show that in vivo growth of MM cells is blocked by treatment of mice with the SHIP1 inhibitor 3AC. Furthermore, we identify three novel pan-SHIP1/2 inhibitors that efficiently kill MM cells through G2/M arrest, caspase activation and apoptosis induction. Interestingly, in SHIP2-expressing breast cancer cells that lack SHIP1 expression, pan-SHIP1/2 inhibition also reduces viable cell numbers, which can be rescued by addition of exogenous PtdIns(3,4)P2. In conclusion, this study shows that inhibition of SHIP1 and SHIP2 may have broad clinical application in the treatment of multiple tumor types.
Similar content being viewed by others


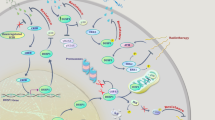
Introduction
Inositol phospholipids play a crucial role in all aspects of cell biology, from cell survival, differentiation and migration, to immune function, organ development and tumor growth. Their production is carefully regulated by a wide range of lipid kinases and phosphatases (1,2). The most studied of these is phosphatidyli-nositol 3-kinase (PI3K), which produces the phosphoinositides (phosphatidyl inositol phosphates [PIPs]) PtdIns(3)P1, PtdIns(3,4)P2 and PtdIns(3,4,5)P3. The last phospholipid [PtdIns(3,4,5)P3] acts as second messenger by binding PH domain-containing proteins such as protein kinase B (PKB/Akt), implicated in cell survival. Many tumors, including breast cancer and hematological malignancies such as the plasma cell neoplasm multiple myeloma (MM), present with constitutive activation of the PI3K-Akt pathway (3). Activating mutations in the PI3K gene (PIK3CA) have been described, but constitutive PI3K signaling may also occur as a secondary effect (4). Activating mutations in receptor tyrosine kinases or Ras, oncogenic translocation products, and cytokine signaling loops have also been described as contributing factors (5,6). Regardless of the activating mechanism, targeting the PI3K/Akt pathway with specific inhibitors is believed to be a promising approach in treating cancer (7,8).
PI3K signaling is generally thought to be counteracted by lipid phosphatases. One of these is the phosphatase and tensin homolog (PTEN), which hydrolyzes PtdIns(3,4,5)P3 to PtdIns(4,5)P2, thereby decreasing the PI3K-induced survival signal and acting as a tumor suppressor (reviewed in [9]). Others are the SH2-domain containing inositol 5′ polyphosphatases: SH2 domaincontaining inositol-5′-phosphatase 1 (SHIP1) and SHIP2, which dephosphorylate inositol lipids at the 5D position of the inositol ring, when a phosphate is present at the 3D position, thereby generating PtdIns(3,4)P2. SHIP1 and SHIP2 share 38% sequence homology, but may have different cellular functions, since SHIP2 is ubiquitously expressed, whereas SHIP1 is predominantly found in cells from the hematopoietic lineage and bone-forming osteolineage cells (10,11).
Recently, a growing body of literature disputes the dogma of lipid phosphatases acting only as tumor suppressors (2,9). In particular, the role of SHIP1 and SHIP2 has become less obvious. It is now clear that both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 are capable of enhancing Akt phosphorylation (12). PtdIns(3,4)P2 may actually be necessary for full activation of Akt, since PtdInd(3,4)P2 is more efficient than PtdIns(3,4,5)P3 at binding the PH domain of Akt, resulting in its phosphorylation on Ser473 and hence its full membrane activity (13,14). Indeed, knockdown of SHIP2 in myoblasts, resulting in increased PtdIns(3,4,5)P3 levels, nevertheless decreased Akt phosphorylation and increased cell death (15). Intriguingly, enhanced SHIP2 expression levels correlating with poor prognosis have been observed in primary breast cancer, and growth of a human breast cancer cell line in a murine model was inhibited by SHIP2 knockdown (16,17). In addition, increased PtdIns(3,4)P2 levels were found in leukemic cells (18). Definitive evidence of SHIP1 enzyme activity and its product PtdIns(3,4)P2 being required for cancer cell survival was provided by our recent description of a SHIP1 inhibitor that selectively causes apoptosis of SHIP1-expressing blood cancer cells, which are in turn protected from this inhibitor by introduction of exogenous PtdIns(3,4)P2 (19). These findings do not rule out a contribution of PtdIns(3,4,5)P3 to cancer cell survival, but simply indicate that both PtdIns(3,4,5)P3 and PtdInd(3,4)P2 are likely required for a cell to achieve and sustain a malignant state, which can be formulated as the “Two PIP hypothesis” (2). The recent demonstration that the 4′ inositol polyphosphatases inositol polyphosphate phosphatase 4A (INPP4A) and INPP4B, which specifically hydrolyze PtdIns(3,4)P2 to PtdIns(3)P, prevent tumor formation and cell transformation further highlights the critical role of the SHIP1/2 product PtdIns(3,4)P2 in malignancy (20,21).
Because a role for SHIP1/2 enzymes as proto-oncogenes is emerging, the use of SHIP inhibitors in SHIP1 and SHIP2 expressing tumors becomes a highly attractive prospect. We recently described a novel selective SHIP1 inhibitor that showed cytotoxicity toward malignant hematologic cancers (19). In the current study, we show that reduced cell growth upon SHIP1 inhibition is a common characteristic in MM cells, further delineate the molecular mechanisms involved in this and demonstrate that SHIP1 inhibitor treatment of mice reduces MM cell growth in vivo. In addition, we describe three novel pan-SHIP inhibitors that are capable of activating caspase 9 and 3 and effectively kill both MM and SHIP2-expressing breast cancer cells.
Materials and Methods
Cell Lines and Material
MM cell lines RPMI8226, U266 and OPM2 (ATCC, Rockville, MD, USA) were routinely maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) (ATCC) supplemented with 10% fetal calf serum (Mediatech, Manassas, VA, USA), whereas MDA-MB-231 and MCF-7 cells were cultured in Eagle’s minimal essential medium (EMEM) with 10% fetal calf serum and l-glutamine. MG-132 was from Sigma Aldrich (St. Louis, MO, USA). PtdIns(3,4,5)P3 and PtdIns(3,4)P2 shuttle PIP kits were purchased from Echelon Biosciences (Salt Lake City, UT, USA) and used per the manufacturer’s instructions.
Detection of Phosphatase Enzymatic Activity
Fluorescent polarization assay (Echelon Biosciences) was used as described previously (19). In short, recombinant SHIP1 or SHIP2 is mixed with its substrate PtdIns(3,4,5)P3 in the presence of potential chemical inhibitors. The reaction product is mixed with PtdIns(3,4)P2 detector protein and a fluorescent PI(3,4)P2 probe. Newly synthesized PtdIns(3,4)P2 displaces the detector protein, thereby enhancing an unbound fluorescent probe in the mixture and decreasing mean polarization units. Thus, identified SHIP inhibitors, (2-phenyl-benzo[/t]quinolin-4-yl)-[2]piperidyl-methanol hydrochloride (1PIE), 1-[(chlorophenyl)methyl]-2-methyl-5-(methylthio)-1H-indole-3-ethanamine hydrochloride (2PIQ) and (2-adamantan-1-yl-6,8-dichloro-quinolin-4-yl)-pyridin-2-yl-methanol hydrochloride (6PTQ) were subsequently tested for inhibition of free phosphate production by recombinant SHIP1 or SHIP2 (Echelon Biosciences) by Malachite Green assay (Echelon Biosciences) as described before (19) or by fluorescent polarization assay. To demonstrate selectivity of the compounds for SHIP1 and SHIP2 over other phosphatases, SHIP1 and the inositol 5-phosphatase oculocerebrorenal syndrome of Lowe (OCRL) were immunoprecipitated from OPM2 cells. For this purpose, OPM2 cells were lysed in IP-lysis buffer (20 mmol/L Tris, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Triton × 100, 1 mmol/L phenylmethylsulfonyl fluoride and Halt protease inhibitor), and SHIP1 or OCRL were immunoprecipitated by using mouse IgG antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Beads were washed four times with immunoprecipitation (IP) lysis buffer and once with Trisbuffered saline (TBS)/MgCl2 (10 mmol/L) and resuspended in TBS/MgCl2. SHIP inhibitors (200 µmol/L) were added to the beads for 5 min, after which immunoprecipitated SHIP1 was incubated in the presence of 100 µmol/L PtdIns(3,4,5)P3 (Echelon Biosciences), whereas immunoprecipitated OCRL was incubated in the presence of 100 µmol/L PtdIns(4,5)P2 for 30 min. Malachite Green solution was added according to the manufacturer’s instructions, and the plate was read after 20 min. Identification of 3α-aminocholestane (3AC) was described previously (19).
Cell Viability Assay
Cells were treated in triplicate or more with increasing concentrations of compounds. Cell viability was determined with a Cell Counting Kit (Dojindo Molecular Technologies, Rockville, MD, USA) per the manufacturer’s instructions. The odds density (OD) of compound-treated cells was divided by the OD of their vehicle control, and the viability was expressed as a percentage of untreated cells. Results are expressed as mean ± standard error of the mean (SEM) of three individual experiments. For PIP add-back experiments, MCF-7 cells were treated for 2 h with 10 µmol/L SHIP inhibitors, after which cells were washed and fresh medium was added. Cells were subsequently cultured in the absence (0 µmol/L) or presence (10 or 20 µmol/L) of either PtdIns(3,4)P2-diC16 (P-3416; Echelon Biosciences) or PtdIns(3,5)P2-diC16 (P-3516; Echelon Biosciences) for 36 h, after which cell viability was determined by the Dojindo Cell Counting Kit.
Staining with Bromodeoxyuridine
Exponentially growing MM cells were treated with compounds as indicated and 10 µmol/L BrdU was added for the last 14 h of treatment. Cells were stained with anti-5-bromo-2′-deoxyuridine-allophycocyanin (anti-BrdU-APC) using the BrdU Flow Cytometry Kit according to the manufacturer’s instructions (BD Bioscience, San Jose, CA, USA) and analyzed by flow cytometry.
Annexin V Staining
Annexin V/propidium iodide (PI) staining was performed using the Annexin V-APC apoptosis detection kit from BD Pharmingen (Sharon, MA, USA) per the manufacturer’s instructions. In short, cells were treated with SHIP inhibitors for 36 h. Cells were harvested, washed twice with ice-cold phosphate-buffered saline and stained with Annexin-APC and PI in binding buffer for 15 min. Fluorescence was determined by flow cytometry (LSRII; Becton Dickinson Medical Systems, Sharon, MA, USA). Results are expressed as mean ± SEM.
OPM2 Tumor Challenge Studies
NOD/SCID/γcIL2R (NSG) mice (The Jackson Laboratory, Bar Harbor, ME, USA) were injected intraperitoneally with 1 × 107 OPM2 cells and 6 h later received an initial injection of 3AC or vehicle. 3AC was suspended in a 0.3% Klucel/H2O solution at 11.46 mmol/L and administered by intraperitoneal injection of 100-µL solution. Vehicle-treated mice received 100-µL injection of 0.3% Klucel/H2O solution. The final concentration of 3AC in the treated mice was 60 µmol/L. The mice were then treated with 3AC or vehicle daily for the next 6 d and then twice per week in the remaining 15 wks of the survival study. In some instances, tumors from the vehicle- or 3AC-treated hosts were excised and single-cell suspensions were made for Western blot analysis of SHIP2 expression after mice were deemed to be moribund and recommended for humane euthanasia by veterinary staff.
Enzyme-Linked Immunosorbent Assay for Human Igλ Light Chain in Mouse Peripheral Blood
Mice were bled into a serum collection tube (Microvette 300Z; Sarstedt, Numbrecht, Germany) 4 wks after the OPM2 challenge, and serum was obtained after pelleting of blood cells at 5,000g for 5 min. Human Igλ light chain amounts were determined using an Ig light chain detection kit from Biovendor (Chandler, NC, USA) per the manufacturer’s instructions.
Detection of Circulating OPM2 Cells in Mouse Blood
Mice were bled into a blood collection tube (Microvette 300Z, Sarstedt, Numbrecht, Germany) 4 wks after OPM2 challenge and red cells were lysed. White blood cells were incubated with anti-CD16/32 to block Fc receptor binding and then stained with antibodies against human HLA-ABC, clone W6/32. Samples were acquired on an LSRII cytometer (Becton Dickinson), and dead cells were excluded from the analysis after cytometer acquisition by exclusion of cells that stained positively for DAPI (di aminido phenyl indol).
Western Blot Analysis
Cells were treated as described and lysed in cell Laemmli buffer. Protein concentration was determined by RC/DC protein assay (Pierce, Rockford, IL, USA) according to the manufacturer’s description. Immunoblotting was performed as described (22). Detection was performed according to the manufacturer’s guidelines (ECL, Pierce, Rockford, IL, USA). All phospho-antibodies were from Cell Signaling Technology (Beverly, MA, USA). SHIP1 P1C1 and actin antibodies were from Santa Cruz Biotechnology. For quantitative Western blot analysis, gels were blotted on Immobilon-FL transfer membrane (Millipore, Billerica, MA, USA). Anti-rabbit or anti-mouse IRDye-conjugated secondary antibodies were used according to the manufacturer’s directions, and blots were scanned by Odyssey infrared imaging (LI-COR Biosciences, Lincoln, NE, USA). Analysis of results was done using Odyssey 3.0 software.
Statistical Analysis
Statistical analysis was performed using either GraphPad Prism 5 or SPSS 17 software. The effect of inhibitors on cell viability was determined by Student t test for paired samples, and comparisons between inhibitors were performed with an independent samples t test. Increases in Annexin V-positive cells upon treatment with inhibitors was calculated by a Student t test for paired samples. Mouse survival curves were compared by log-rank (Mantel-Cox) test. Statistical analysis of comparison of serum Igλ free chain and percentage of circulating OPM2 cells in 3AC- and vehicle-treated mice were performed by an independent samples t test.
All supplementary materials are available online at www.molmed.org .
Results
Inhibition of SHIP1 Reduces Cell Viability of MM Cells Through Different Mechanisms
Activation of SHIP1 has been shown to have antitumorigenic effects in MM cells. However, because both the SHIP substrate PtdIns(3,4,5)P3 and its product PtdIns(3,4)P2 are capable of activating the Akt survival pathway in MM cells (Figure 1A), it is conceivable that inhibition of SHIP1 may also lead to cell death. Indeed, we previously demonstrated a cell growth inhibitory effect of SHIP1 inhibition on human MM OPM2 cells. Because MM is a heterogeneous disease, we tested whether other SHIP1-expressing MM cell lines would be equally affected. As shown earlier, OPM2 cell viability was effectively reduced by 3AC treatment. RPMI8226 and U266 cells showed significantly less sensitivity to 3AC treatment when compared with OPM2 cells, although viability was decreased significantly at concentrations of ≥12.5 µmol/L (Figure 1B).

SHIP1 inhibition reduces viable cell numbers and either G2/M or G0/G1 cell cycle arrest. (A) U266 MM cells were treated for 10 min with increasing concentrations of PtdInd(3,4)P2 or PtdIns(3,4,5)P3, using shuttle PIP kit as described. Cells were subsequently lysed and proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Blots were probed with antibodies against phosphorylated Akt. Equal loading was confirmed by reprobing the blots with antibodies against actin. (B) OPM2, U266 and RPMI8226 cells were treated for 48 h with increasing concentrations of 3AC, and viable cell numbers were determined by a cell-counting kit. The means of at least three independent experiments are shown. Asterisks indicate significant differences between OPM2 cells and U266 and RPMI8226 cells. —●—, OPM2; —■—, RPMI8226; —▲—, U266. (C) MM cell lines were cultured in the presence or absence of 12.5 µmol/L 3AC of 48 h and 10 µmol/L BrdU for 14 h. Cells were subsequently labeled with APC-anti-BrdU and 7AAD, and cell cycle status was analyzed by flow cytometry. A representative example of two independent experiments is shown. conc, concentration. ▦, sub G0/G1; ▩, G0/G1; ▤, G2/M; ▥, S.
To investigate the molecular mechanisms responsible for the reduced viable cell numbers upon 3AC treatment, we determined the effect of SHIP1 inhibition on cell cycle progression by BrdU incorporation and 7-amino-actinomycin D (7AAD) staining. OPM2 cells are highly proliferative, with up to 75% of cells undergoing S phase during the 14 h of BrdU exposure. Treatment with 3AC for 36 h severely reduced the percentage of cells in the S phase, which was accompanied by an increase of cells in the G2/M phase (Figure 1C). In contrast, in the less proliferative RPMI8226 and U266 cells, cell cycle progression is blocked in the G0/G1 phase upon 3AC treatment, in conjunction with a reduced percentage of cells undergoing the S phase. These results indicate that inhibition of SHIP1 affects different MM cells in different stages of their cell cycle, leading to a reduced number of viable cells upon 3AC treatment.
SHIP1 Inhibition Causes Caspase Activation and Apoptosis in MM Cells
Next, we investigated the potential of 3AC to induce programmed cell death (apoptosis) in MM cells, which may be one of the consequences of cell cycle arrest. As shown in Figure 2A, a 2.7-fold increase in the percentage of Annexin V-positive cells was observed in OPM2 cells upon 36 h of 3AC exposure (268% ± 47%, P = 0.03), whereas little induction of apoptosis was detected in U266 (160% ± 3%, P = 0.01) and RPMI8226 cells (103% ± 12%, P = 0.8). These differences were also reflected in a biochemical analysis of apoptosis. Intrinsic apoptosis is mediated through release of mitochondrial cytochrome c, which activates a caspase cascade, starting with the cleaving of procaspase 9 to its active form and resulting in the cleaving and activation of caspase 3. Poly (ADP-ribose) polymerase (PARP), involved in DNA repair in response to environmental stress, is one of the main cleavage targets of caspase 3 and acts as an important hallmark of apoptosis. Consistent with our cell viability and apoptosis data, cleaving of caspase 9, caspase 3 and PARP was evident in OPM2 cells upon SHIP1 inhibition (Figure 2B). In contrast, 3AC treatment did not increase levels of cleaved PARP in RPMI8226 and U266 cells, although some cleaving of caspase 3 was observed in the latter. Interestingly, we also observed a conversion of light chain 3 (LC3)B-I to LC3B-II upon 3AC treatment of MM cells, which is considered to be a hallmark of autophagy.

SHIP1 inhibition affects apoptosis induction differently in MM cell lines. (A) MM cells were treated with increasing concentrations of 3AC for 48 h and Annexin V-positive cells were determined by fluorescence-activated cell sorter (FACS) analysis. Results represent the mean increase in apoptotic cells in two independent experiments. ▤, U266; ▦, RPMI8226; ▩, OPM2. (B) MM cells were treated for 48 h with increasing concentrations of 3AC. After cell lysis, proteins were separated by SDS-PAGE and blots were probed with antibodies against hallmark proteins for apoptosis or autophagy (upper panel: cleaved caspase 3, cleaved caspase 9, LC3B-II; lower panel: cleaved PARP). Equal loading was confirmed by reprobing blots with antibodies against actin. conc, concentration.
SHIP1 Inhibition Results in Proteasome-Dependent Degradation of SHIP1 Protein
To further elucidate the molecular mechanism by which 3AC may affect cell signaling and viability, we investigated the expression of SHIP1, SHIP2 and PTEN protein in 3AC-treated MM cells (Figure 3A). Surprisingly, prolonged exposure of MM cells to 3AC reduced SHIP1 protein levels, without affecting either SHIP2 or PTEN in U266 or RPMI8226 cells. In OPM2 cells, SHIP2 levels were slightly upregulated, possibly as a compensatory mechanism. Cotreatment of cells with the proteasome inhibitor MG-132 prevented the reduction in SHIP1 expression, suggesting that proteasomal degradation of ubiquitinated SHIP1 is enhanced by 3AC treatment (Figure 3B). Thus, treatment of MM cells with the SHIP1 inhibitor results in the downregulation of SHIP1 in a proteasome-dependent fashion, which may contribute to the cytotoxic effects on MM cells after long-term exposure to this compound.

Treatment of MM cells with 3AC results in proteasomal degradation of SHIP1. (A) MM cells were treated for 48 h with increasing concentrations of 3AC. After cell lysis, proteins were separated by SDS-PAGE and blots were probed with antibodies against SHIP1, SHIP2 and PTEN. Equal loading was confirmed by reprobing blots with antibodies against actin. (B) MM cells were treated for 48 h with 10 µmol/L 3AC in the absence or presence of 10 µmol/L MG-132. After cell lysis, proteins were separated by SDS-PAGE and blots were probed with antibodies against SHIP1. Equal loading was confirmed by reprobing blots with antibodies against actin. conc, concentration.
SHIP1 Inhibition Reduces MM Cell Growth In Vivo
We previously showed that 3AC treatment reduces basal- and insulinlike growth factor (IGF)-1-induced activation of Akt in OPM2 cells (19) and now demonstrate that SHIP1 inhibition results in cell death through apoptosis. We sought then to determine if the observed in vitro effects on cancer cell survival were sufficiently sustained and robust to enable abrogation of MM growth in vivo. For this analysis, we used a tumor xenograft model of MM. We challenged NOD/SCID/ γcIL2R (NSG) mice with OPM2 cells and then initiated SHIP1 inhibitor or vehicle treatment. Mice received an initial injection of either 3AC or vehicle 6 h after injection of MM cells and then daily for 1 wk (induction phase) followed by biweekly injections of 3AC or vehicle for the subsequent 15 wks of the study (maintenance phase). OPM2 is an IgGλ-secreting cell line, and the amount of free human Igλ light chain in the serum of mice is therefore a clear indication of tumor burden. We found that 3AC results in reduced MM growth in vivo, as determined by quantitation of free human Igλ light chain in the plasma after OPM2 challenge (Figure 4A). In addition, reduced numbers of circulating OPM2 cells, as determined by human HLA-ABC staining, was observed in peripheral blood from 3AC-treated mice compared with vehicle controls (Figures 4B, C). Most importantly, 3AC treatment results in significantly enhanced survival of mice after tumor challenge (Figure 4D). Thus, treatment with a SHIP1 selective inhibitor (3AC) significantly abrogated MM tumor growth in vivo as well as in vitro. In 3AC-treated mice that resisted treatment, we found that MM tumors exhibited an upregulation of SHIP2 (Figure 4E), reminiscent of in vitro treatment of OPM2 cells (see Figure 3A) and suggesting that SHIP1 inhibition may select for tumor cells with increased SHIP2 expression.

3AC treatment inhibits MM cell growth in vivo. NOD/SCID/γcIL2R (NSG) mutant mice were challenged with 1 × 107 MM cells (OPM2) by intraperitoneal injection. Six h later, the mice received their first injection of 3AC, followed by a daily dose for the next 7 d and then a biweekly dose for the ensuing 15 wks. (A) Enzyme-linked immunosorbent assay quantitation of human (Hu) λ light chain present in the serum of OPM2 challenged mice (3AC- or vehicle-treated) and unchallenged mice as indicated. (B) Representative FACS detection and quantitation of human MM cells in the circulation of NSG mice is indicated. (C) Box and whisker plots showing the mean, range and standard deviation for MM detected by flow cytometry in the indicated electronic gate in (B). (D) Survival of 3AC and vehicle treatment (Veh) groups that were challenged with MM cells (n = 11/group). (E) Western blot analysis of SHIP2 expression in MM tumors recovered from vehicle-treated (Veh) or 3AC-treated mice. PBMC, peripheral blood mononuclear cells.
Novel Pan-SHIP1/2 Inhibitors Reduce Cell Viability MM Cells
With the exception of primary osteolineage cells, SHIP1 expression is limited to cells from the bone marrow and blood. To investigate a potential wider use for SHIP inhibitors in the treatment of cancer, we searched for inhibitors of SHIP2. Our initial high throughput screening of chemical libraries established 3AC as a selective inhibitor of SHIP1, but also identified three other compounds that were capable of inhibiting SHIP1 activity as determined by fluorescence polarization assays (Figures 5A–C, panels i and ii) (that is, 1PIE, 2PIQ and 6PTQ). However, whereas 3AC showed high selectivity for SHIP1 as determined by Malachite Green phosphatase assay (19), these latter compounds also showed high inhibitory activity against SHIP2 (Figures 5A–C, panel iii). The SHIP1/2 half-maximal inhibitory concentration (IC50) values for 1PIE, 2PIQ and 6PTQ are 30 µmol/L/30 µmol/L, 500 µmol/L/500 µmol/L and 63 µmol/L/35 µmol/L, respectively. To demonstrate selectivity of 1PIE, 2PIQ and 6PTQ over other lipid phosphatases, we precipitated the inositol 5-phosphatase OCRL from OPM2 cells and tested its ability to dephosphorylate PtdIns(4,5)P2 in the presence of pan-SHIP inhibitors by Malachite Green assay. In parallel, SHIP1 was precipitated from OPM2 cells, and its capacity for PtdIns(3,4,5)P3 dephosphorylation was measured in the absence or presence of 1PIE, 2PIQ and 6PTQ. As shown in Figure 5D, 1PIE, 2PIQ and 6PTQ were highly selective for recombinant SHIP2 and precipitated SHIP1, with little effect on OCRL phosphatase activity.

Identification of pan-SHIP inhibitors. (A–C) (i) Structures of 1PIE (A), 2PIQ (B) and 6PTQ (C). (ii) Fluorescence polarization assay shows inhibition of recombinant SHIP1 activity by 100 µmol/L 1PIE (A), 2PIQ (B) and 6PTQ (C). The decrease in mean polarization units without compound (−) is inhibited by adding 1PIE, 2PIQ or 6PTQ to the reaction mix (+). (iii) Comparison of efficiency of recombinant SHIP1 and SHIP2 inhibition by increasing concentrations of inhibitors, as determined by either Malachite Green assay (2PIQ, B) or fluorescence polarization assay (1PIE [A] and 6PTQ [C]). (D) Recombinant SHIP2 and SHIP1 precipitated from OPM2 cell lysates were allowed to dephosphorylate PtdIns(3,4,5)P3 in the absence (DMSO) or presence of inhibitors. OCRL, precipitated from OPM2 cell lysates, was allowed to dephosphorylate PtdIns(4,5)P2 in the presence of inhibitors or DMSO control. Free phosphate groups were detected with Malachite Green reaction. The percentage inhibition of phosphatase activity in two independent immunoprecipitation experiments is represented (means ± SEM).
Using these compounds, we then set out to investigate the effect of pan-SHIP1/2 inhibition on tumor cell growth. As shown in Figures 6A–C, a significant reduction in viable MM cell numbers was observed with all compounds. Although cell killing reached statistical significance by 7.5 µmol/L for all inhibitors used, 2PIQ was less effective in all MM cell lines tested. RPMI8226 cells showed a significantly lower sensitivity to all pan-SHIP inhibitors when compared with OPM2 and U266 cells, which behaved essentially identical. As may be expected, MM cell lines showed a higher sensitivity to the pan-SHIP inhibitors, compared with the SHIP1-selective compound 3AC (compare Figures 6A–C with Figure 1B).

Pan-SHIP1/2 inhibitors reduce viable cell numbers of SHIP2-expressing MM cells. RPMI8226 (A), U266 (B) and OPM2 (C) cells were treated with increasing concentration of pan-SHIP inhibitors for 36 h, and cell viability was determined with a cell counting kit. The mean of three independent experiments is shown. Asterisks indicate the concentration (conc) at which cell killing became significant for each compound. Double asterisks indicate at which concentrations 2PIQ showed significantly less cell killing than 1PIE or 6PTE. —●—, 1PIE; —■—, 6PTQ; —▲—2PIQ. (D) Untreated MM (RPMI8226, U266 and OPM2) cells and breast cancer cells (MCF-7 and MDA-MB-231) were lysed, and proteins were separated by SDS-PAGE, after which the blots were probed with antibodies against SHIP1, SHIP2 and PTEN. Equal loading was confirmed by reprobing the blots with antibodies against actin. conc, concentration.
Cell Cycle Arrest and Apoptosis Induction by Pan-SHIP Inhibition in MM Cells
Next, we aimed to investigate the molecular effect of pan-SHIP inhibition on MM cells. We chose to focus these studies on 1PIE, since this compound is one of the most efficient cytotoxic agents and showed high selectivity toward SHIP2. Treatment of OPM2, RPMI8226 and U266 cells with 10 µmol/L 1PIE resulted in a G2/M cell cycle arrest in all cell lines studied, thereby reducing the amount of cells undergoing the S phase (Figure 7A). In addition, we observed a drastic increase in sub-G0/G1 phase cells, an indication of considerable onset of apoptosis. This result was confirmed by Annexin V staining of 1PIE-treated MM cells (Figure 7B). Consistent with cell survival data, an increase in apoptotic cells was found in all cell lines tested. RPMI8226 cells, which were the least susceptible to pan-SHIP1/2 inhibitors in cell viability assays, also showed the least increase in percentage of Annexin V-positive cells (191% ± 41%, P = 0.16), followed by U266 (555% ± 70%, P = 0.02) and OPM2 cells (603% ± 90%, P = 0.03). Initiation of apoptosis was accompanied by an increased presence of cleaved caspase 9, caspase 3 and PARP, implying activation of the intrinsic apoptosis pathways in all these cell lines in response to exposure to increasing concentrations of 1PIE (Figure 7C). In addition, cleaving of LC3 was observed, indicative of autophagic cell death mechanisms (see Figure 7C).

Pan-SHIP inhibitor 1PIE causes cell cycle arrest and induces apoptosis in MM cells. (A) MM cell lines were cultured in the presence or absence of 10 µmol/L 1PIE for 48 h and 10 µmol/L BrdU for 14 h. Cells were subsequently labeled with APC-anti-BrdU and 7AAD, and cell cycle status was analyzed by flow cytometry A representative example of two independent experiments is shown. ▩, sub G0/G1; ▦ G0/G1; ▤, G2/M; ▥, S. (B) MM cells were treated with increasing concentrations of 1PIE for 48 h and Annexin V-positive cells were determined by FACS analysis. Results represent the mean increase in apoptotic cells in two independent experiments. ▤, U266; ▩, RPMI8226; ▦, OPM2. (C) MM cells were treated for 48 h with increasing concentrations of 1PIE. After cell lysis, proteins were separated by SDS-PAGE, and blots were probed with antibodies against cleaved caspase 3, cleaved caspase 9, cleaved LC3B-II and cleaved PARP. Equal loading was confirmed with antibodies against actin. conc, concentration.
Taken together, these data show that inhibition of SHIP1/2 in MM cells results in a G2/M cell cycle arrest, followed by caspase cascade activation and ending in extensive apoptosis.
Pan-SHIP Inhibitors Reduce Cell Viability of SHIP2 Expressing Breast Cancer Cells
We next sought to determine whether inhibition of SHIP may have broader applicability in cancer treatment. For this purpose, we studied the effect of pan-SHIP inhibition on cell viability in SHIP2-expressing breast cancer cells. Two well-described cell lines, MDA-MB-231 and MCF-7, which do not express SHIP1 (Figure 6D), were resistant to treatment with 3AC (Figures 8A, B), consistent with the SHIP1 selectivity of this compound. Excitingly, however, cell viability was severely reduced by treatment of breast cancer cells with increasing concentrations of 1PIE, 2PIQ or 6PTQ, showing that targeting SHIP2 activity may be a viable objective for the treatment of breast cancer.

Pan-SHIP inhibitors reduce viable cell numbers of SHIP2-expressing breast cancer cells. MDA-MB-231 (A) and MCF-7 (B) cells were treated with indicated concentrations of pan-SHIP inhibitors for 2 h. Cells were washed twice, and fresh medium was added. Viable cell number was determined with a cell counting kit after 24 h, and results represent the mean of at least two independent experiments. —●—, 3AC; —■—, 1PIE; —▲—, 6PTQ; —▼—, 2PIQ. (C–E) After treatment of MCF-7 cells with 10 µmol/L 1PIE, 2PIQ or 6PTQ for 2 h, cells were subsequently cultured for 36 h in the absence or presence of the indicated concentrations of PtsIns(3,4)P2 or PtdIns(3,5)P2, after which cell viability was determined with a cell counting kit. The mean of three independent experiments is shown. —●—, PI(3,4)P2; —■—, PI(3,5)P2. (F) MCF-7 cells were treated with 10 µmol/L 3AC for 2 h, after which the medium was replaced and cells were cultured in the absence or presence of indicated concentrations of PtsIns(3,4)P2 or PtdIns(3,5)P2 for 36 h. Cell viability was determined with a cell counting kit. —●—, PI(3,4)P2; —■—, PI(3,5)P2.
Exogenous PtdIns(3,4)P2 Rescues Breast Cancer Cells from Pan-SHIP Inhibitors
As for 3AC treatment of OPM2 MM cells (19), we observed a diminished IGF-1-induced phosphorylation of Akt in the presence of 1PIE, 2PIQ or 6PTQ in MDA-MB-231 cells and to a lesser degree in MCF-7 cells (Supplementary Figure 1). To prove an involvement of PtdIns(3,4)P2 in the reduced viability of breast cancer cell lines by pan-SHIP inhibitors, MCF-7 breast cancer cells were cultured in the presence of 10 µmol/L 1PIE, 2PIQ or 6PTQ, thereby reducing viable cell levels to around 20%. Adding increasing concentrations of exogenous PtdIns(3,4)P2 rescued cells from treatment with all pan-SHIP inhibitors, albeit to a different extent for each compound (Figures 8C–E). In contrast, exogenous addition of the phospholipid PtdIns(3,5)P2, which is not involved in PI3K signaling, did not rescue MCF-7 cells from treatment with either 1PIE, 2PIQ or 6PTQ. In addition, exogenous PtdIns(3,4)P2 or PtdIns(3,5)P2 had little effect on MCF-7 growth when SHIP2 was not inhibited, that is, in the presence of the selective SHIP1 inhibitor 3AC (Figure 8F). Together, these results confirm the importance of reduced PtdIns(3,4)P2 levels in the cytotoxic effect of pan-SHIP inhibition in breast cancer cells.
Discussion
The prevalent belief that the lipid phosphatases SHIP1 and SHIP2 act solely to counteract PI3K-induced survival signals (23) is slowly being challenged (2,19). PtdIns(3,4)P2 was demonstrated to have a higher affinity for Akt than PtdIns(3,4,5)P3, (14), and in accordance with this finding, several studies have shown that reduction of SHIP2 activity may result in decreased, rather then increased, Akt activity, leading to reduced proliferation and enhanced cell death (15,24). SHIP2 overexpression has been described in primary breast cancer, and SHIP2 is required for the prosurvival signal initiated by epidermal growth factor receptor in this tumor type (25). We now describe three novel pan-SHIP inhibitors, which were able to effectively kill two breast cancer cell lines in addition to three MM cell lines. Although we are not the first to identify SHIP2 inhibitors, to the best of our knowledge, we are the first to present data suggesting SHIP2 enzymatic inhibition may indeed be a useful tool in the treatment of breast cancer. Because breast cancer remains one of the most commonly diagnosed tumor types, and depending on disease stage, remains incurable in 7–85% of patients (www.cancer.org, national cancer database), our findings may present a step forward in the ongoing search for novel, more effective treatments.
A role for SHIP inhibition in MM was suggested by our previous studies showing cell killing of a MM cell line with the SHIP1 inhibitor 3AC (19). We now extend these findings to a wider cohort of MM cell lines and show the ability of 3AC to inhibit MM cell growth in an in vivo mouse model. Furthermore, we demonstrate that in addition to SHIP1, SHIP2 may present a target for treatment. MM, a clonal plasma cell neoplasm leading to renal failure, hypercalcemia and skeletal destruction, presents with very heterogeneous clinical behavior (26). It is therefore interesting to note that 3AC treatment exerts different mechanistic effects on different MM cell lines. We find a G2/M cell cycle block in OPM2 cells, with caspase cascade activation, PARP cleavage and apoptosis induction. In contrast, RPMI8226 and U266 cells were blocked in the G0/G1 phase of cell cycle, but showed less induction of apoptosis. These results are reminiscent of a previous study in which treatment of myeloid leukemia cells with the natural compound apigenin resulted in G2/M arrest followed by classic apoptosis, whereas the same compound caused a G0/G1 block in an erythroid leukemia cell line (27), which corresponded to autophagy rather than apoptosis. Autophagy, or autophagocytosis, is a reversible process in which intracellular components are sequestered and degraded in double membrane autophagosomes (28). This strategy may serve to temporarily protect cells from cytotoxic stimuli, but can progress to apoptosis after prolonged cellular stress (29). The number of autophagosomes correlates to the amount of lipidated LC3B-II protein found in the cell and is generally considered to be a hallmark of autophagy (30). We found increased LC3B-II expression in all MM cells upon treatment with 3AC and pan-SHIP inhibitors. Interestingly, PTEN positively regulates autophagy, and loss of PTEN has been shown to inhibit autophagy without affecting LC3B conversion (31). It is therefore conceivable that OPM2 cells, which are PTEN deficient (Figures 3A and 6D), are incapable of properly initiating the autophagic process, thus directly diverting to apoptosis, whereas the PTEN-expressing U266 and RPMI8226 cells are relatively more resistant to 3AC-induced cell death by engaging the autophagic pathway. Additionally, another explanation might be found in Maiso et al. (32), who found that inhibition of IGF signaling, resulting in reduced Akt activity and G0/G1 cell cycle arrest, leads to caspase 3−, 8− and 9-independent apoptosis in PTEN-competent MM cell lines.
PTEN deficiency in OPM2 cells results in increased constitutive Akt phosphorylation compared with RPMI8226 and U266 cells (33 and our own unpublished observations). It is possible that as a result, this cell line has become more dependent on Akt-activated survival signals and is therefore more susceptible to perturbations in the levels of phospholipids governing this pathway. Consistent with the “Two PIP hypothesis,” the ratio and absolute amounts of both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 may be critical to cancer cell survival and growth, where a change in the level of either PIP species could cause cell death. This may also explain why stimulation of SHIP1 activity with a specific agonist also caused apoptosis in MM cell lines, including OPM2 and RPMI8226 (34).
Our in vitro studies with chemical inhibition of SHIP1 in blood cancer cells predicted that SHIP inhibition, because of its negative impact on PtdIns(3,4)P2 levels and Akt/PKB activation in cancer cells, is sufficiently cytotoxic for MM cells such that MM tumor growth in vivo might be abrogated. Here we demonstrate that chemical inhibition of SHIP1 can eliminate residual disease in a lethal MM tumor model. Our results with chemical inhibition of SHIP suggest that SHIP has a tumor-promoting role in human myeloid leukemias and MM via its synthesis of PtdIns(3,4)P2, whereas other recent studies indicate SHIP can serve as a tumor suppressor in murine virally induced erythroid leukemia (35) and murine B cell lymphoma (36). In these latter models, it will be critical to determine whether inappropriate induction of s-SHIP or SHIP2 expression has occurred in these SHIP1 mutant tumor models and thus provided a compensatory enzymatic source of PtdIns(3,4)P2. Our finding that increased SHIP2 expression is selected in 3AC-resistant MM tumors is consistent with a “paralogue compensation” model. Intriguingly, viral infection can induce inappropriate expression of s-SHIP in lineage committed hematopoietic cells (37), suggesting that “isoform compensation” by s-SHIP may also be possible in virally induced leukemia models. Alternatively, survival of erythroid leukemias and B lymphomas may be more dependent on the presence of PtdIns(3,4,5)3 than PtdIns(3,4)P2, whereas myeloid leukemias and MM survival may be PtdIns(3,4)P2 biased. In all likelihood, most cancers must sustain a certain threshold of both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 to maintain their malignant state—the “Two PIP hypothesis” (2).
Aside from being a phosphatase, SHIP1 also functions to mask receptor tails to prevent recruitment of other signaling proteins (38,39), or as an adaptor protein for proteins such as Shc, DOK1 and Grb2, and as such has been proposed to reduce Ras signaling (40). Theoretically, it is possible that while blocking phosphatase activity with 3AC, these other functions of SHIP1 may not be affected. However, we observed a decrease in SHIP1 protein expression in MM cells upon prolonged treatment with 3AC, suggesting that these scaffolding functions may no longer play a role. It has recently been shown that SHIP-1 is ubiquitinated and targeted for proteasomal degradation upon its phosphorylation (41). However, we did not observe a difference in IGF-1-stimulated SHIP1 phosphorylation in MM cells after pretreatment with 3AC (unpublished observations, GM Fuhler). Hence, the reason for the proteasomal degradation of SHIP1 upon 3AC treatment remains unclear.
Conclusion
In conclusion, we describe three novel pan-SHIP inhibitors and describe the cell-killing effects of SHIP1/2 inhibition in MM and breast cancer models. The widespread expression of SHIP2 suggests that inhibition of SHIP2 may find a broader clinical application, whereas the use of SHIP1 inhibitors may have the benefit of surpassing innocent bystander effects and therefore be more readily used in the treatment of hematological malignancies.
Disclosure
WG Kerr is the inventor or coinventor on patents, both issued and pending, related to modulation of SHIP expression and activity for therapeutic purposes. JD Chisholm is a coinventor on a pending patent related to the use of 3AC derivatives in cancer.
References
Yuan TL, Cantley LC. (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene. 27:5497–510.
Kerr WG. (2011) Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann. N. Y. Acad. Sci. 1217:1–17.
Alkan S, Izban KF. (2002) Immunohistochemical localization of phosphorylated AKT in multiple myeloma. Blood. 99:2278–79.
Troxell ML, et al. (2010) High prevalence of PIK3CA/AKT pathway mutations in papillary neoplasms of the breast. Mod. Pathol. 23:27–37.
Dey JH, et al. (2010) Targeting fibroblast growth factor receptors blocks PI3K/AKT signaling, induces apoptosis, and impairs mammary tumor outgrowth and metastasis. Cancer Res. 70:4151–62.
Montero JC, Lopez-Perez R, San Miguel JF, Pandiella A. (2008) Expression of c-Kit isoforms in multiple myeloma: differences in signaling and drug sensitivity. Haematologica. 93:851–9.
Bunney TD, Katan M. (2010) Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat. Rev. Cancer. 10:342–52.
Harvey RD, Lonial S. (2007) PI3 kinase/AKT pathway as a therapeutic target in multiple myeloma. Future Oncol. 3:639–47.
Ruela-de-Sousa RR, Queiroz KC, Peppelenbosch MP, Fuhler GM. (2010) Reversible phosphorylation in haematological malignancies: potential role for protein tyrosine phosphatases in treatment? Biochim. Biophys. Acta. 1806:287–303.
Hazen AL, et al. (2009) SHIP is required for a functional hematopoietic stem cell niche. Blood. 113:2924–33.
Rohrschneider LR, Fuller JF, Wolf I, Liu Y, Lucas DM. (2000) Structure, function, and biology of SHIP proteins. Genes Dev. 14:505–20.
Scheid MP, et al. (2002) Phosphatidylinositol (3,4,5)P3 is essential but not sufficient for protein kinase B (PKB) activation; phosphatidylinositol (3,4)P2 is required for PKB phosphorylation at Ser-473: studies using cells from SH2-containing inositol-5-phosphatase knockout mice. J. Biol. Chem. 277:9027–35.
Ma K, Cheung SM, Marshall AJ, Duronio V. (2008) PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell Signal. 20:684–94.
Franke TF, Kaplan DR, Cantley LC, Toker A. (1997) Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bispho-sphate. Science. 275:665–8.
Mandl A, Sarkes D, Carricaburu V, Jung V, Rameh L. (2007) Serum withdrawal-induced accumulation of phosphoinositide 3-kinase lipids in differentiating 3T3-L6 myoblasts: distinct roles for Ship2 and PTEN. Mol. Cell. Biol. 27:8098–112.
Prasad NK, et al. (2008) High expression of obesity-linked phosphatase SHIP2 in invasive breast cancer correlates with reduced disease-free survival. Tumour Biol. 29:330–41.
Prasad NK. (1996) SHIP2 phosphoinositol phosphatase positively regulates EGFR-Akt pathway, CXCR4 expression, and cell migration in MDA-MB-231 breast cancer cells. Int. J. Oncol. 34:97–105.
Jain SK, et al. (1996) PI 3-kinase activation in BCR/abl-transformed hematopoietic cells does not require interaction of p85 SH2 domains with p210 BCR/abl. Blood. 88:1542–50.
Brooks R, et al. (2010) SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J. Immunol. 184:3582–9.
Gewinner C, et al. (2009) Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 16:115–25.
Ivetac I, et al. (2009) Regulation of PI(3)K/Akt signalling and cellular transformation by inositol polyphosphate 4-phosphatase-1. EMBO Rep. 10:487–93.
Fuhler GM, et al. (2004) Disturbed granulocyte macrophage-colony stimulating factor priming of phosphatidylinositol 3,4,5-trisphosphate accumulation and Rac activation in fMLP-stimulated neutrophils from patients with myelodysplasia. J. Leuk. Biol. 76:254–62.
Hamilton MJ, et al. (2011) Role of SHIP in cancer. Exp. Hematol. 39:2–13.
Artemenko Y, Gagnon A, Sorisky A. (2009) Catalytically inactive SHIP2 inhibits proliferation by attenuating PDGF signaling in 3T3-L1 preadipocytes. J. Cell Physiol. 218:228–36.
Prasad NK, Tandon M, Badve S, Snyder PW, Nakshatri H. (2008) Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis. 29:25–34.
Pantou D, et al. (2005) Cytogenetic manifestations of multiple myeloma heterogeneity. Genes Chromosomes Cancer. 42:44–57.
Ruela-de-Sousa RR, et al. (2010) Cytotoxicity of apigenin on leukemia cell lines: implications for prevention and therapy. Cell Death Dis. 1:e19.
Mehrpour M, Esclatine A, Beau I, Codogno P. (2010) Overview of macroautophagy regulation in mammalian cells. Cell Res. 20:748–62.
Pardo R, et al. (2010) Gemcitabine induces the VMP1-mediated autophagy pathway to promote apoptotic death in human pancreatic cancer cells. Pancreatology. 10:19–26.
Kabeya Y, et al. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19:5720–8.
Ueno T, et al. (2008) Loss of Pten, a tumor suppressor, causes the strong inhibition of autophagy without affecting LC3 lipidation. Autophagy. 4:692–700.
Maiso P, et al. The insulin-like growth factor-I receptor inhibitor NVP-AEW541 provokes cell cycle arrest and apoptosis in multiple myeloma cells. Br. J. Haematol. 141:470–82.
Shi Y, et al. (2002) Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 62:5027–34.
Kennah M, et al. (2009) Activation of SHIP via a small molecule agonist kills multiple myeloma cells. Exp. Hematol. 37:1274–83.
Lakhanpal GK, et al. (2009) The inositol phosphatase SHIP-1 is negatively regulated by Fli-1 and its loss accelerates leukemogenesis. Blood. 116:428–36.
Miletic AV, et al. (2010) Coordinate suppression of B cell lymphoma by PTEN and SHIP phosphatases. J. Exp. Med. 207:2407–20.
Avota E, Harms H, Schreider-Schaulies S. (2006) Measles virus inducec expression of SIP110, as constitutively membrane clustered lipid phosphatase, which inhibits T cell proliferation. Cell. Microbiol. 8:1826–39.
Wahle JA, et al. (2007) Inappropriate recruitment and activity by the Src homology region 2 domain-containing phosphatase 1 (SHP1) is responsible for receptor dominance in the SHIP-deficient NK cell. J. Immunol. 179:8009–15.
Peng Q, et al. (2010) TREM2- and DAP12-dependent activation of PI3K required DAP10 and is inhibited by SHIP1. Sci. Signal. 3:ra38.
Gratacap MP, Severin S, Chicanne G, Plantavid M, Payrastre B. (2008) Different roles of SHIP1 according to the cell context: the example of blood platelets. Adv. Enzyme Regul. 48:240–52.
Ruschmann J, et al. (2010) Tyrosine phosphorylation of SHIP promotes its proteasomal degradation. Exp. Hematol. 38:392–402.
Acknowledgments
This work was supported in parts by grants from the National Institutes of Health (RO1 HL72523, RO1 HL095580, RO1 HL107127-01) and the Paige Arnold Butterfly Run. WG Kerr is the Murphy Family Professor of Children’s Oncology Research. GM Fuhler was supported by the Dutch Cancer Society (grant 2010-4737) and the Vereniging Trustfonds, Rotterdam, the Netherlands.
All in vivo studies were performed with approval from the Committee on the Humane Use of Animals at SUNY Upstate Medical University.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (http://creativecommons.org/licenses/by-nc-nd/4.0/)
About this article
Cite this article
Fuhler, G.M., Brooks, R., Toms, B. et al. Therapeutic Potential of SH2 Domain-Containing Inositol-5′-Phosphatase 1 (SHIP1) and SHIP2 Inhibition in Cancer. Mol Med 18, 65–75 (2012). https://doi.org/10.2119/molmed.2011.00178
Received:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00178