
Abstract
Dysferlinopathies are autosomal recessive disorders caused by mutations in the dysferlin (DYSF) gene, encoding the dysferlin protein. DYSF mutations lead to a wide range of muscular phenotypes, with the most prominent being Miyoshi myopathy (MM) and limb girdle muscular dystrophy type 2B (LGMD2B) and the second most common being LGMD. Symptoms generally appear at the end of childhood and, although disease progression is typically slow, walking impairments eventually result. Dysferlin is a modular type II transmembrane protein for which numerous binding partners have been identified. Although dysferlin function is only partially elucidated, this large protein contains seven calcium sensor C2 domains, shown to play a key role in muscle membrane repair. On the basis of this major function, along with detailed clinical observations, it has been possible to design various therapeutic approaches for dysferlin-deficient patients. Among them, exon-skipping and minigene transfer strategies have been evaluated at the preclinical level and, to date, represent promising approaches for clinical trials. This review aims to summarize the pathophysiology of dysferlinopathies and to evaluate the therapeutic potential for treatments currently under development.
Similar content being viewed by others


Introduction
Dysferlinopathies are autosomal recessive muscular dystrophies caused by mutations in the gene encoding dysferlin (DYSF; Online Mendelian Inheritance in Man [OMIM] gene number 603009, 2p13, GenBank NM_003494.2) (1,2). Dysferlinopathies are rare muscular dystrophies, as the number of adult patients whose symptoms meet the full diagnostic criteria is estimated between 1/100,000 to 1/200,000 (3). Disease-causing mutations in DYSF mainly cause limb girdle muscular dystrophy type 2B (LGMD2B; OMIM number 253601) (1) and distal Miyoshi myopathy (MM; OMIM number 254130) (2). However, other phenotypes associated with mutations in DYSF have been identified, including the distal anterior compartment myopathy (DACM) (alternatively called distal myopathy with anterior tibial onset [DMAT]; OMIM number 606768) and proximo-distal phenotypes (PD) (4,5). LGMD2B is characterized by proximal weakness, initially affecting the muscles of the pelvic and shoulder girdles, with age of onset in the late teens or early adulthood. For MM, however, the muscles initially affected are distal, in particular, the gastrocnemius. Age of onset is at the end of childhood (as with LGMD2B), and progression is typically slow. Proximodistal forms of dysferlinopathies are interesting as they display a range of symptoms, with a hypothesized influence of both genetic and environmental factors contributing to the phenotypic variation. At onset, nearly 35% of patients present with a mixed phenotype that combines both proximal and distal myopathies of the lower limbs. However, in every case, by late stages of disease progression, all limb-girdle muscles are affected.
The dysferlin gene is located on 2p13 and encodes several transcripts. The most common transcript is composed of 55 exons, and alternative splicing events (such as exon 1 for DYSF_υ1 [GenBank DQ267935], exon 5a [GenBank DQ976379] and exon 40a [GenBank EF015906]) generate transcript diversity. Fourteen different splice isoforms, containing exon 5a, 17 or 40a, have been described for human DYSF (6). Seven of these isoforms are transcribed from the “canonical” DYSF promoter (GenBank accession number AF075575), whereas the remaining seven are transcribed from the alternative DYSF_υ1 promoter (GenBank accession number DQ267935). Isoform 8 (NM_003494.2) (which excludes 5a and 40a, but includes exon 17) is expressed at high levels in skeletal muscle and is commonly used to describe the DYSF gene. In addition to these splice forms, >400 different DYSF sequence variations were reported in the Leiden Muscular Dystrophy database (https://doi.org/www.dmd.nl), including deleterious mutations, as well as nonpathogenic polymorphisms. Most of the reported disease-causing variants are point mutations and small insertions/deletions (7), but large exonic deletions and duplications have also been described (8). To date, no mutations have been identified in the alternatively spliced exon 1 of DYSF_υ1, 5a or 40 (9).
Translation of isoform 8 results in a 237-kDa transmembrane protein, named dysferlin. This protein belongs to the ferlin family, which includes otoferlin, myoferlin, fer-1-like 5 (FER1L5) and the originally identified Caenorhabditis elegans Fer-1. Fer-1 mediates calcium-dependent membrane fusion during spermatogenesis (10). Otoferlin is involved in vesicle exocytosis and is important for synaptic vesicle-plasma membrane fusion (mediated by Ca2+) at the inner hair-cell ribbon synapse (11). As such, mutations in otoferlin were shown to cause nonsyndromic deafness (deafness, autosomal recessive 9 [DFNB9; OMIM number 601071]) (12). Myoferlin is involved in both myoblast-myoblast fusion (13) and myoblast-myofiber fusion during muscle differentiation and maturation, respectively (14). Myoferlin has also been implicated in vesicle recycling during myoblast fusion because of its interaction with EH-containing domain 2 (EHD2) (15). Data published recently suggest that FER1L5 can interact, as previously described for myoferlin, with EHD1 and EHD2 to engage myoblast fusion (16). However, FER1L5 has not been associated with any human pathology to date.
All members of this gene family encode type II transmembrane proteins that contain several C2 domains (also called calcium sensors). They are involved in many biological processes including phospholipid interactions (13,14) and Ca2+-dependent interactions. Other proteins that contain the C2 domain are typically involved in active membrane dynamics and membrane repair (17). To understand ferlin function more precisely, it is important to determine ferlin C2 domain topology. Different three-dimensional shapes adopted by these protein domains allow ferlin members to interact with binding partners; therefore, C2 structural data would be very informative (18,19). Within the ferlin family, dysferlin is particularly well conserved among mammals (90% similarity between human and mouse [20,21]) and contains seven C2 domains. Multiple copies of C2 domains are frequently found in this particular protein family (22). Recently, their topology was described (C2A, C2B and C2E are type 1 C2 domains, while all the others are type 2), and it appears that C2 secondary structures are important for protein function (18,19). The C2 domains of dysferlin are similar to the well-characterized C2 domain of synaptotagmin VII, a protein predominantly involved in vesicle exocytosis in neurons (23,24). Besides C2 domains, two other motifs are found in this protein family: dysferlin sequences (3 of 6 human ferlin family members contain this sequence) and ferlin sequences (carried by all the members of the ferlin family). Both domains are of unknown function (20). Moreover, dysferlin sequences are conserved between orthologs. In addition, dysferlin also has several nuclear localization sequences (although there is no evidence for its nuclear localization). Finally, several proline, glutamic acid, serine and threonine (PEST) sequences, involved in protein-protein interaction, or in protein ubiquitinylation and degradation by the proteasome, have been described (25).
The DYSF gene, although expressed in a variety of tissues including brain, lung and placenta, is most highly enriched in skeletal and heart muscles and monocytes/macrophages (1,2,26,27). Dysferlin is expressed in skeletal muscle as early as 5–6 weeks of human embryonic development (28), where it is found mainly in the intracellular network of muscle fiber (T-tubule and in the plasma membrane). Furthermore, it was demonstrated that dysferlin is not an important regulator of the dystrophin-glycoprotein complex (DGC), since in dysferlin null mice, no alteration of the DGC complex was evident (29,30). These data clearly indicate that mechanisms involved in dysferlinopathies must be different from the ones caused by DGC deficiencies. Interestingly, a role of dysferlin in the maintenance of muscle reparation capabilities has been described. In dysferlin-deficient mice, muscle recovery after injuries is measurably slower than in wild-type mice, since recovery in the mutant tissue involves myogenesis rather than membrane repair (31). In fact, dysferlin was directly implicated in sarcolemmal resealing of muscle fiber damage resulting from mechanical stress, two-photon laser lesions or muscle elongation in humans and mice (29,32–35). The involvement of dysferlin in membrane repair is also supported by the numerous abnormalities in muscles of LGMD2B/MM patients, mainly subsarcolemmal vesicle accumulation (36,37). It was shown in vitro that Dysf−/− cultures have delayed expression of myogenin, resulting in defective muscle differentiation (38). Finally, two additional functions for dysferlin were described recently: (i) dysferlin is required for T-tubule homeostasis in humans (39) and (ii) dysferlin is associated with inflammation (40,41).
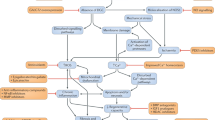
Because of its size and its numerous protein domains, dysferlin likely interacts with numerous partners, some of which have already been described. Identified interacting proteins are involved in diverse physiological processes including membrane maintenance (annexin 1 and 2, affixin and caveolin-3), cytoskeleton regulation (calpain-3, AHNAK [desmoyokin]) and membrane repair (mitsugumin 53 [MG53]) (Figure 1). Indeed, it is well documented that dysferlin interacts with caveolin-3, a protein concentrated at the intersection between the T-tubule network and the sarcolemma (42) and whose mutation results in LGMD1C (OMIM 607801) (43–45). Caveolin-3 may serve as a chaperone for dysferlin, directing it to the plasma membrane. Annexins I and II (which bind phospholipids and are involved in Ca2+ channel formation) (46,47) and affixin (or ß-parvin, a focal adhesion protein) have each been shown to interact with dysferlin in human skeletal muscles (48). Calpain-3 (mutated in LGMD2A) also interacts with dysferlin (49,50) (OMIM 253600). Calpain-3 is a calcium-dependent protease for which substrates are present in sarcolemme/sarcomeres and function as a regulator in muscle remodeling (51). Finally, AHNAK has been shown to interact with dysferlin. Interestingly, along with annexin I & II and affixin, AHNAK has not been directly implicated in a human disease (52–54). AHNAK is a large protein found at the enlargeosome (large-scale vesicle) surface and is involved in cellular processes including cell membrane differentiation, repair and signal transduction through its protein-protein interactions (52,54–56). According to Huang et al. (57), AHNAK and dysferlin colocalize at the sarcolemmal membrane, where dysferlin was shown to interact with α-tubulin (58). A recent publication clearly demonstrated that AHNAK is also found in the costameric network and is not present in the T-tubule system, leading to the hypothesis that AHNAK is involved in fiber stiffness (59).

Binding partners of dysferlin. In normal myofibers, dysferlin is anchored by its C-terminal domain to the plasma membrane and membrane network to cytosolic vesicles. In these contexts, it associates with different partners according to its localization. Schematized here are the binding partners of dysferlin and their localization within the myofiber. ch, Channel.
Finally, dysferlin has binding partners that play direct and indirect roles in membrane repair, such as MG53 (also called TRIM72). MG53 associates with phosphatidylserine in cell membranes and acts as a sensor for the entry of extracellular oxidative molecules. As a result, MG53-containing vesicles are attracted to sites of injury (due to the presence of an oxidative environment), thereby recruiting dysferlin into this niche. However, the interaction between dysferlin and MG53, along with caveolin-3, has only been described in vitro by coimmunoprecipitation experiments (60). There is still no direct evidence of an in vivo interaction between these proteins, but it is likely that they are part of the same complex. Although dysferlin and MG53 (60) are the only proteins known to be involved in muscle repair, their precise function in this context remains largely unknown.
Dysferlin colocalizes with the dihydropyridine receptor in T-tubules, suggesting a role for dysferlin in T-tubulogenesis (39). The T-tubule system is an extensive internal membrane invagination that passes transversally from the sarcolemma across a myofibril of striated muscle. T-tubules are involved in sarcomere stimulation by triggering the action potential deep in the fiber, allowing the release of calcium ions from the sarcoplasmic reticulum (61). Because of binding partners such as the annexins and caveolin-3, dysferlin could play a role in “invagination/evagination” of the membrane. Interestingly, T-tubule biogenesis requires both fusion and “invagination/evagination” of the membrane (62) (see Figure 1). Recent work has shown that dysferlin’s plasma membrane localization is transitory; membrane anchorage never exceeds 3 hours, and the protein’s half-life is only 5 hours. The syntaxin-4-associated endocytosis pathway plays a critical role in regulating this process (63).
From Symptoms to Diagnosis
Dysferlinopathies, for which onset generally occurs early in the second decade of life, are characterized by atrophy and weakness of the gastrocnemius muscle and/or the anterior tibial muscles. If a patient presents with these symptoms, the first step toward diagnosis is to measure serum creatine kinase (which is elevated in most cases of LGMD2B). In some cases, this test is supplemented with a skeletal muscle computed tomography scan or muscle magnetic imagery analysis. The second diagnostic step involves immunoblot analysis of a muscle biopsy or blood monocytes. Reduction or absence of dysferlin supports the dysferlinopathy diagnosis. It should be mentioned that there are secondary dysferlinopathies, where low dysferlin levels by Western blot result from mutations in other genes, for instance, CAPN3 (LGMD2A). This step increases the complexity of dysferlinopathy diagnosis. Histology of affected muscles shows when extensive fiber degeneration-regeneration has taken place, revealed by centronuclear fibers of varying sizes and immune infiltration (predominantly CD8+ cells and macrophages). No B cells are detected, probably because intracellular material is released after membrane lesion (64).
What are the Mutations Involved in Dysferlinopathies?
Because the discovery that DYSF dysfunction underlies both LGMD2B (1) and MM (2), a wide spectrum of DYSF mutations has been identified, including missense, nonsense, frameshift deletions/insertions, splicing mutations and large deletions (1,2,7,8,46,65,66). Although no mutational hotspots were identified (UMD-DYSF database, Leiden muscular dystrophy pages: https://doi.org/www.dmd.nl), five founder mutations were reported: c.2372C>G (p.Pro791Arg) for a native Canadian population, c.4872_4876delin-sCCCC (p.Glu1624AspfsX9) for a Libyan Jewish population, c.2875C>T (p.Arg959Trp) for an Italian population, c.5713C>T (p.Arg1905X) for a Spanish population (region of Sueca) and c.2779delG (p.Ala927LeufsX21) for a Caucasian Jewish population (67–71).
Loss of dysferlin is frequently associated with nonsense or frameshift mutations (7,66). However, except for rare cases (for example, exon 32 deletion causes mild dysferlinopathy [see below, 72]), no measurable phenotype to genotype correlation was established. In general, the dysferlin gene has a high level of polymorphisms, once again increasing the complexity for dysferlinopathy molecular diagnosis.
There are several DYSF mutations that lead to mRNA instability and degradation through the nonsense-mediated mRNA decay (NMD) process (73). At the protein level, Fujita et al. (2007) have described two major pathways for degradation of misfolded dysferlin: the endoplasmic reticulum-associated degradation (ERAD) system and autophagy (74). ERAD is used to clear nonaggregated native or mutant dysferlin, whereas autophagy removes aggregated versions of the mutated protein. In situations where unfolded-dysferlin degradation is misregulated, the pathophysiology could be explained by the formation of amyloidlike aggregates (75). In silico analysis of dysferlin predicted a bi-partite nuclear localization sequence (NLS) near its N-terminus (https://doi.org/www.genscript.com/psort/psort2.html). Whereas endogenous dysferlin is typically not found in the nucleus, this motif could become relevant for truncated proteins that lack transmembrane domains. Indeed, truncated dysferlins may be directed to inappropriate cellular compartments, in particular the nucleus, because of the NLS sequence. Unfortunately, this result has not yet been observed because of the C-terminal specificity of the Hamlet antibody. Every DYSF mutation reported to date affects protein expression levels in skeletal muscle, except for one (TG573/574AT; pVal67Asp), which affects dysferlin calcium-binding properties (76). Before effective therapeutic approaches can be developed, it is critical to fully characterize DYSF sequence variations that result in pathologies.
Therapeutic Approaches for Dysferlinopathies
Today, no curative treatment for dysferlinopathies exists. In the following section, several validated approaches for dysferlinopathy therapy will be discussed, including synthesis of functional protein after adeno-associated virus (AAV) vector transfer, gene surgery (exon skipping, trans-splicing) and pharmacological or immunological preclinical treatments. As always, therapeutic approaches must first be evaluated in cells and/or animal models. Recently, fibroblasts derived from human patients have been successfully converted into myoblasts (77). Several mouse cell culture systems have been developed recently (dysf−/− C2C12 cells, single-cell cloned shRNA cell line and immortalized cells from A/J mice), which will be helpful when assessing novel therapeutic strategies. In addition to these cell culture systems, several animal model systems also exist. In particular, two natural mice strains (SJL/J and A/J) were identified that have dystrophic phenotypes. The SJL/J mouse strain has a splicing mutation in the dysferlin gene, making it a natural model for LGMD2B and MM (21,78). Genomic analysis identified the mutation as a 141-bp genomic deletion involving a tandem repeat and altering the 3′ splicing site of exon 45, therefore leading to the splicing of this 171-bp long exon. Another strain of mice carrying a deficient dysferlin was fortuitously discovered in The Jackson Laboratory. The A/J strain presents mild progressive muscular dystrophy. Genetic analysis of this strain demonstrated that an early transposon (ETn) retrotransposon inserted itself in the intron 4 of the dysferlin gene. However, research involving these strains is not easy because of a mild phenotype and the absence of a matched control strain. Thus, genetically engineered and backcrossed mice (dysf−/−, C57BL/10.SJL-Dysf, B6.129-Dysftm1Kcam/Mmmh, B6.A/J-Dysfprmd) were created (29,78–80). Issues still remain with these dysferlinopathy models, since none truly recapitulate the human disease. Although not perfect, these models will serve as valuable tools to develop and test novel therapeutic approaches.
Transfer of Full-Length Dysferlin
The ultimate goal is to alleviate the symptoms associated with dysferlin deficiency. Currently, gene transfer approaches are the main strategy tested to treat recessive diseases such as dysferlinopathy. Molkentin demonstrated that the restoration of dysferlin only in skeletal muscle fibers would be sufficient to rescue the dysferlin-deficient mice (81). The AAV vector is the most common viral vector used for muscle gene therapy (82). For dysferlinopathy, however, there are major technical issues associated with using AAV in therapeutic approaches. The encapsidation size is limited to approximately 4 kb, the equivalent of three C2 domains (83). To solve this problem, researchers used the concatemerization property of AAV (84,85). Two independent AAV vectors are used: one carrying 5′ dysferlin cDNA fused with an intronic sequence that carries a donor splice site and the other containing the 3′ cDNA-bearing intronic sequence and a splice acceptor site. This approach allows expression, after concatemerization and splicing, of a full-length dysferlin transcript (a scheme representing this strategy, as seen in Figure 2). This process can be used to reverse membrane repair deficits associated with dysferlin mutant myofibers. Functionally, this treatment results in improved locomotor activity in injected mice, which is a promising result. However, the use of two AAV vectors and the low levels of expression are issues that must be addressed before use as a viable therapy (80). Brown and his colleagues have also tried to use one AAV to encode full-length dysferlin (using the AAV5 serotype) (86). They conclude that this strategy allows expression of dysferlin at detectable levels; however, dysferlin expression could be in part the result of a recombination event between different parts of the transgene carried by AAVs (87).

Therapeutic strategies currently being tested. Dysferlinopathies are an ensemble of pathologies, due to mutations in the DYSF gene. Wide spectrums of mutations are responsible for the pathologies, including missense, nonsense, frameshift, deletion, insertion and splicing mutations. Currently, two main types of strategies are envisaged: gene transfer (AAV concatemerization, or minigene) and mRNA surgery (exon-skipping and transsplicing strategies). Black arrows indicate strategies available for all gene mutations, whereas dashed arrows indicate mutation-specific strategies (mutations in exon 32 for exon skipping, 3′ mutations for trans-splicing). Inverted terminal repeats (ITRs) are the adeno-associated vector backbone.
Transfer of Dysferlin Minigenes
Recently, a minidysferlin was discovered by our team in a patient with a moderately severe muscular phenotype, and this discovery has opened up new therapeutics prospects (88) (see Figure 2). This minidysferlin, which contains only the last two C2 domains (out of seven) and the transmembrane domain, is expressed in muscle biopsies and correctly targeted to the plasma membrane in this patient. In collaboration with I Richard’s team (88), we injected AAV encoding minidysferlin into dysferlin-deficient mice and observed membrane insertion of minidysferlin, which restored membrane repair. Although this minidysferlin prevented the appearance of severe dysferlinopathies, it was not completely functional, since no histological improvements were seen in treated mice (88). Therefore, our experiments demonstrate that the minidysferlin retains membrane repair function, although functions associated with missing C2 domains are lacking. For example, the C2A domain interacts with phospholipids, AHNAK and α-tubulin (58). C2A (as well as C2B, C2C, C2D and C2E) is absent from the natural minidysferlin expressed in this patient. This result certainly explains why restoring minidysferlin is not sufficient to rescue all aspects of the dysferlin phenotype. Nevertheless, these results indicate that both the AAV injection and the level of expression of a functional minidysferlin are not toxic. This is important information for future clinical developments, since we have shown that the overall function of dysferlin can be, at least partially, carried by a large deleted amino acid sequence protein. This result also pinpoints the necessity to develop genomic explorations in patients affected by muscular dystrophies to identify new natural truncated mutated proteins.
Messenger Surgery by Exon Skipping
In dysferlinopathies, correlations between genotype and phenotype are rare (as previously mentioned). However, the Sinnreich group identified a family in which two severely affected sisters were homozygous for a dysferlin null mutation (72, Figures 2 and 3). Their mother was heterozygous for the same null mutation and had a lariat branch point mutation in intron 31 (leading to an inframe skipping of exon 32) in trans. This patient presented a mild phenotype, so it is likely that the exon 32 skipped-allele partially complemented the null mutation. In collaboration with the Garcia group, we have considered this discovery as a “natural” proof of concept for an exon-skipping therapeutic approach for dysferlinopathies. The use of an exon-skipping strategy was first applied in Duchenne muscular dystrophies (89), and clinical trials using antisense oligonucleotides (AONs) are underway (90). The results obtained in preclinical trials are promising and pave the way for similar approaches for dysferlinopathies (91). In particular, using an exon-skipping strategy to treat DYSF mutations lying in exon 32 is promising. According to the UMD database, approximately 4% of dysferlin patients have disease-causing mutations in this exon. AON directed against splicing regulatory elements of exon 32 were tested on myotubes transdifferentiated from patient fibroblasts previously infected by a lentivirus encoding MyoD. The efficiency of exon skipping induced by these AONs was validated by reverse transcription-polymerase chain reaction. Indeed, a fragment corresponding to a sequence deleted of exon 32 was observed in samples from AON-treated cells (92,93). Even if this technology proves promising, it will be important to improve efficiency of AON delivery and to determine the efficiency of this “quasi-dysferlin” before switching to clinical trials. The different approaches described in this section have been schematically represented in Figure 3.

A natural proof of exon skipping. Depicted is a family studied by Sinnreich et al. (72), in which the index case (identified by an asterisk) carries two mutant alleles. The family tree (A) and the nature of the two mutant alleles (B) are shown.
Pharmacological and Immunological Approaches
Pharmacological or immunological approaches have also been envisaged to treat dysferlinopathies. Recently, PTC124® (also known as Ataluren™) was shown to read through nonsense mutations. Wang et al. (94) showed that treating myotubes from a patient with a premature stop codon allowed the expression of a sufficient functional dysferlin to rescue myotube membrane blebbing. However, in dysferlinopathies, less than one-third of mutations are nonsense mutations.
In 2007, the use of Dantrolen™ as a potential treatment for dysferlinopathies was described. CPK levels were reduced in response to Dantrolen (95); however, no amelioration of the muscle phenotype has been seen to date. In 2001, the presence of a complement deposit in nonnecrotic muscle fibers deficient for dysferlin (96) was observed, and it was hypothesized that the complement contributed to the deleterious effects of the dysferlin absence. A clinical trial was launched to try to inhibit accumulation of this deposit by the administration of intravascular immunoglobulin (IV-IG); however, the initially encouraging results were not pursued (reviewed in 97). Recently, promising results were published regarding the increase of muscle strength for dysferlinopathy patients treated with Rituximab™ (a human/murine chimeric monoclonal antibody directed against CD20-positive B cells) (98). The authors suggest a possible role for B cells in dysferlin pathophysiology. After the transcriptomics studies performed in EP Hoffman’s laboratory, demonstrating the upregulation of proteins involved in rho signaling, they started preclinical studies using an inhibitor of a rho kinase (Fasudil™) (99). This preclinical treatment demonstrated some benefits: notably, a lower level of inflammatory cells in the muscles and an increase in body weight, but no impact in another physiological process such as muscle fiber degeneration/regeneration or in muscle force (100). Nevertheless, this study is a starting point for drug-testing trials in dysferlin-deficient mice, by a standardized measurement of dysferlin functions that will be useful for high-throughput screening.
Conclusion
It is essential to characterize dysferlin at the molecular level to functionally understand this sarcolemmal protein in both normal and pathological conditions. This knowledge is absolutely required if adapted therapeutic strategies are to be developed for dysferlinopathies (and possibly other forms of muscular dystrophies), where enhancement of sarcolemmal resealing could be of benefit. Researchers are still working to define functional roles for each C2 domain and to accurately describe dysferlin activity in a variety of physiological processes, such as T-tubule homeostasis and membrane repair. As molecular diagnostic technologies improve, a better understanding of these diseases will emerge, enabling the development of appropriate therapeutic strategies on the basis of clinical observations.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Bashir R, et al. (1998) A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 20:37–42.
Liu J, et al. (1998) Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 20:31–6.
Moore SA, et al. (2006) Limb-girdle muscular dystrophy in the United States. J. Neuropathol. Exp. Neurol. 65:995–1003.
Nguyen K, et al. (2007) Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch. Neurol. 64:1176–82.
Ueyama H, et al. (2002) Clinical heterogeneity in dysferlinopathy. Intern. Med. 41:532–6.
Pramono ZA, et al. (2009) Identification and characterisation of human dysferlin transcript variants: implications for dysferlin mutational screening and isoforms. Hum. Genet. 125:413–20.
Krahn M, et al. (2009) Analysis of the DYSF mutational spectrum in a large cohort of patients. Hum. Mutat. 30:E345–75.
Krahn M, et al. (2009) Identification of different genomic deletions and one duplication in the dysferlin gene using multiplex ligation-dependent probe amplification and genomic quantitative PCR. Genet. Test. Mol. Biomarkers. 13:439–42.
Krahn M, Labelle V, Borges A, Bartoli M, Levy N. (2010) Exclusion of mutations in the dysferlin alternative exons 1 of DYSF-v1, 5a, and 40a in a cohort of 26 patients. Genet. Test. Mol. Biomarkers. 14:153–4.
Washington NL, Ward S. (2006) FER-1 regulates Ca2+-mediated membrane fusion during C. elegans spermatogenesis. J. Cell Sci. 119:2552–62.
Roux I, et al. (2006) Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 127:277–89.
Yasunaga S, et al. (1999) A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat. Genet. 21:363–9.
Davis DB, Doherty KR, Delmonte AJ, McNally EM. (2002) Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J. Biol. Chem. 277:22883–8.
Doherty KR, et al. (2005) Normal myoblast fusion requires myoferlin. Development. 132:5565–75.
Doherty KR, et al. (2008) The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion. J. Biol. Chem. 283:20252–60.
Demonbreun AR, et al. (2010) Myoferlin is required for insulin-like growth factor response and muscle growth. FASEB J. 24:1284–95.
Zhang D, Aravind L. (2010) Identification of novel families and classification of the C2 domain superfamily elucidate the origin and evolution of membrane targeting activities in eukaryotes. Gene. 469:18–30.
Nalefski EA, Falke JJ. (1996) The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 5:2375–90.
Therrien C, Di Fulvio S, Pickles S, Sinnreich M. (2009) Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides. Biochemistry. 48:2377–84.
Lek A, Lek M, North KN, Cooper ST. (2010) Phylogenetic analysis of ferlin genes reveals ancient eukaryotic origins. BMC Evol. Biol. 10:231.
Vafiadaki E, et al. (2001) Cloning of the mouse dysferlin gene and genomic characterization of the SJL-Dysf mutation. Neuroreport. 12:625–9.
Cho W, Stahelin RV. (2006) Membrane binding and subcellular targeting of C2 domains. Biochim. Biophys. Acta. 1761:838–49.
Shao X, Davletov BA, Sutton RB, Sudhof TC, Rizo J. (1996) Bipartite Ca2+-binding motif in C2 domains of synaptotagmin and protein kinase C. Science. 273:248–51.
Sutton RB, Davletov BA, Berghuis AM, Sudhof TC, Sprang SR. (1995) Structure of the first C2 domain of synaptotagmin I: a novel Ca2+/phospholipid-binding fold. Cell. 80:929–38.
Rechsteiner M, Rogers SW. (1996) PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21:267–71.
Ho M, et al. (2002) A novel, blood-based diagnostic assay for limb girdle muscular dystrophy 2B and Miyoshi myopathy. Ann. Neurol. 51:129–33.
Vandre DD, et al. (2007) Dysferlin is expressed in human placenta but does not associate with caveolin. Biol. Reprod. 77:533–42.
Anderson LV, et al. (1999) Dysferlin is a plasma membrane protein and is expressed early in human development. Hum. Mol. Genet. 8:855–61.
Bansal D, et al. (2003) Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 423:168–72.
Matsuda C, et al. (1999) Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology. 53:1119–22.
Roche JA, Lovering RM, Bloch RJ. (2008) Impaired recovery of dysferlin-null skeletal muscle after contraction-induced injury in vivo. Neuroreport. 19:1579–84.
Bansal D, Campbell KP. (2004) Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 14:206–13.
Doherty KR, McNally EM. (2003) Repairing the tears: dysferlin in muscle membrane repair. Trends Mol. Med. 9:327–30.
Meldolesi J. (2003) Surface wound healing: a new, general function of eukaryotic cells. J. Cell. Mol. Med. 7:197–203.
von der Hagen M, et al. (2005) The differential gene expression profiles of proximal and distal muscle groups are altered in pre-pathological dysferlin-deficient mice. Neuromuscul. Disord. 15:863–77.
Cenacchi G, Fanin M, De Giorgi LB, Angelini C. (2005) Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 58:190–5.
Piccolo F, Moore SA, Ford GC, Campbell KP. (2000) Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb: girdle muscular dystrophies. Ann. Neurol. 48:902–12.
de Luna N, et al. (2006) Absence of dysferlin alters myogenin expression and delays human muscle differentiation “in vitro.” J. Biol. Chem. 281:17092–8.
Klinge L, et al. (2010) Dysferlin associates with the developing T-tubule system in rodent and human skeletal muscle. Muscle Nerve. 41:166–73.
Roche JA, et al. (2010) Extensive mononuclear infiltration and myogenesis characterize recovery of dysferlin-null skeletal muscle from contraction-induced injuries. Am. J. Physiol. Cell Physiol. 298:C298–312.
De Luna N, et al. (2010) Role of thrombospondin 1 in macrophage inflammation in dysferlin myopathy. J. Neuropathol. Exp. Neurol. 69:643–53.
Hernandez-Deviez DJ, et al. (2006) Aberrant dysferlin trafficking in cells lacking caveolin or expressing dystrophy mutants of caveolin-3. Hum. Mol. Genet. 15:129–42.
Capanni C, et al. (2003) Dysferlin in a hyperCK-aemic patient with caveolin 3 mutation and in C2C12 cells after p38 MAP kinase inhibition. Exp. Mol. Med. 35:538–44.
Matsuda C, et al. (2001) The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum. Mol. Genet. 10:1761–6.
Walter MC, et al. (2003) Variable reduction of caveolin-3 in patients with LGMD2B/MM. J. Neurol. 250:1431–8.
Cagliani R, et al. (2005) Mutation finding in patients with dysferlin deficiency and role of the dysferlin interacting proteins annexin A1 and A2 in muscular dystrophies. Hum. Mutat. 26:283.
Lennon NJ, et al. (2003) Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 278:50466–73.
Matsuda C, et al. (2005) Dysferlin interacts with affixin (beta-parvin) at the sarcolemma. J. Neuropathol. Exp. Neurol. 64:334–40.
Anderson LV, et al. (2000) Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul. Disord. 10:553–9.
Huang Y, et al. (2008) Calpain 3 is a modulator of the dysferlin protein complex in skeletal muscle. Hum. Mol. Genet. 17:1855–66.
Duguez S, Bartoli M, Richard I. (2006) Calpain 3: a key regulator of the sarcomere? FEBS J. 273: 3427–36.
Borgonovo B, et al. (2002) Regulated exocytosis: a novel, widely expressed system. Nat. Cell. Biol. 4:955–62.
Haase H, et al. (1999) Signaling from betaadrenoceptor to L-type calcium channel: identification of a novel cardiac protein kinase A target possessing similarities to AHNAK. FASEB J. 13:2161–72.
Hohaus A, et al. (2002) The carboxyl-terminal region of ahnak provides a link between cardiac L-type Ca2+ channels and the actin-based cytoskeleton. FASEB J. 16:1205–16.
Benaud C, et al. (2004) AHNAK interaction with the annexin 2/S100A10 complex regulates cell membrane cytoarchitecture. J. Cell Biol. 164: 133–144.
Gentil BJ, et al. (2001) The giant protein AHNAK is a specific target for the calcium- and zincbinding S100B protein: potential implications for Ca2+ homeostasis regulation by S100B. J. Biol. Chem. 276:23253–61.
Huang Y, et al. (2007) AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 21:732–42.
Azakir BA, Di Fulvio S, Therrien C, Sinnreich M. (2010) Dysferlin interacts with tubulin and microtubules in mouse skeletal muscle. PLoS One. 5:e10122.
Marg A, Haase H, Neumann T, Kouno M, Morano I. (2010) AHNAK1 and AHNAK2 are costameric proteins: AHNAK1 affects transverse skeletal muscle fiber stiffness. Biochem. Biophys. Res. Commun. 401:143–8.
Cai C, et al. (2009) Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 284:15894–902.
Franzini-Armstrong C. (1991) Simultaneous maturation of transverse tubules and sarcoplasmic reticulum during muscle differentiation in the mouse. Dev. Biol. 146:353–63.
Klinge L, et al. (2007) From T-tubule to sarcolemma: damage-induced dysferlin translocation in early myogenesis. FASEB J. 21:1768–76.
Evesson FJ, et al. (2010) Reduced plasma membrane expression of dysferlin mutants is attributed to accelerated endocytosis via a syntaxin-4-associated pathway. J Biol. Chem. 285:28529–39.
Gallardo E, et al. (2001) Inflammation in dysferlin myopathy: immunohistochemical characterization of 13 patients. Neurology. 57:2136–8.
Aoki M, et al. (2001) Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. 57:271–8.
Nguyen K, et al. (2005) Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum. Mutat. 26:165.
Weiler T, et al. (1999) Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum. Mol. Genet. 8:871–7.
Argov Z, et al. (2000) Muscular dystrophy due to dysferlin deficiency in Libyan Jews: clinical and genetic features. Brain. 123:1229–37.
Cagliani R, et al. (2003) Molecular analysis of LGMD-2B and MM patients: identification of novel DYSF mutations and possible founder effect in the Italian population. Neuromuscul. Disord. 13:788–95.
Vilchez JJ, et al. (2005) Identification of a novel founder mutation in the DYSF gene causing clinical variability in the Spanish population. Arch. Neurol. 62:1256–9.
Leshinsky-Silver E, et al. (2007) Dysferlinopathy in the Jews of the Caucasus: a frequent mutation in the dysferlin gene. Neuromuscul Disord 17:950–4.
Sinnreich M, Therrien C, Karpati G. (2006) Lariat branch point mutation in the dysferlin gene with mild limb-girdle muscular dystrophy. Neurology. 66:1114–6.
Wenzel K, et al. (2006) Novel sequence variants in dysferlin-deficient muscular dystrophy leading to mRNA decay and possible C2-domain misfolding. Hum. Mutat. 27:599–600.
Fujita E, et al. (2007) Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Hum. Mol. Genet. 16:618–29.
Spuler S, et al. (2008) Dysferlin-deficient muscular dystrophy features amyloidosis. Ann. Neurol. 63:323–8.
Therrien C, Dodig D, Karpati G, Sinnreich M. (2006) Mutation impact on dysferlin inferred from database analysis and computer-based structural predictions. J. Neurol. Sci. 250:71–8.
Chaouch S, et al. (2009) Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: validation of an exonskipping approach to restore dystrophin in Duchenne muscular dystrophy cells. Hum. Gene Ther. 20:784–90.
Bittner RE, et al. (1999) Dysferlin deletion in SJL mice (SJL-Dysf) defines a natural model for limb girdle muscular dystrophy 2B. Nat. Genet. 23: 141–2.
Ho M, et al. (2004) Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 13:1999–2010.
Lostal W, et al. (2010) Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet. 19:1897–907.
Millay DP, et al. (2009) Genetic manipulation of dysferlin expression in skeletal muscle: novel insights into muscular dystrophy. Am. J. Pathol. 175:1817–23.
Daniele N, Richard I, Bartoli M. (2007) Ins and outs of therapy in limb girdle muscular dystrophies. Int. J. Biochem. Cell. Biol. 39:1608–24.
Bueler H. (1999) Adeno-associated viral vectors for gene transfer and gene therapy. Biol. Chem. 380:613–22.
Yan Z, Zhang Y, Duan D, Engelhardt JF. (2000) Trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc. Natl. Acad. Sci. U. S. A. 97:6716–21.
Xu Z, et al. (2004) Trans-splicing adenoassociated viral vector-mediated gene therapy is limited by the accumulation of spliced mRNA but not by dual vector coinfection efficiency. Hum. Gene Ther. 15:896–905.
Allocca M, et al. (2008) Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J. Clin. Invest. 118:1955–64.
Hirsch ML, Agbandje-McKenna M, Samulski RJ. (2010) Little vector, big gene transduction: fragmented genome reassembly of adeno-associated virus. Mol. Ther. 18:6–8.
Krahn M, et al. (2010) A naturally occurring human minidysferlin protein repairs sarcolemmal lesions in a mouse model of dysferlinopathy. Sci. Transl. Med. 2:50ra69.
Brun C, et al. (2003) U7 snRNAs induce correction of mutated dystrophin pre-mRNA by exon skipping. Cell. Mol. Life Sci. 60:557–66.
van Deutekom JC, et al. (2007) Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 357:2677–86.
Kinali M, et al. (2009) Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 8:918–28.
Levy N, et al. (2010) Therapeutic exon ‘switching’ for dysferlinopathies? Eur. J. Hum. Genet. 18:969–70; author reply 971.
Wein N, et al. (2010) Efficient bypass of mutations in dysferlin deficient patient cells by anti-sense-induced exon skipping. Hum. Mutat. 31:136–42.
Wang B, et al. (2010) Membrane blebbing as an assessment of functional rescue of dysferlin-deficient human myotubes via nonsense suppression. J. Appl. Physiol. 109:901–5.
Hattori H, et al. (2007) A novel compound heterozygous dysferlin mutation in Miyoshi myopathy siblings responding to dantrolene. Eur. J. Neurol. 14:1288–91.
Selcen D, Stilling G, Engel AG. (2001) The earliest pathologic alterations in dysferlinopathy. Neurology. 56:1472–81.
Albrecht DE, et al. (2009) 3rd Annual Dysferlin Conference 2–5 June 2009, Boston, Massachusetts, USA. Neuromuscul. Disord. 19:867–73.
Lerario A, et al. (2010) Effects of rituximab in two patients with dysferlin-deficient muscular dystrophy. BMC Musculoskelet. Disord. 11:157.
Kesari A, et al. (2008) Dysferlin deficiency shows compensatory induction of Rab27A/Slp2a that may contribute to inflammatory onset. Am. J. Pathol. 173:1476–87.
Rayavarapu S, et al. (2010) Characterization of dysferlin deficient SJL/J mice to assess preclinical drug efficacy: fasudil exacerbates muscle disease phenotype. PLoS One. 5:e12981.
Acknowledgments
We warmly acknowledge the constant support of the Association Française contre les Myopathies (AFM) and the Jain Foundation. F Barthélémy and N Wein received PhD fellowship grants from AFM and the Fondation pour la Recherche Médicale (FRM), respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Barthélémy, F., Wein, N., Krahn, M. et al. Translational Research and Therapeutic Perspectives in Dysferlinopathies. Mol Med 17, 875–882 (2011). https://doi.org/10.2119/molmed.2011.00084
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2011.00084