
Abstract
Chronic hepatitis C (CHC) is generally a slowly progressive disease, but some factors associated with rapid progression have been identified. Steatosis, independently of its metabolic or viral origin, leads to liver injury and fibrosis. It is suggested that hepatitis C virus may contribute to a wide spectrum of metabolic disturbances—namely, steatosis, insulin resistance, increased prevalence of impaired glucose tolerance, type 2 diabetes mellitus and lipid metabolism abnormalities. Adipokines, which are produced mainly by adipose tissue, may influence the inflammatory response and insulin sensitivity and contribute to the development of metabolic abnormalities in CHC and also regulate fibrogenesis and angiogenesis. Visfatin was described as an adipokine with immunomodulating and proinflammatory properties that promotes B-cell maturation and enhances activation of leukocytes, synthesis of adhesion molecules and production of proinflammatory cytokines. Visfatin exerts insulin-mimetic effects, decreases plasma glucose levels and regulates cell energy balance. Chemerin stimulates chemotaxis of dendritic cells, macrophages and natural killer (NK) cells toward the site of inflammation. On the other hand, it inhibits synthesis of proinflammatory mediators and enhances adiponectin production, influences adipocyte differentiation and maturation and regulates glucose uptake in adipocytes. Vaspin expression in human adipose tissue seems to be a compensatory mechanism associated with obesity and insulin resistance. Vaspin suppresses leptin, tumor necrosis factor (TNF)-α and resistin expression. Leptin protects against liver steatosis but accelerates fibrosis progression and exacerbates the inflammatory process. In contrast, adiponectin exerts a hepatoprotective effect. In this report, data indicating a possible role of these adipokines in the pathogenesis of chronic hepatitis are summarized.
Similar content being viewed by others


Metabolic Aspects of Chronic Hepatitis C
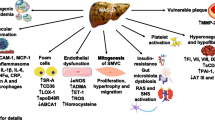
Chronic hepatitis C (CHC) has a number of features that suggest that it should be recognized not only as a viral disease but also as a metabolic liver disease that encompasses insulin resistance (IR), liver steatosis, impaired glucose tolerance or type 2 diabetes mellitus (T2DM) and disturbances in lipid metabolism (Figure 1). Hepatitis C virus (HCV) has been shown to induce IR by direct action and by promotion of inflammatory processes and/or fibrosis (Figure 2). The observation that successful treatment improves insulin sensitivity supports a direct causal role of HCV in IR development (1). Increasing data suggest that IR is closely related to the extent of steatosis and inflammatory activity in the liver (2,3). IR is an independent predictor of the progression of fibrosis (2,3). A clinically important factor is the negative influence of IR on the rate of sustained virological response to antiviral treatment (4,5). IR also appears to increase the risk of developing hepatocellular carcinoma (6). Liver steatosis is closely related to IR. It appears more frequently in patients with CHC than in the general population (7). Data indicate that steatosis, independently of its metabolic or viral origin, contributes to liver injury and faster progression of fibrosis (8,9).

Metabolic aspects of CHC.

Pathogenesis of IR in CHC.
Adipose tissue acts as a store of energy and as an active endocrine organ. Adipokines (adipocytokines)—agents secreted primarily by adipocytes—modulate lipid and glucose metabolism and insulin sensitivity (10). In addition to their well-established role in controlling adipose tissue physiology, adipokines have been shown to be involved in regulation of the inflammatory response, angiogenesis and fibrogenesis (11,12). As a result, adipokines together with IR seem to play a distinct role in the pathogenesis of liver disease. CHC is another disease in which adipokines may represent a link between viral infection, steatosis, metabolic disturbances and disease progression. It has been suggested that some adipokines exert a protective effect and others a negative effect in CHC (11–13). Recent studies have shown that obesity is associated with a chronic, low-grade inflammatory state that induces dysregulation of adipocytokines and contributes to IR and T2DM (14,15). Hepatic cirrhosis is more prevalent in obese individuals than in the general population, and obesity is an independent risk factor for liver fibrosis in nonalcoholic steatohepatitis (NASH), alcohol-induced liver disease and CHC and development of hepatocellular carcinoma (HCC) (16–22).
Characteristics and Action of Adipokines
The role of adipokines in CHC has not yet been clearly defined (11,12,23). The family of adipokines is still growing. Novel adipokines such as visfatin, chemerin and vaspin were recently described. The better known adipokines are adiponectin and leptin, but their role in CHC is confusing and the results of studies are contradictory (11,12). Given the properties of adipokines mentioned above, they are likely to play a pivotal role in CHC.
A better understanding of the pathogenic role of novel adipokines in the inflammatory process and in mechanisms underlying IR development and fibrosis progression in CHC may have a prophylactic implication in preventing progression of liver fibrosis and improving response to antiviral therapy.
Visfatin Characteristics
Visfatin, also known as nicotinamide phosphoribosyltransferase (Nampt) and pre-B-cell colony-enhancing factor 1 (PBEF-1), has multiple biological functions and is produced by a variety of cells. The main sources of visfatin are lymphocytes, monocytes, neutrophils, hepatocytes, adipocytes and pneumocytes. Increased levels of visfatin are found in both acute and chronic inflammatory diseases (24,25). Visfatin was originally cloned as a putative cytokine shown to enhance the maturation of B-cell precursors in the presence of interleukin (IL)-7 and stem cell factor. It was therefore named PBEF (26). Visfatin is an adipokine with immunomodulating and proinflammatory properties. It was reported to be a cytokine that promotes B-cell maturation and inhibits neutrophil apoptosis (26,27). Visfatin enhances activation of leukocytes, synthesis of adhesion molecules and production of proinflammatory cytokines (24,25). Visfatin also stimulates proangiogenic activity (28). On the other hand, visfatin is reported to exert insulin-mimetic effects in cultured cells and to lower plasma glucose levels in mice by binding to and activating the insulin receptor (29). However, the physiological relevance of visfatin is still uncertain because its plasma concentration is 40- to 100-fold lower than that of insulin despite having similar receptor-binding affinity (29,30). Visfatin exerts a cardioprotective effect during myocardial infarction and has been suggested to play a protective role in nonalcoholic fatty liver disease (NAFLD) (31,32). Fukuhara et al. (29) reported that visfatin is enriched in the visceral fat of both humans and mice and that its plasma levels increase during the development of obesity. However, the relationship between the amount of adipose tissue and obesity is still unresolved (33–39). Visfatin has the ability to regulate the cell cycle and carcinogenesis (24,25). Finally, visfatin is a nicotinamide phosphoribosyltransferase (Nampt) enzyme that catalyzes the first step in the biosynthesis of nicotinamide adenine dinucleotide (NAD) from nicotinamide. Therefore, visfatin plays a pivotal role as regulator of cell energy balance (24,25,40). The action of visfatin is shown in Figure 3.

Action of visfatin. ROS, reactive oxygen species.
Visfatin and Necro-Inflammatory Activity in Chronic Hepatitis
Serum visfatin concentration in patients with CHC infected with genotype 1b was found to be significantly higher than in healthy controls (38). There was no association between the serum visfatin level and body mass index (BMI). Interestingly, visfatin serum concentration was significantly higher in patients with CHC patients with a lower BMI (<25 kg/m2) than in overweight patients with a BMI ≥25 kg/m2 (38). Another study showed that there was no difference in visfatin serum levels between patients infected with HCV genotype 1 and those infected with genotype 3 (41).
Serum visfatin was found to be negatively related to the grade of necro-inflammatory activity in CHC (38), suggesting that visfatin may be a regulator of the inflammatory process in CHC. The highest levels were seen in subjects with minimal inflammatory activity. Significantly lower levels were found in patients with moderate or severe inflammatory activity, but were still twice as high as in the control group (38). These results indicate possible protective properties of visfatin in CHC. A similar protective effect of visfatin against hepatocyte injury was described in NAFLD. Serum visfatin in patients with NAFLD was significantly increased compared with both lean and obese healthy controls (32). Visfatin levels decreased markedly when NASH was diagnosed (32,42). However, it was still significantly higher than in both lean and obese healthy controls (32). In another study, Gaddipati et al. (43) showed that visceral visfatin levels decreased significantly in patients with NASH compared with patients with simple or moderate steatosis. Aller et al. (42) found that serum visfatin in patients with NAFLD was related to the grade of portal inflammation and predicted that the presence of portal inflammation, as in CHC, was not related to BMI. However, there was no association between serum visfatin and intensity of lobular inflammation in NAFLD (42). Moreover, the visceral visfatin levels were higher in non-NAFLD subjects (43). The observed decrease of visceral visfatin levels was independent of BMI and IR. On the basis of these findings, the authors pointed to the protective role of visfatin in NAFLD (43). The positive correlation between serum visfatin and γ-globulins found in patients with CHC additionally points to its involvement in the inflammatory process (38).
Visfatin was found to induce the synthesis of IL-6 in peripheral blood mononuclear cells and dendritic cells (25). IL-6 stimulates hepatocytes to produce various proinflammatory cytokines (44). On the other hand, IL-6 plays a pivotal role in liver regeneration and has a protective role against hepatocyte injury during the ongoing inflammatory process in the liver parenchyma (45). These observations may additionally support a protective role of visfatin against the liver injury.
A study by Dahl et al. (46) in patients with NAFLD showed that liver visfatin expression and its serum level were markedly decreased, with no difference between simple steatosis and NASH. Within the liver, visfatin was located to hepatocytes. An intriguing finding of this study was that visfatin inhibited apoptosis of hepatocytes in vitro. The antiapoptotic effect of visfatin in hepatocytes involved enzymatic synthesis of NAD (46). Because hepatocyte apoptosis is an important feature of chronic hepatitis, downregulation of visfatin in advanced inflammatory processes has possible pathogenic consequences and also suggests a hepatoprotective role for visfatin.
TNFα is a proinflammatory cytokine/adipokine that is elevated and positively associated with the inflammatory activity grade and fibrosis stage in CHC (47,48). Visfatin increases TNFα production in human peripheral blood mononuclear cells and in murine liver hepatocytes (25). TNFα initiates apoptosis in hepatocytes (49) and upregulates expression of vascular adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) in liver endothelial cells, facilitating migration of leukocytes to the inflammation site (50). Visfatin may also induce VCAM-1 and ICAM-1 synthesis directly in endothelial cells and leukocytes by activation of nuclear factor (NF)-κB (25,51). Both these adhesion molecules are significantly increased in CHC (47,52), and serum ICAM-1 concentration is associated with the inflammatory activity grade (47). These findings suggest that visfatin directly, together with TNFα, or through induction of TNFα, may enhance production of adhesion molecules and therefore may have a pivotal role in the regulation of the necro-inflammatory process in the liver and facilitates migration of immune cells to the site of inflammation. On the other hand, in patients with NAFLD, TNFα levels in visceral adipose tissue were shown to be inversely associated with visceral visfatin levels, suggesting that TNFα downregulates visfatin expression (43).
These interesting but contradictory observations indicate that further studies are necessary to elicit the exact role of visfatin in liver tissue inflammation (24,25,32,38).
Visfatin and Angiogenesis in Chronic Hepatitis
Angiogenesis is another phenomenon observed in CHC, which influences disease progression (53,54). In CHC, the angiogenesis is markedly increased and positively associated with necro-inflammatory activity and fibrosis stage (55). It has not been resolved as to whether angiogenesis merely represents a homeostatic mechanism aimed at ensuring an adequate oxygen supply to the site of inflammation or whether it has an additional pathogenic role leading to liver tissue damage facilitating fibrogenesis (54). Development of fibrosis results in constriction of fibroblasts, sinusoid capillarization and disturbance of the liver architecture and, together with accumulation of inflammatory cells that occur in viral hepatitis, may increase resistance of liver tissue to blood flow and oxygen supply (56). Under these circumstances, an angiogenesis switch occurs, leading to an increase in the proangiogenic factors contributing to vascular remodeling and formation of new vessels (56). On the other hand, the process of liver chronic wound healing typical of fibrogenic chronic liver diseases is characterized by an overexpression of the same proangiogenic growth factors (54). Also, the exact role of the virus in pathogenesis of angiogenesis has not been clearly defined. Vascular endothelial growth factor (VEGF) and hepatocyte growth factor are the main proangiogenic agents (54), levels of which were found to be significantly increased in CHC (13,57). Matrix metalloproteinases (MMPs) and their tissue inhibitors also play a key role in angiogenesis development and progression. They regulate remodeling and degradation of the extracellular matrix and therefore facilitate proliferation and migration of endothelial cells (ECs), which results in the formation of new blood vessels (58).
Visfatin was found to induce expression of genes and proteins for MMP-2, MMP-9 and VEGF and its receptor (VEGF-R2) in human umbilical vein endothelial cells in a dose-dependent manner (28). Simultaneously, visfatin inhibits expression of genes and proteins for tissue inhibitors of matrix metalloproteinases (TIMP)—TIMP-1 and TIMP-2. Inhibition of VEGF and VEFG-R2 results in downregulation of the expression of MMPs induced by visfatin (28). Visfatin increases proliferation, migration of ECs and formation of new blood vessels in a dose-dependent manner. Moreover, it decreases apoptosis of ECs (28).
Visfatin influences the angiogenic process by activation of phosphatidylinositol 3-kinase (PI3K), protein kinase B (PKB/Akt) and extracellular signal-regulated kinase 1/2 (ERK1/2) (p42/p44 mitogen-activated protein kinase [MAPK]) (28). Because angiogenesis and enlarged extracellular matrix production promote fibrosis, the ability of visfatin to increase MMPs, VEGF and its receptor and to inhibit TIMP synthesis illustrates its potential involvement in the pathogenesis of these processes in chronic hepatitis. However, further investigations are necessary to determine the exact role of visfatin in these processes.
Visfatin, Insulin Resistance and Steatosis in Chronic Hepatitis
Recently, more attention has been focused on the metabolic aspects of CHC. IR is a hallmark of metabolic disturbances. HCV may evoke IR both directly, with its core protein, or indirectly, by induction of cytokines (1). The mechanism of IR in CHC is complex and still not clearly defined (1,59,60) and is described in Figure 2. Exacerbation of IR augments fibrosis progression and inflammatory activity (1–3,59). Markedly reduced phosphorylation of some mediators of the insulin pathway—for example, PI3K, PKB/Akt and MAPK, observed in CHC, provokes disturbances of carbohydrate and lipid metabolism (1,60). Visfatin increases phosphorylation of all of these mediators (28). Moreover, visfatin increases phosphorylation of insulin receptor substrate (IRS)-1 (61), which is inhibited by proinflammatory cytokines and direct action of the virus (1,60). This observation shows that the potential protective action of visfatin against IR is enhanced by HCV. Additionally, visfatin improves insulin receptor sensitivity (29) and owing to its action as nicotinamide phosphoribosyltransferase (Nampt) increases synthesis of NAD and nicotinamide mononucleotide, enhancing pancreatic β cells and improving insulin production and secretion (24). Alongside the direct effect of the virus, TNFα and IL-6 play an important role in IR development. Levels of both of these agents are significantly increased in CHC (47,48). The ability of visfatin to induce their synthesis might suggest its adverse effect on insulin sensitivity (25). A further observation pointing to the unfavorable role of visfatin in glucose metabolism is its influence on NF-κB synthesis and release of reactive oxygen species (25,51). Further investigations are necessary to delineate the precise role of visfatin in regulation of IR, not only in CHC.
The relationship between visfatin and liver steatosis in CHC is also unresolved. No association was found between the grade of liver steatosis and serum visfatin concentration in patients with CHC infected with genotype 1b (38). It should be mentioned that steatosis was present in 35% of patients with CHC and, in the majority, it encompassed <33% of the lobule area. The small area of steatosis was a limitation of this study, impeding clear interpretation of the results obtained (38). Similar results were found by Baranova et al. (41) in patients with CHC infected with genotype 1b or 3. On the other hand, Aller et al. found that serum visfatin was not related to steatosis grade and did not differ between patients with low-grade (<33% of the lobule area) and high-grade (>33% of lobule area) steatosis in overweight and obese patients with NAFLD, but IR was significantly increased in a patient with NAFLD with high-grade steatosis (42). In another study, Gaddipati et al. (43) showed that a significant decline in the visceral adipose tissue visfatin level was associated with the grade of steatosis in patients with NAFLD (43).
Visfatin was found to decrease the serum cholesterol level and increase peroxisome proliferator-activated receptor (PPAR)-γ expression (61). However, Chang et al. (37) reported that visfatin mRNA expression in visceral adipose tissue was positively correlated with fasting triglycerides, total cholesterol levels and steady-state plasma glucose measured with a modified insulin suppression test, but not with BMI in obese men and women (BMI 42.01 and 38.89 kg/m2, respectively) (37). Moreover, mRNA visfatin expression was strongly correlated with the TNFα gene in subcutaneous and visceral fat. A study by Catalan et al. (62) found that total cholesterol, high-density lipoprotein (HDL) cholesterol and triglycerides were significant and independent determinants of circulating concentrations of visfatin in obese patients (BMI 44.9 kg/m2). A positive correlation after BMI adjustment was found with the hepatic enzymes alanine aminotransferase (ALT), aspartate aminotransferase and γ-glutamyltransferase, which are commonly increased in obese patients with fatty liver disease.
Visfatin as a regulator of cell energy controls NAD synthesis. NAD is a coenzyme with important roles in a variety of biological processes, partly through activation of sirtuin-1 involved in control of the metabolic processes (63). It has been suggested recently that an increase of sirtuin-1 exerts protective effects against the development of NAFLD in rats, preventing lipid accumulation in the liver (64). Presuming that steatosis results from IR and lipid abnormalities, visfatin is probably an important participant in the pathogenesis of liver steatosis in CHC.
Visfatin and Fibrosis in Chronic Hepatitis
Many studies point to an essential role for some adipokines in the pathogenesis of liver fibrosis (11,12). Circulating visfatin levels are significantly decreased in liver cirrhosis of different origin—namely, posthepatic, alcohol and biliary cirrhosis, compared with healthy controls, presumably owing to decreased hepatic expression and production (65). The different underlying etiologies of liver cirrhosis had no significant impact on plasma visfatin levels or on hepatic visfatin production. Patients in the early clinical stages of cirrhosis—child class A liver cirrhosis— already had decreased plasma visfatin levels that were, however, significantly higher than those of patients with child class B or C liver cirrhosis. Plasma visfatin in cirrhosis is not associated with IR and plasma glucose but correlates with hepatic glucose production and the arterial ketone body ratio, indicating a potential link between the NAD-generating properties of visfatin and metabolism (65). In patients with NAFLD, there was no difference between individuals with and without fibrosis, but there were no data clarifying whether any of the patients analyzed had cirrhosis (42).
Similarly, there was no association between fibrosis stage and serum visfatin level in patients with CHC, either those infected with genotype 1b (38) or genotype 3. Nevertheless, the levels of visfatin were significantly higher than in healthy volunteers (41). Visfatin concentration did not differ between patients with portal, periportal or bridging fibrosis. However, the lack of cirrhotic patients in the investigated group limits useful interpretation of the results. IR is a well-defined risk factor for liver fibrosis (1–3), but hyperglycemia per se also induces fibrosis progression (66). Additionally, regulation of synthesis and activity of MMPs and their tissue inhibitors by visfatin (28) suggest that visfatin may influence liver fibrosis progression. Visfatin, with its ability to reduce the glucose level and increase insulin sensitivity (61), might potentially inhibit the fibrotic process. Moreover, activation of MMPs might facilitate removal of the extracellular matrix and suppress fibrosis progression.
The potential positive and negative aspects of visfatin action in liver pathology are summarized in Table 1. In view of the multifunctional properties of visfatin, further investigations are required to resolve its role in the pathogenesis of chronic hepatitis.
Chemerin Characteristics
Another member of the growing adipokine family is chemerin, also known as tazarotene-induced gene 2 (TIG2) or retinoic acid receptor responder protein 2 (RARRES2). Chemerin is a chemoattractant protein that acts as a ligand for the G-protein-coupled receptor: chemokine receptor-like 1 (CMKLR1) (also known as chemerin receptor 23 [ChemR23]). Chemerin is a protein secreted in an inactive form as prochemerin and activated through C-terminal cleavage by inflammatory and coagulation serine proteases (67). In humans, chemerin mRNA is highly expressed in white adipose tissue, liver and lungs, while its receptor, CMKLR1, is predominantly expressed in immune cells as well as adipose tissue (68).
On the one hand, chemerin was found to stimulate chemotaxis of dendritic cells, macrophages and NK cells toward the site of inflammation (10,69–71), and on the other hand, it was found to inhibit synthesis of proinflammatory mediators and to enhance adiponectin production (10,71,72). Chemerin has been associated with autocrine/paracrine signaling for adipocyte differentiation and maturation. It regulates glucose uptake in adipocytes and stimulates lipolysis (68,73,74). Studies using mature human adipocytes, 3T3-L1 cells and in vivo studies in mice showed that chemerin stimulates the phosphorylation of MAPK, ERK1 and ERK2, which are involved in mediating lipolysis and the insulin-signaling pathway (73). Blocking chemerin or its receptor induces synthesis of IL-6 and the insulin receptor while decreasing expression of glucose transporter 4 (GLUT4) and adiponectin (73). Serum chemerin levels in humans are related to BMI, concentration of triglycerides and total cholesterol, levels of blood pressure and IR (68,73–76). The action of chemerin is shown in Figure 4.

Action of chemerin.
Chemerin and Necro-Inflammatory Activity in Chronic Hepatitis
The only available study investigating serum chemerin in CHC showed that its serum concentrations were higher in patients with CHC than in the control group (77). Similarly to visfatin, serum chemerin levels were negatively associated with necro-inflammatory activity grade. The highest concentration was observed in patients with minimal inflammation, whereas the lowest was in individuals with moderate/severe inflammation. However, the concentration was still more than twice as high as in healthy volunteers (77). These results suggest that chemerin is an adipokine involved in the inflammatory process in CHC. The decrease of serum chemerin when inflammation is more severe may be explained by the fact that chemerin may bind to its receptor on activated inflammatory cells and migrate to the site of inflammation, aggravating the inflammatory response and hepatocyte injury. NK cells play a pivotal role in innate immunity against HCV infection in acute hepatitis C, helping to eradicate the virus. The ability of chemerin to activate NK cells shows its potential involvement in the antiviral response in acute hepatitis C. The relationship between necro-inflammatory activity and chemerin was also observed in NAFLD. The study showed that serum chemerin was significantly increased in patients with NASH compared with those with simple steatosis. Moreover, the NAFLD activity score was positively associated with serum chemerin (76).
On the other hand, chemerin inhibits production of proinflammatory TNFα and IL-6 (72). As mentioned above, both these cytokines are upregulated in CHC (44,47). Chemerin restricts the harmful proinflammatory activity of these cytokines by inhibiting their synthesis (10,72) and may exert a protective effect against liver injury. These findings suggest that chemerin may, on the one hand, initiate and strengthen the acute phase of the inflammatory response, but on the other, it may facilitate the extinguishing or reduction of chronic inflammation.
Chemerin and Insulin Resistance in Chronic Hepatitis
Inhibition of TNFα and IL-6 by chemerin upregulates IRS-1 phosphorylation and enhances adipocytes insulin sensitivity (72,74). These observations may point to a possible action of chemerin in regulation of IR. However, another study showed that chemerin induces IR in human skeletal muscle cells (78). Chemerin influences phosphorylation of IRS-1, PKB/Akt and glycogen synthase kinase 3. Additionally, it activates p38MAPK, NF-κB or ERK1/2. Serum chemerin was positively correlated with the level of C-reactive protein, fasting insulin, triglycerides, ALT activity and the homeostasis model of insulin resistance (HOMA-IR) in patients with type 2 diabetes (79) and nondiabetic obese patients (80). There was no relationship between serum chemerin and HOMA-IR, waist circumference, serum lipids concentrations, fasting insulin and ALT activity in patients with NAFLD (76). Similarly, in CHC, there was no association between serum chemerin and HOMA-IR, waist circumference, serum lipids concentration or fasting insulin (77). Serum chemerin did not differ significantly between CHC patients with HOMA-IR ≥3 and <3. The limitation of the study was that it did not include obese patients with CHC, and the average BMI of the patients studied was 25.0 kg/m2 (77). Therefore, definitive analysis of the influence of chemerin on insulin sensitivity and association with BMI in patients with CHC is difficult.
Chemerin and Fibrosis in Chronic Hepatitis
The observation of a higher chemerin level in hepatic venular blood than in systemic arterial and portal blood suggests that the liver is a pivotal source of this adipokine (79). Moreover, its concentration in hepatic venular blood was higher in patients with child A liver cirrhosis than in those with child B or C liver fibrosis (79). In patients with CHC, there was no association between serum chemerin and fibrosis stage (76). However, serum chemerin concentration tended to be higher in patients with more advanced fibrosis. Clear interpretation of the results was limited because the study included patients with portal and periportal fibrosis but not patients with cirrhosis (76). Definitive exclusion of an association between chemerin and liver fibrosis is not possible because of the ability of chemerin to enhance synthesis of transforming growth factor (TGF)-β (72). Chemerin activates the pathway dependent on PI3K/Akt and MAPK in ECs, activating angiogenesis and synthesis of MMPs (81). The ability of chemerin to induce production of MMPs suggests its possible involvement in the pathogenesis of liver fibrosis and points to its potential antifibrogenic effect.
The potential positive and negative aspects of chemerin action in the liver are summarized in Table 2.
Vaspin Characteristics
Vaspin (visceral adipose tissue-derived serine protease inhibitor) is an adipokine that has been isolated from both visceral and subcutaneous white adipose tissue. Visceral vaspin expression significantly correlated with BMI, percentage of body fat and the level of plasma glucose after 2 h of oral glucose tolerance testing, whereas its subcutaneous expression significantly correlated with waist-to-hip ratio, fasting plasma insulin concentration and glucose infusion rate during the steady state of a euglycemic-hyperinsulinemic clamp. Insulin sensitivity, together with percentage of body fat, appeared to be the strongest determinant of subcutaneous vaspin expression. Some studies indicated that the induction of vaspin mRNA expression in human adipose tissue might be a compensatory mechanism associated with obesity and IR (82,83). Vaspin suppresses leptin, TNFα and resistin expression (10). Administration of recombinant vaspin significantly improved insulin sensitivity and glucose tolerance (84). The characteristics of vaspin action are presented in Figure 5.

Action of vaspin.
Vaspin in Chronic Hepatitis
Contrary to serum visfatin and chemerin, vaspin concentration decreased significantly in patients with CHC and was not associated with inflammatory activity (77). Vaspin correlated positively with fasting glucose in patients with CHC (77). This result supports other findings that the induction of vaspin mRNA expression in human adipose tissue might be a compensatory mechanism associated with obesity and IR (82,83). As mentioned above, vaspin suppresses leptin, TNFα and resistin expression (10). Amelioration of the inflammatory process may help to improve IR. The lower levels of vaspin found in patients with CHC (77) may result in TNFα overexpression and IR development and therefore more pronounced disease progression.
There was no association between the severity of hepatic fibrosis and the level of vaspin. Nevertheless, serum vaspin was higher, although not significantly, when fibrosis was more advanced (77). That study group did not include patients with CHC with cirrhosis, which limits data interpretation and may influence the association between cytokine levels found in that study and the stage of fibrosis (77).
However, the possibility of an association of vaspin with fibrogenesis cannot be excluded. Vaspin decreases production of a profibrogenic factor (leptin [10]), but in an analyzed group of patients with CHC, no association was found between vaspin and leptin serum concentrations. Moreover, leptin concentration was not related to the stage of fibrosis (77). Additionally, upregulation of vaspin as a compensatory mechanism in IR may also protect against fibrosis development and progression.
Similarly, a study on patients with NAFLD showed that their vaspin serum level was lower than that in healthy controls (76). Levels of vaspin were significantly upregulated in patients with NASH compared with patients with simple steatosis. There was no difference in vaspin concentration between NAFLD patients with different grades of lobular and portal inflammation or with various fibrosis stages. Vaspin was positively associated with hepatocyte ballooning degeneration (76). On the other hand, a study by Aktas et al. (85) showed that vaspin was a predictor of liver fibrosis in NAFLD, independent of potential confounders, including metabolic parameters. Serum vaspin levels showed a statistically significant association with liver fibrosis. After stepwise linear regression analysis, serum vaspin levels were the only independent predictor of liver fibrosis scores in patients with NAFLD (85). All these results suggest a possible involvement of vaspin in liver fibrogenesis, but further investigations are necessary to elucidate its exact role in liver fibrosis.
In human studies, Youn et al. (86) found sexual dimorphism in the level of circulating vaspin, with a higher concentration in women than in men only in normal glucose-tolerant patients but not in patients with T2DM. Elevated serum vaspin was associated with obesity and impaired insulin sensitivity in normal glucose-tolerant patients, whereas T2DM seemed to abolish this correlation (86). Similarly, Seeger et al. (87) found that vaspin serum concentration was significantly higher in women and that gender was an independent predictor of circulating vaspin. There was no difference in vaspin serum concentration between men and women in both NAFLD and CHC (76,77). Vaspin levels were not associated with IR or BMI in CHC (77). HOMA-IR was significantly higher in patients with CHC, but there was no difference in vaspin level between patients with differing HOMA-IR values (77). Hepatitis C virus may induce IR by direct action on the insulin-signaling pathways (1,84), therefore influencing our results.
Leptin Characteristics
Leptin is a protein encoded by the obese (ob) gene (88). Expression of leptin is predominant in adipose tissue and is determined mainly by the status of energy stores in white adipose tissue and the size of adipocytes (89). Leptin receptors (ObR) are expressed in a broad range of peripheral tissues, including the liver, and have isoforms as a result of alternative splicing. The level of secretion of leptin is associated with the fat mass and provides antiobesity signals, regulating food intake, sympathetic tone and energy expenditure in conditions of energy excess. Obese patients have an increased leptin concentration. This is the result of leptin resistance in these subjects, which is caused by abnormal leptin transport and disturbances in ObR signaling, including overexpression of the suppressor of cytokine signaling-3 (SOCS3), an agent that inhibits leptin signaling (90). Leptin influences innate and adaptive immunity. Leptin-deficient mice are protected from injury in models of autoimmune disease and T-cell-mediated hepatitis induced by concanavalin A injection (91,92). On the other hand, leptin-deficient mice are more susceptible to bacterial and viral infections, and this reflects increased hepatotoxicity after administration of endotoxin (93). Leptin generally acts as a proinflammatory agent and participates in protection from bacterial and viral infections. In the damaged liver, leptin exacerbates the inflammatory response, via NF-κB-mediated stimulation of chemokine expression such as monocyte chemoattractant protein-1 (94).
Leptin in Chronic Hepatitis
Since Potter et al. (95) first demonstrated that rat hepatic stellate cells (HSCs) express leptin during the process of transactivation, more attention has been focused on its role in hepatic fibrosis. Leptin appeared to be an essential prerequisite for the development of liver fibrosis (95,96). In mice, deficiency of leptin or impaired leptin receptor signaling reduces fibrogenesis, whereas administration of recombinant leptin during acute or chronic liver injury increases fibrogenesis (97,98). Fibrogenesis in the liver results from the activation of different cell types. Leptin promotes fibrogenesis indirectly by activation of Kupffer cells, macrophages and sinusoidal endothelial cells through upregulation of TGFβ1 production (11,12,96,99,100) and directly by activation of HSCs (100,101). Leptin increases expression of procollagen I, TGFβ1 and α-smooth muscle actin in activated HSCs (96,100). It promotes fibrogenesis by stimulation of TIMPs and inhibition of MMP-1 (102,103). Leptin seems to increase proliferation and to inhibit apoptosis of HSCs through ERK and the Akt-dependent pathway (104). The involvement of leptin in liver fibrosis regulation is summarized in Figure 6.

Leptin and liver fibrosis. NADPH, nicotinamide adenine dinucleotide phosphateoxidase; ROS, reactive oxygen species; SEC, sinusoidal endothelial cell.
Activated HSCs lead to leptin overexpression, whereas low leptin levels in quiescent HSCs are related to increased synthesis of adiponectin (105). An additional action of leptin that is important in fibrogenesis is its regulation of hepatic angiogenesis (106). Activation of ObRs in HSCs contributes to increased production of VEGF and angiopoietin-1 and up-regulates monocyte chemoattractant protein-1 (94). Leptin also acts on ECs, stimulating their proliferation and production of reactive oxygen species (107). Recently, leptin has been shown to promote HCC development, both directly and through upregulation of angiogenesis (11,108). ObRs are expressed at higher levels in HCC, especially in poorly differentiated HCCs, which exert higher vascularization and ObR expression. Moreover, leptin promotes proliferation, migration and invasiveness of HCC cells (109). Therefore, in chronic liver diseases, leptin may facilitate HCC development by promotion of fibrogenesis, induction of angiogenesis and direct stimulation of the proliferation of cancer cells via the ERK/MAPK and PI3K/Akt pathways (108,109).
The role of leptin in steatosis, inflammation, fibrosis and IR in CHC is still not clearly understood. Increased levels of leptin have been found in patients with CHC compared with healthy controls in some studies (13,77,110), whereas in other studies, comparable (111) or even lower leptin levels have been described (112). The estimation of a possible role of leptin in fibrogenesis in CHC has produced conflicting results. A relationship with fibrosis severity was found in some (113,114) but not in other reports (13,77,115). Serum leptin was found to be higher in patients with cirrhosis during the course of chronic viral hepatitis (20 due to chronic hepatitis B and 15 due to CHC) (116). Moreover, another study showed a significant association between serum leptin and fibrosis stage in HCV- and hepatitis B virus-infected patients (117).
Leptin protects against fatty liver directly by activation of adenosine monophosphate-activated protein kinase (AMPK) (10) and also by lowering the expression of sterol regulatory element binding protein (SREBP)-1 (91). Fatty liver in obese patients with higher levels of leptin may result from IR. The association of steatosis with leptin concentration in CHC is also unclear (118). In one study, leptin levels were correlated with steatosis grade only in univariate analysis (119). Another study showed an association but only in patients infected with genotype 1 but not with genotype 3 HCV (118). Other reports did not demonstrate changes in leptin concentrations with changes in steatosis grade (77,111,114).
In summary, these studies indicate that although serum leptin levels may be altered in patients with CHC, results for an association between serum leptin and liver histology in these patients are conflicting. Further studies must be performed to ascertain more precisely the role of leptin in CHC. The influence of leptin on liver tissue histology is described in Figure 7.

Influence of leptin on liver tissue alterations in chronic hepatitis. FFA, free fatty acid; ROS, reactive oxygen species.
Adiponectin Characteristics
Adiponectin circulates in complexes of different size, but its metabolic activities are mostly associated with the high-molecular weight form (10). Adiponectin binds to two specific receptors, AdiopoR1 and AdipoR2. AdiopR2 is mainly expressed in the liver, whereas AdipoR1 is found in other tissues, including skeletal muscles (120). The main downstream effector for AdiopR2 is PPARα, whereas for AdipoR1, it is AMPK (106). Adiponectin serum levels are inversely related to body fat mass and are decreased in obesity and T2DM. Adiponectin exerts an insulin-sensitizing effect in adipose tissue, skeletal muscles and liver (10,121).
Adiponectin in Chronic Hepatitis
Adiponectin has a hepatoprotective and antifibrogenic effect in cases of liver injury and protects against liver steatosis (122,123). In NAFLD and alcoholic hepatitis, adiponectin attenuates inflammatory activity and steatosis by downregulating SREBP-1 activity and suppressing TNFα production (124,125). Moreover, adiponectin increases PPARα expression in the liver (11,12,124,126). A modest increase in adiponectin concentration in ob / ob mice leads to an increase of PPARγ expression in the adipose tissue and ameliorates necro-inflammatory activity. In this model, adiponectin provides signals that transport triglycerides to the adipose tissue, sparing the liver and the skeletal muscles, and reduces IR (11,12,127).
Adiponectin also affects glucose metabolism via AMPK activation, which downregulates glucose synthesis in the liver (128). Moreover, adiponectin inhibits protein tyrosine phosphatase 1B, which results in an improvement of insulin sensitivity (100,128).
Adiponectin and leptin both counteract ectopic fat deposition, but they have opposite effects on the inflammatory process. Adiponectin ameliorates inflammation because it increases synthesis of antiinflammatory IL-10, decreases production of TNFα and IL-6 and inhibits the activation of NF-κB (124,129). On the other hand, the inflammatory process downregulates adiponectin synthesis and decreases its level in serum. Studies on leptin-deficient mice have shown that ob / ob mice are protected from T-cell-mediated hepatitis, whereas lipodystrophic mice, which lack both adiponectin and leptin, are not, suggesting a pivotal role for adiponectin in protection from liver damage. Moreover, adiponectin administration protected both lipodystrophic and ob / ob mice, whereas leptin administration worsened their condition (11,12,130). Adiponectin also blocks Fas-mediated hepatocyte apoptosis, which is implicated in CHC pathogenesis (131).
Adiponectin reduced fibrogenesis in mice treated with CCl4. This effect was independent of its metabolic action and was associated with modulation of HSCs, which express both adiponectin receptors (105). A pivotal step in the antifibrogenic action of adiponectin is mediated by activation of AMPK (11,12,132). Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis by upregulating SOCS3 in mice. Adiponectin stimulates MMP-1 activity and blocks leptin-induced TIMP-1 expression in mice HSCs (133). The hepatoprotective activity of adiponectin is described in Figure 8.

Influence of adiponectin on liver tissue alterations in chronic hepatitis. FFA, free fatty acid; ROS, reactive oxygen species.
Studies of adiponectin in CHC have produced conflicting results. Some investigations showed that serum adiponectin was not altered in CHC (134,135), but others showed that adiponectin levels were significantly lower in CHC (136,137) and, additionally, were associated with high viral load and genotype 2 infection (134). CHC with metabolic syndrome was associated with lower adiponectin levels and higher IR. IR and visceral obesity were independently associated with adiponectin levels (136).
Results are contradictory also for the relationship between serum adiponectin concentrations and histopathological alteration in the liver in CHC (138). Petit et al. (137) found a correlation between serum adiponectin concentration and hepatic steatosis, but in the study by Jonsson et al. (139), such a relationship was found only in men with CHC. In the study by Durente-Mangoni et al. (140), hypoadioponectinemia in CHC was associated with the grade of hepatic steatosis, and a changed balance between adiponectin and TNFα was suggested. Patients with HCV genotype 4 infection with steatosis showed lower serum adiponectin levels and higher levels of leptin. Low adiponectin levels in these patients were independently and inversely associated with the grade of steatosis and HOMA-IR (141). In patients infected with HCV genotype 3, adiponectin levels were reduced, independently of the extent of steatosis. Adiponectin levels are increased after successful antiviral treatment (142). Moreover, patients with HCV genotype 1 infection had significantly higher IR than patients with genotype 2 infection and lower serum adiponectin concentration (134,143). This observation points not only to the direct influence of HCV on adiponectin synthesis, but also to a genotype-specific difference in inducing IR and adiponectin level. There are two possible explanations: the influence of the degree of histopathological changes in the liver on adiponectin concentration (143) or the existence of HCV-specific differences in AdipoR1 and AdipoR2 gene expression (134).
Additionally, low adiponectin levels are associated with poor response to antiviral therapy (144). In the study by Palmer et al. (145) high concentrations of total and high-molecular weight adiponectin were related to the presence of a cellular immune response against HCV, suggesting a pivotal role for adiponectin in the regulation of immunity in CHC. The study by Latif et al. (146) showed that serum adiponectin levels inversely correlate not only with the grade of steatosis and histological activity index, but also with the stage of fibrosis (146).
Adiponectin together with IR seems to be implicated in hepatocarcinogenesis. Serum levels of leptin and insulin, HOMA and BMI were significantly higher, and serum adiponectin was significantly lower, in patients with HCC at the time of tumor detection than in control patients >5 years after sustained virological response (147). Hepatic fibrosis may be closely related to the emergence of HCC after sustained virological response. IR and adipocytokine disorders may be implicated in hepatocarcinogenesis after sustained virological response, in part by promoting hepatic fibrosis (147). The protective action of adiponectin in chronic hepatitis against fibrosis, inflammation and steatosis is described in Table 3.
In conclusion, adipokines are important participants in the pathogenesis of chronic hepatitis. However, many aspects of their action and influence on liver disease pathogenesis need to be extensively investigated and elucidated.
Summary
1. CHC is associated with metabolic abnormalities.
2. Metabolic abnormalities and obesity promote progression of chronic liver disease in CHC and NAFLD.
3. Studies on adipokines as pivotal regulators of metabolic processes and inflammatory response carried out over the past few years suggest that adipokines play a role in pathogenesis of inflammatory reaction and fibrogenesis in the liver during chronic hepatitis, including CHC and NAFLD. Adipokines play a role in a pathogenesis of chronic hepatitis indirectly by regulation of metabolic processes and directly by interfering with the inflammatory process and fibrosis.
4. Regulation of insulin sensitivity by adipokines seems to play a key role in fibrosis progression.
5. Adiponectin and leptin are the best known adipokines. Adiponectin exerts hepatoprotective action, whereas leptin promotes fibrosis progression.
6. Recent studies point to the potential role of novel adipokines (visfatin, chemerin and vaspin) in chronic hepatitis pathogenesis.
7. Data relating to visfatin, chemerin and vaspin in CHC and NAFLD are limited. Further investigations involving a larger number of patients are required to better determine the exact role of novel adipokines in CHC and NAFLD. Precise evaluation of the role of adipokines in chronic hepatitis would facilitate management and new therapeutic approaches.
Disclosure
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Bernsmeier C, Heim MH. (2009) Insulin resistance in chronic hepatitis C: mechanisms and clinical relevance. Swiss Med. Wkly. 139:678–84.
Hui JM, et al. (2003) Insulin resistance is associated with chronic hepatitis C virus infection and fibrosis progression. Gastroenterology 125, 1695–704.
Fartoux L, et al. (2005) Insulin resistance is a cause of steatosis and fibrosis progression in chronic hepatitis C. Gut 54:1003–8.
Romero-Gomez M, et al. (2005) Insulin resistance impairs sustained response rate to peginterferon plus ribiavirin in chronic hepatitis C patients. Gastroenterology. 128:636–41.
Lecube A, et al. (2007) Glucose abnormalities are an independent risk factor for non-response to antiviral treatment in chronic hepatitis C. Am. J. Gastroenterol. 102:2189–95.
Veldt BJ, et al. (2008) Increased risk of hepatocellular carcinoma among patients with hepatitis C cirrhosis and diabetes mellitus. Hepatology. 47:1856–62.
Negro F, Sanyal AJ. (2009) Hepatitis C virus, steatosis and lipid abnormalities: clinical and pathogenic data. Liver Int. 29 Suppl 2:26–37.
Matos C, et al. (2006) Steatosis in chronic hepatitis C: relationship to the virus and host risk factors. J. Gastroenterol. Hepatol. 21:1236–39.
Asselah T, Rubbia-Brandt L, Marcellin P, Negro F. (2006) Steatosis in chronic hepatitis C: why does it really matter? Gut 55:123–30.
Rabe K, et al. (2008) Adipokines and insulin resistance. Mol. Med. 14:741–51.
Marra F, Bertolani C. (2009) Adipokines in liver diseases. Hepatology. 50:957–69.
Bertolani C, Marra F. (2008) The role of adipokines in liver fibrosis. Pathophysiology. 15:91–101.
Zwirska-Korczala K, et al. (2005) Leptin, neopterin and hepatocyte growth factor as markers of fibrosis and inflammatory activity in chronic hepatitis C. Exp. Clin. Hep. 1:OR60–5.
Shoelson SE, Lee J, Goldfine AB. (2006) Inflammation and insulin resistance. J. Clin. Invest. 116:1793–801.
Greenberg AS, Obin MS. (2006) Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 83:461S–465S.
Kukla M, et al. (2007) Liver tissue alterations in morbidly obese patients undergoing bariatric surgery. Exp. Clin. Hep. 3:OR12–8.
Rector RS, Thyfault JP, Wei Y, Ibdah JA. (2008) Non-alcoholic fatty liver disease and the metabolic syndrome: an update. World J. Gastroenterol. 14:185–92.
Adams LA, Angulo P. (2005) Recent concepts in non-alcoholic fatty liver disease. Diabet. Med. 22:1129–33.
Gabriel A, Kukla M, Ziolkowski A. (2008) Histopathological features and current scoring systems for semiquantitative assessment of nonalcoholic fatty liver disease. Exp. Clin. Hep. 4:RA48–54.
Naveau S, et al. (1997) Excess weight is a risk factor for alcoholic liver disease. Hepatology. 25:108–11.
Hourigan LF, et al. (1999) Fibrosis in chronic hepatitis C correlates significantly with body mass index and steatosis. Hepatology. 29:1215–9.
Wolk A, et al. (2001) A prospective study of obesity and cancer risk (Sweden). Cancer Causes Control. 12:13–21.
Kamada Y, Takehara T, Hayashi N. (2008) Adipocytokines and liver disease. J. Gastroenterol. 43:811–22.
Luk T, Malam Z, Marshall JC. (2008) Pre-B cell colony-enhancing factor (PBEF)/visfatin: visfatin novel mediator of innate immunity. J. Leukoc. Biol. 83:804–16.
Moschen AR, et al. (2007) Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 178:1748–58.
Samal B, et al. (1994) Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol. 14:1431–37.
Jia SH, et al. (2004) Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Invest. 113:1318–27.
Adya R, et al. (2008) Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 78:356–65.
Fukuhara A, et al. (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 307:426–30.
Arner P. (2006) Visfatin: a true or false trail to type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 91:28–30.
Lim SY, et al. (2008) The novel adipocytokine exerts direct cardioprotective effects. J. Cell. Mod. Med. 12:1395–403.
Jarrar MH, et al. (2008) Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol. Ther. 27:412–21.
Berndt J, et al. (2005) Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes. 54:2911–16.
Filippatos TD, Deremezis CS, Kiortsis DN, Tselepis AD, Elisaf MS. (2007) Increased plasma levels of visfatin/pre-B cell colony-enhancing factor in obese and overweight patients with metabolic syndrome. J. Endocrinol. Invest. 30:323–6.
Pagano C, et al. (2006) Reduced plasma visfatin/pre-B cell colony-enhancing factor in obesity is not related to insulin resistance in humans. J. Clin. Endocrinol. Metab. 91:3165–70.
Chen MP, et al. (2006) Elevated plasma level of visfatin/pre-B cell colony-enhancing factor in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 91:295–9.
Chang YC, Chang TJ, Lee WJ, Chuang LM. (2010) The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance and plasma lipids. Metab. Clin. Exp. 59:93–9.
Kukla M, et al. (2010) Serum visfatin in chronic hepatitis C. J. Viral. Hepat. 17:254–60.
Zwirska-Korczala K, et al. (2008) Postprandial response of ghrelin and PYY and indices of low-grade chronic inflammation in lean young women with polycystic ovary syndrome. J. Phys-iol. Pharmacol. 59 Suppl 2:161–7
Wang T, et al. (2006) Structure of Nampt/PBEF/visfatin, a mammalian NAD+ biosynthetic enzyme. Nat. Struct. Mol. Biol. 13:661–2.
Baranova A, et al. (2010) Association of serum adipocytokines with hepatic steatosis and fibrosis in patients with chronic hepatitis C. Digestion. 83:32–40.
Aller R, et al. (2009) Influence of visfatin on histopathological changes of non-alcoholic fatty liver disease. Dig. Dis. Sci. 54:1772–7.
Gaddipati R, et al. (2010) Visceral adipose tissue visfatin in nonalcoholic fatty liver disease. Ann. Hepatol. 9:266–70.
Ramadori G, Christ B. (1999) Cytokines and the hepatic acute-phase response. Semin. Liver. Dis. 19:141–55.
Selzner N, et al. (2002) Cold ischemia decreases liver regeneration after partial liver transplantation in the rat: a TNF-α / IL-6-dependent mechanism. Hepatology. 36:812–8.
Dahl TB, et al. (2010) Intracellular nicotinamide phosphoribosylotransferase protects against hepatocyte apoptosis and is down-regulated in nonalcoholic fatty liver disease. J. Clin. Endocrinol. Metab. 95:3039–47.
Kukla M, et al. (2008) Serum levels of sICAM-1, TNFα, sTNF-R1, and sTNF-R2 in patients with chronic hepatitis C treated with pegylated interferon a and ribavirin. Exp. Clin. Hep. 4:OR12–20.
Jonsson JR, et al. (2008) Obesity and steatosis influence serum and hepatic inflammatory markers in chronic hepatitis C. Hepatology. 48:80–7.
Ding WX, Yin XM. (2004) Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J. Cell. Mol. Med. 8:445–54.
Wolf D, et al. (2001) TNF-alpha-induced expression of adhesion molecules in the liver is under the control of TNFR-1-relevance for concanavalin A-induced hepatitis. J. Immunol. 166:1300–7.
Kim SR, et al. (2008) Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim. Biophys. Acta. 1783:886–95.
Kukla M, et al. (2009) sPECAM-1 and sVCAM-1: role in pathogenesis and diagnosis of chronic hepatitis C and association with response to antiviral therapy. Ther. Adv. Gastroenterol. 2:79–90.
Kukla M, et al. (2009) Angiogenesis in chronic viral hepatitis. Gastroenterol. Pol. 16:304–9.
Fernandez M, et al. (2009) Angiogenesis in liver disease. J. Hepatol. 50:604–20.
Gabriel A, et al. (2009) Angiogenesis in chronic hepatitis C is associated with inflammatory activity grade and fibrosis stage. Pathol. Res. Pract. 205:758–64.
Amarapurkar AD, et al. (2007) Angiogenesis in chronic liver disease. Ann. Hepatol. 6:170–3.
Salcedo X, et al. (2005) The potential of angiogenesis soluble markers in chronic hepatitis C. Hepatology. 42:696–701.
Jackson C. (2002) Matrix metalloproteinases and angiogenesis. Curr. Opin. Nephrol. Hypertens. 11:295–9.
Brensmeier C, et al. (2008) Virus-induced over-expression of protein phosphatase 2A inhibits insulin signalling in chronic hepatitis C. J. Hepatol. 49:429–40.
Aytug S, et al. (2003) Impaired IRS-1/PI3-kinase signaling in patients with HCV: a mechanism for increased prevalence of type 2 diabetes. Hepatology. 38:1384–92.
Sun Q, et al. (2009) Overexpression of visfatin/PBEF/Nampt alters whole-body insulin sensitivity and lipid profile in rats. Ann. Med. 41:311–20.
Catalan V, et al. (2011) Association of increased visfatin/PBEF/Nampt circulating concentrations and gene expressions levels in peripheral blood cells with lipid metabolism and fatty liver in human morbid obesity. Nutr. Metab. Cardiovasc. Dis. 21:245–53.
Hara N, et al. (2007) Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem. 282:24574–82.
Deng XQ, Chen LL, Li NX. (2007) The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 27:708–15.
de Boer JF, Bahr MJ, Böker KH, Manns MP, Tietge UJ. (2009) Plasma levels of PBEF/Nampt/visfatin are decreased in patients with liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 296:G196–201.
Ratziu V, et al. Fibrogenic impact of high serum glucose in chronic hepatitis C. J. Hepatol. 39:1049–55.
Zabel BA, Allen SJ, Kulig P. (2005) Chemerin activation by serine proteases of the coagulation fibrinolytic, and inflammatory cascades. J. Biol. Chem. 280:34661–6.
Bozaoglu K, et al. (2007) Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology. 148:4687–94.
Wittamer V, et al. (2003) Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198:977–85.
Moretta A, et al. (2008) NK cells at the interface between innate and adaptive immunity. Cell. Death Differ. 15:226–33.
Yoshimura T, Oppenheim JJ. (2008) Chemerin reveals a chimeric nature. J Exp. Med. 205:2187–90.
Cash JL, et al. (2008) Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J Exp. Med. 205:767–75.
Gorlaski L. (2007) Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 282:28175–88.
Takahasi M, et al. (2008) Chemerin enhances insulin signaling and potentiates insulin-stimulated glucose uptake in 3T3-L1 adipocytes. FEBS Lett. 582:573–8.
Stejskal D, et al. (2008) Chemerin is an independent marker of the metabolic syndrome in a Caucasian population: a pilot study. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 152:217–21.
Kukla M, et al. (2010) Serum chemerin and vaspin in nonalcoholic fatty liver disease. Scan. J. Gastroenterol. 45:235–42.
Kukla M, et al. (2010) Chemerin, vaspin and insulin resistance in chronic hepatitis C. J. Viral. Hepat. 17:661–7.
Sell H, et al. (2009) Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes. 58:2731–40.
Weigert J, et al. (2010) Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin. Endocrinol. (Oxf). 72:342–8.
Bozaoglu K, et al. (2009) Chemerin is associated with metabolic syndrome phenotypes in Mexican-American population. J. Clin. Endocrinol. Metab. 98:3085–9.
Kaur J, et al. (2010) Identification of chemerin receptor (ChemR23) in human endothelial cells: chemerin-induced endothelial angiogenesis. Biochem. Biophys. Res. Commun. 391:1762–8.
Klöting N, et al. (2006) Vaspin gene expression in human adipose tissue: association with obesity and type 2 diabetes. Biochem. Biophys. Res. Commun. 339:430–6.
Wada J. (2008) Vaspin: a novel serpin with insulin-sensitizing effects. Expert Opin. Investig. Drugs. 17:327–33.
Narita R, et al. (2004) Insulin resistance and insulin secretion in chronic hepatitis C virus infection. J. Hepatol. 41:132–8.
Aktas B, et al. (2011) Serum levels of vaspin, obestatin, and apelin-36 in patients with nonalcoholic fatty liver disease. Metabolism. 60:544–9.
Youn BS, et al. (2008) Serum vaspin concentration in human obesity and type 2 diabetes. Diabetes. 57:372–7.
Seeger J, et al. (2008) Serum levels of the adipokine vaspin in relation to metabolic and renal parameters. J. Clin. Endocrinol. Metab. 93:247–51.
Weisberg SP, et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112:1796–808.
Friedman JM, Halaas JL. (1998) Leptin and the regulation of body weight in mammals. Nature. 395:763–70.
Myers MG, Cowley MA, Munzberg H. (2008) Mechanisms of leptin action and leptin resistance. Ann. Rev. Physiol. 70:537–56.
Kakuma T, et al. (2000) Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc. Natl. Acad. Sci. U. S. A. 97:8536–41.
Faggioni R, et al. (2000) Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicology: role of tumor necrosis factor alpha and IL-18. Proc. Natl. Acad. Sci. U. S. A. 97:2367–72.
Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. (1997) Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. U. S. A. 94:2557–62.
Aleffi S, et al. (2005) Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 42:1339–48.
Potter JJ, Womack L, Mezey E, Anania FA. (1998) Transdifferentiation of rat hepatic stellate cells results in leptin expression. Biochem. Biophys. Res. Commun. 244:178–82.
Wang J, Brymora J, George J. (2008) Role of adipokines in liver injury and fibrosis. Expert. Rev. Gastroenterol. Hepatol. 2:47–57.
Ikejima K, et al. (2001) Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology. 34:288–97.
Leclerq IA, et al. (2002) Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J. Hepatol. 37:206–13.
Ikejima K, et al. (2002) Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 122:1399–410.
Bertolani C, Marra F. (2010) Role of adipocytokines in hepatic fibrosis. Curr. Pharm. Des. 16:1929–40.
Marra F. (2007) Leptin and liver tissue repair: do rodent models provide the answers? J. Hepatol. 46:12–8.
Cao Q, Mak KM, Ren C, Lieber CS. (2004) Leptin stimulates tissue inhibitor of metalloptoteinase-1 tissue inhibitor in human hepatic stellate cells: respective roles of the JAK/STAT and Jak-mediated H2O2-dependent MAPK pathways. J. Biol. Chem. 279:4292–304.
Cao Q, Mak KM, Lieber CS. (2007) Leptin repress matrix metalloproteinase-1 gene expression in LX2 human hepatic stellate cells. J. Hepatol. 46:124–33.
Saxena NK, et al. (2004) Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 18:1612–4.
Ding X, et al. (2005) The roles of leptin and adiponectin: a novel paradigm in adipocytikine regulation of liver fibrosis and stellate cell biology. Am. J. Pathol. 166:1655–69.
Anagnostoulis S, et al. (2008) Human leptin induces angiogenesis in vivo. Cytokine 42:353–57.
Rahmouni K, Haynes WG. (2005) Endothelial effects of leptin: implications in health and diseases. Curr. Diab. Rep. 5:260–6.
Kitade M, et al. (2006) Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology. 44:983–91.
Saxena NK, et al. (2007) Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 67:2497–507.
Tiftikci A, et al. (2009) Serum levels of adipokines in patients with chronic HCV infection: relationship with steatosis and fibrosis. Arch. Med. Res. 40:294–8.
Giannini E, et al. (2000) Leptin has no role in determining severity of steatosis and fibrosis in patients with chronic hepatitis C. Am. J. Gastroenterol. 95:3211–7.
Testa R, et al. (2000) Serum leptin levels in patients with viral chronic hepatitis or liver cirrhosis. J. Hepatol. 33:33–7.
Piche T, et al. (2004) The severity of liver fibrosis is associated with high leptin levels in chronic hepatitis C. J. Viral. Hepat. 11:91–6.
Crespo J, et al. (2002) Plasma leptin and TNF-alpha levels in chronic hepatitis C patients and their relationship to hepatic fibrosis. Dig. Dis. Sci. 47:1604–1.
Myers RP, Messous D, Poynard T, Imbert-Bismut F. (2007) Association between leptin, metabolic factors and liver histology in patients with chronic hepatitis C. Can. J. Gastroenterol. 21:289–94.
Bolukbas FF, et al. (2004) Child-Pugh classification dependent alterations in serum leptin among cirrhotic patients: a case controlled study. BMC Gastroenterol. 23:4–23.
Manolakopoulos S, et al. (2007) An assessment of serum leptin levels in patients with chronic viral hepatitis: a prospective study. BMC Gastroenterol. 7:17.
Myers RP, Messous D, Poynard T, Imbert-Bismut F. (2007) Association between leptin, metabolic factors and liver histology in patients with chronic hepatitis C. Can. J. Gastroenterol. 21:289–94.
Gordon A, McLean CA, Pedersen JS, Bailey MJ, Roberts SK. (2005) Hepatic steatosis in chronic hepatitis B and C: predictors, distribution and effect on fibrosis. J. Hepatol. 43:38–44.
Kadowaki T, et al. (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 116:1784–92.
Pittas AG, Joseph NA, Greenberg AS. (2004) Adipokines and insulin resistance. J. Clin. Endocrinol. Metab. 89:447–52.
Xu A, et al. (2003) The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver disease. J. Clin. Inves. 112:91–100.
Bastard JP, et al. (2006) Recent advances in the relationship between obesity, inflammation and insulin resistance. Eur. Cytokine Netw. 17:4–12.
Purohit V, Gao B, Song BJ. (2009) Molecular mechanisms of alcoholic fatty liver. Alcohol Clin. Exp. Res. 33:191–205.
Xu A, et al. (2003) The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest. 112:91–100.
Masaki T, et al. (2004) Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology. 40:177–84.
Kim JY, et al. (2007) Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Invest. 117:2621–37.
Andreelli F, et al. (2006) Liver adenosine monophosphate-activated kinase-alpha2 catatylic subunit is a key target for the control of hepatic glucose production by adiponectin and leptin but not insulin. Endocrinology. 147:2432–41.
Tilg H, Moschen AR. (2006) Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 6:772–83.
Sennello JA, et al. (2005) Regulation of T-cell-mediated hepatic inflammation by adiponectin and leptin. Endocrinology. 146:2157–64.
Wedemeyer I, et al. (2009) Adiponectin inhibits steatotic CD95/Fas up-regulation by hepatocytes: therapeutic implications for hepatitis C. J. Hepatol. 50:140–9.
Testa R, et al. (2000) Serum leptin levels in patients with viral chronic hepatitis or liver cirrhosis. J. Hepatol. 33:33–7.
Handy JA, et al. (2010) Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J. Cell Biochem. 110:1195–207.
Liu CJ, et al. (2005) Serum adiponectin correlates with viral characteristics but not histologic features in patients with chronic hepatitis C. J. Hepatol. 43:235–42.
Sato S, et al. (2005) Serum adiponectin concentration in patients with hepatitis C virus. J. Clin. Gastroenterol. 39:744–5.
Grigorescu M, et al. (2008) Metabolic syndrome, insulin resistance and adiponectin levels in chronic hepatitis C. J. Gastrointestin. Liver Dis. 17:147–54.
Petit JM, et al. (2005) Decreased plasma adiponectin concentration are closely related to steatosis in hepatitis C virus infected patients. J. Clin. Endocrinol. Metab. 90:2240–3.
Cua IH, et al. (2007) Insulin resistance and liver injury in hepatitis C is not associated with virus-specific changes in adipocytokines. Hepatology. 46:66–73.
Jonsson JR, Barrie HD, O’Rourke P, Clouston AD, Powell EE. (2008) Obesity and steatosis influence serum and hepatic inflammatory markers in chronic hepatitis C. Hepatology. 48:80–7.
Durente-Mangoni E, et al. (2006) Hepatic steatosis and insulin resistance are associated with serum imbalance of adiponectin/tumour necrosis factor-alpha in chronic hepatitis C patients. Aliment Pharmacol. Ther. 24:1349–57.
Ashour E, Samy N, Sayed M, Imam A. (2010) The relationship between serum adiponectin and steatosis in patients with chronic hepatitis C genotype-4. Clin. Lab. 56:103–10.
Wang AY, et al. (2005) High molecular weight adiponectin correlates with insulin sensitivity in patients with hepatitis C genotype 3, but not genotype 1 infection. Am. J. Gastroenterol. 100:2717–23.
Hsu CS, et al. (2008) High hepatitis C viral load is associated with insulin resistance in patients with chronic hepatitis C. Liver Int. 28:271–7.
Zografos TA, et al. (2008) Adiponectin: a new independent predictor of liver steatosis and response to IFN-alpha treatment in chronic hepatitis C. Am. J. Gastroenterol. 103:605–14.
Palmer C, Hampartzoumian T, Lloyd A, Zekry A. (2008) A novel role for adiponectin in regulating the immune responses in chronic hepatitis C virus infection. Hepatology. 48:374–84.
Latif HA, Assal HS, Mahmoud M, Rasheed WI. (2011) Role of serum adiponectin level in the development of liver cirrhosis in patients with hepatitis C virus. Clin. Exp. Med. 11:123–9.
Fukushima N, et al. (2010) Adipocytokine involvement in hepatocellular carcinoma after sustained response to interferon for chronic hepatitis C. Hepatol. Res. 40:911–22.
Acknowledgments
The authors thank Professor Fabio Marra for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kukla, M., Mazur, W., Bułdak, R.J. et al. Potential Role of Leptin, Adiponectin and Three Novel Adipokines—Visfatin, Chemerin and Vaspin—in Chronic Hepatitis. Mol Med 17, 1397–1410 (2011). https://doi.org/10.2119/molmed.2010.00105
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2010.00105