
Abstract
Diseases of mucosal inflammation represent important causes of morbidity and mortality, and have led to Intense research efforts to understand the factors that lead to their development. It is well accepted that a breakdown of the normally impermeant epithelial barrier of the intestine, the lung, and the kidney is associated with the development of inflammatory disease in these organs, yet significant controversy exists as to how this breakdown actually occurs, and how such a breakdown may lead to inflammation. In this regard, much work has focused upon the role of the epithelium as an “innocent bystander,” a target of a leukocyte-mediated inflammatory cascade that leads to its destruction in the mucosal inflammatory process. However, recent evidence from a variety of laboratories indicates that the epithelium is not merely a passive component in the steps that lead to mucosal inflammation, but is a central participant in the process. In addressing this controversy, we and others have determined that epithelial cells express Toll-like receptors (TLRs) of the innate immune system, and that activation of TLRs by endogenous and exogenous ligands may play a central role in determining the balance between a state of “mucosal homeostasis,” as is required for optimal organ function, and “mucosal injury,” leading to mucosal inflammation and barrier breakdown. In particular, activation of TLRs within intestinal epithelial cells leads to the development of cellular injury and impairment in mucosal repair in the pathogenesis of intestinal inflammation, while activation of TLRs in the lung and kidney may participate in the development of pneumonitis and nephritis respectively. Recent work in support of these concepts is extensively reviewed, while essential areas of further study that are required to determine the significance of epithelial TLR signaling during states of health and disease are outlined.
Similar content being viewed by others


The Clinical and Scientific Importance of Mucosal Inflammation
Standing at the interface between the host and the environment, mucosal-lined surfaces represent the first line of defense against potential pathogens. This defensive role is particularly relevant to the mucosa of the gastrointestinal tract, the pulmonary system, and the urinary tract, each of which is particularly susceptible to the development of inflammatory diseases due to their role as a barrier that must not only protect, but also serve the physiological function of each of the organ systems. In the case of the gastrointestinal tract, mucosal inflammation is manifest as inflammatory bowel disease (including Crohn’s disease and ulcerative colitis) (1–3) or necrotizing enterocolitis (NEC), a leading cause of death in preterm infants (4). In the case of the pulmonary system, mucosal inflammation may be manifest as pneumonitis, pneumonia, or asthma (5–7), acute and chronic pulmonary conditions that have a high degree of morbidity and potential mortality. And in the case of the urinary tract, mucosal inflammation may be manifest as interstitial nephritis, cystitis, and urethritis (8–10), causes of significant morbidity in patients of all ages. To elucidate the pathogenesis of mucosal inflammatory diseases, research over the past several decades has focused on the role of the immune system in their development—in particular the relationship between mucosal lymphocytes, macrophages, and neutrophils, and the effects of their cellular by-products on mucosal integrity and function (11–13). However, recent work has shed light upon the important role that the epithelia itself may play as a primary regulator of the immune response in the development of mucosal inflammation. No longer an innocent bystander, the epithelial-lined mucosa at each of these sites has been shown to possess all of the required armamentarium to allow an effective response to invading challenges, and to lead the battle to neutralize potential microbial threats (14–16). Not only is the epithelium able to respond to potentially dangerous microbial products, it also may sense endogenous molecules that are released during conditions of stress, hypoxia, or injury—so-called danger molecules that may play a critical role in the development of mucosal inflammation (17–19). In order, therefore, to understand the pathogenesis of mucosal inflammation and to assist in the rational design of anti-inflammatory strategies, it is necessary to define the receptors and signaling pathways that mediate the inflammatory response with respect to the epithelium itself.
The innate immune system consists of a series of receptors and their associated signaling molecules that is present both on leukocytes and epithelial cells through the body, and which initiates an immune response by responding directly to pre-formed ligands ([20] provides a recent review). The innate immune system lies in contradistinction to the adaptive immune system, a set of cellular and molecular interactions that must first “learn” how to deal with a potential pathogen, and then respond through the release of antibodies or other cellular derived products. The innate immune system includes pattern recognition receptors such as the TLRs, the NOD-like receptors (NLRs), the RIG-like receptors (RLRs), and C-type lectins, and their role in inflammatory signaling in leukocytes has been extensively reviewed (21–23,186). Relatively few reports have focused on the ability of the innate immune system to signal within the epithelium, although emerging evidence from a variety of laboratories including our own indicates that innate immune signaling within the epithelium plays a critical role in the pathogenesis of mucosal inflammation. The current review will focus on the TLR family, which has been shown to play a critical role in the response of epithelial cells to bacterial and endogenous ligands in the pathogenesis of various mucosal inflammatory diseases (Table 1).
Defining the Controversies in the Pathogenesis of Mucosal Inflammation
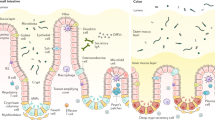
A central controversy in the field of mucosal inflammation may be stated as follows (Figure 1 provides a pictorial representation of this): Is mucosal inflammation a reflection of a leukocyte-driven immune response that has gone awry, resulting in tissue injury and the loss of mucosal barrier function (Figure 1A)? Or is it the mucosa itself, long known to play a role as a primary immune organ that is capable of producing a large number of pro-inflammatory molecules, that somehow has developed an exaggerated response that then leads to mucosal injury (Figure 1B)? Mucosal surfaces, such as the intestinal mucosa and the upper respiratory tract, are constantly exposed to environmental stimuli, such as commensal luminal bacteria in the intestine, as well as endogenous stimuli, or, as is the case in the lower respiratory tract and urinary tract mucosa, may encounter and respond to endogenous and exogenous stimuli in disease states. In either case, the mucosa must be able to mount an effective immune response, resist barrier failure, and coordinate this response with both mucosal and sub-mucosal leukocytes, while avoiding initiation and propagation of an exaggerated inflammatory response. But where does the answer lie in terms of what is initiating the mucosal inflammatory response?

Mechanisms by which TLR signaling leads to mucosal inflammation. As stated in the text, there are two potential mechanisms by which TLR signaling can lead to the development of mucosal inflammation. (A) TLR activation in response to DAMPs (damage associated molecular patterns) and PAMPs (pathogen associated molecular patterns) on leukocytes leads to the release of pro-inflammatory cytokines, resulting in epithelial destruction. (B) TLR activation by DAMPs and PAMPs on the epithelium itself directly impairs epithelial function and initiates the release of cytokines, leading to the development of mucosal inflammation. The development of mucosal inflammation likely arises from a combination of these mechanisms.
In seeking to answer this question, we and others have focused on the innate immune receptors that are present on the mucosa, and have examined the response of these receptors to known ligands in the development of mucosal inflammatory disorders. Such ligands may be broadly grouped into two categories: the so called “danger signals,” a term used by Matzinger (24), also called damage-associated molecular patterns (DAMPs) ([25,26] provide recent reviews); and those ligands on the surface or interior of pathogen-associated molecular patterns (PAMPs) (Table 2). Not surprisingly, there is a great deal of interest in identifying the important receptors for DAMPs and PAMPs expressed by various cell types, so as to accurately define their relative role in the development of inflammation. In this regard, several investigators have established that DAMPs and PAMPs are recognized by TLRs in many cells, including epithelial cells: a list of the TLRs and their cognate ligands appears in Table 1. Although current dogma suggests that circulating leukocytes play a central role in the coordination of the immune response, emerging evidence suggests that the epithelium also plays a key role in the recognition and response to various “danger molecules” (27–31). This review will examine in detail the various roles of epithelial signaling via TLRs in the development of common, and often devastating, mucosal inflammatory conditions. Much of the focus will be on the TLR-initiated signaling in response to PAMPs, as the majority of work has been performed in this area.
Recognizing Danger: TLRs and Friends
Several recent investigators have shed light upon the important role of TLR signaling in the development of mucosal inflammation (31–34). To understand how TLRs may signal within the epithelium, information may be gained by analyzing TLR signaling in other systems, primarily within leukocytes. A full description of the molecular mechanisms by which TLR signaling occurs is beyond the scope of this review; currently accepted concepts with respect to TLR signaling are described below (21,35 have recent reviews).
The structure of each member of the TLR family of receptors provides important clues to how they function. All currently recognized TLRs are homologous with the interleukin-1 (IL-1) receptor, sharing an intracellular signaling domain, known as the Toll/IL-1R (TIR) domain (36). A model that depicts the currently accepted mode of TLR signaling in leukocytes is shown in Figure 2, in which the interaction with TLR4 and its cognate lig-and lipopolysaccharide (LPS, endotoxin) is shown. The interaction of TLR4 with LPS leads to the activation of myeloid differentiation primary response protein 88 (MyD88)-dependent signaling, resulting in the induction of pro-inflammatory genes such as TNF-α, IL-1β, IL-6, and IL-10 (37–39) and MyD88-independent signaling cascades leading to activation of type-1 interferon (40 has a recent review) (38,41). MyD88-depedent signaling occurs as TIR domain-containing adapter protein (TIRAP/Mal) (42,43) and MyD88 (44–46) interact with TLR4 and recruit IL-1 receptor-associated kinase (IRAK) family members IRAK1 and IRAK4 to the signaling heterocomplex consisting of TLR4, MyD88, and TIRAP (46). Subsequent signaling occurs through tumor necrosis factor receptor-associated factor 6 (TRAF6)-mediated (47) activation of transforming growth factor-β-activated protein kinase 1 (TAK1) (48). TAK1 forms a complex with TAK1 binding proteins (TAB), TAB1 (49), TAB2 (50), and TAB3 (51). The TAK1/TAB1/TAB2/TAB3 complex formation leads to the phosphorylation of IκB by IκB kinase (IκK) (52), initiating nuclear factor-kappa B (NF-κB) signaling pathways and parallel activation of several mitogen-activated protein kinases (MAP-kinases) including c-Jun N-terminal kinase (JNK) (53), p38 MAP-kinase (54), and extracellular signal-regulated kinase (ERK) (55), ultimately resulting in pro-inflammatory gene induction (56,57). It is noteworthy that MyD88-dependent signaling is thought to be the predominant signaling pathway for TLR2 (41), TLR5 (58), TLR7 (59), and TLR9 (60,61).

TLR4 signaling pathways. LPS binding to TLR4 requires binding protein (LBP), MD-2, and the co-receptor CD-14 which initiates MyD88-dependent (blue) and MyD88-independent (yellow) signaling pathways. See text for details.
As shown in Figure 2, TLR4 also may signal in the absence of MyD88. Evidence for this was demonstrated by Kawai et al., who described that the activation of NF-κB and MAP-kinases was reduced significantly, but not abolished, in MyD88-deficient mice (38). The MyD88-independent signaling pathway proximally involves the activation of TRAM, a TIR-domain containing adapter molecule. TRAM associates with and activates TRIF, another TIR-domain containing adapter protein (62,63). TRIF then interacts with and activates TANK-binding kinase 1 (TBK1) and IKKε, two IκK homologs, which leads to the phosphorylation of IRF3 (64,65) and translocation of IRF3 to the nucleus where it regulates the expression of various genes, including the type I IFN family of genes (66). TRIF also interacts with TRAF6 and receptor interacting protein 1 (RIP), leading to the activation of NF-κB (67,68). Importantly, TLR3 signals mainly through the MyD88-independent, TRIF-dependent pathway (62).
TLR-Dependent Signaling in the Intestinal Mucosa: A Role in the Pathogenesis of Intestinal Inflammation?
There is a wide and diverse spectrum of diseases that involves the development of inflammation of the intestinal mucosa. Such diseases include inflammatory bowel disease that is, Crohn’s disease and ulcerative colitis, necrotizing enterocolitis (which is a leading cause of death and disability in newborn infants) and a variety of infectious causes of intestinal dysfunction, due to entero-invasive organisms such as Salmonella and Escherichia coli. As shown in Figure 1, signaling via TLRs could lead to the development of intestinal inflammation through direct interaction of TLRs with the intestinal epithelium, or through effects on sub-epithelial and circulating leukocytes whose activation then leads to the initiation and propagation of mucosal inflammation. Although evidence exists to support this latter possibility, the expression of various TLRs in enterocytes (Table 3) suggests the possibility that direct interaction of intestinal TLRs with cognate ligands (see Table 1) may occur. Enteric bacteria in general, and LPS in particular, have been shown to play a critical role in the development of many diseases of intestinal inflammation (69–72), further suggesting the possibility that enterocyte TLR signaling may contribute directly to the development of these diseases.
To address the role(s), if any, of intestinal epithelial TLR signaling in the pathogenesis of intestinal inflammation, we, and others, have focused on TLR4, the receptor for LPS. Multiple enterocyte cell lines, including (IEC-6) rat enterocytes (27,73), primary and cultured (HT-29 and T84) colonocytes (74–76) and (CMT93) mouse rectal cells (75) express TLR4, the TLR4 adapter protein MD-2, and MyD88. In these cell lines, activation by LPS leads to pro-inflammatory signaling (74–76) as well as changes in cellular processes including proliferation (73) and intracellular TLR4 trafficking (77). These findings provide evidence that en-terocytes may respond directly to LPS via TLR4, yet by no means prove the physiological relevance of such a response. However, clinical significance for TLR signaling in the pathogenesis of intestinal mucosa is suggested as patients with inflammatory bowel disease demonstrate an increase in the expression of TLR4 and TLR2 in the intestinal mucosa (75,78), and we have found that TLR4 expression is increased in experimental and human necrotizing enterocolitis (32). Sensitization of the intestinal mucosa through upregulation of TLRs also occurs in other diseases of intestinal inflammation, including inflammatory bowel disease and intestinal celiac disease (75,79–81), suggesting a potential role in the injury response.
In seeking to further understand the role of enterocyte TLR4 in the pathogenesis of intestinal inflammation, our laboratory recently has examined the role of enterocyte TLR4 activation in the pathogenesis of necrotizing enterocolitis (NEC) (32). NEC is the leading cause of death from gastrointestinal disease in preterm infants (71), and, currently, is one of the leading causes of death of newborns in the United States overall with a mortality rate of nearly 15% (82). We have established recently that enterocyte TLR4 activation plays a critical role in the pathogenesis of NEC (32). Specifically, we found that NEC in both mice and humans is associated with increased expression of TLR4 in the intestinal mucosa, and that physiological stressors associated with NEC development, namely exposure to LPS and hypoxia, sensitize the murine intestinal epithelium to LPS through upregulation of TLR4 (32). In support of a critical role for TLR4 in the development of NEC, TLR4-mutant C3H/HeJ mice were protected from the development of NEC compared with wild-type C3H/HeOUJ littermates (32), a finding consistent with previous work by Caplan et al. (33). TLR4 activation in vitro led to increased enterocyte injury by induction of enterocyte apoptosis and reduced epithelial healing, due to an inhibition of enterocyte migration and proliferation. This latter finding suggests a role for enterocyte TLR4 in the regulation of intestinal mucosal repair. In support of this possibility, increased NEC severity in wildtype C3H/HeOUJ mice resulted from increased enterocyte apoptosis and reduced enterocyte restitution and proliferation compared with TLR4-mutant mice. TLR4 signaling also led to increased serine-phosphorylation of intestinal focal adhesion kinase (FAK), a molecule necessary for efficient enterocyte migration. Surprisingly, TLR4 co-immunoprecipitated with FAK in enterocytes, and siRNA-mediated FAK inhibition restored enterocyte migration after TLR4 activation, demonstrating that the FAK-TLR4 association regulates intestinal healing. Taken together, these findings demonstrate a critical role for TLR4 signaling in the intestinal epithelium in the development of NEC through effects on enterocyte injury and repair (32).
In addition to the effects of enterocyte TLR4 activation on the regulation of intestinal injury and repair, our group also has demonstrated a surprising role for enterocyte TLR4 in the regulation of bacterial translocation across the intestinal barrier (see Figure 1A). Translocation of bacteria across the intestinal barrier is important in the pathogenesis of not only intestinal inflammation, but also systemic sepsis, and may be a critical determinant of the development of multisystem organ dysfunction. We recently have shown that enterocyte TLR4 plays a key role in regulating the ability of enterocytes to internalize Gram-negative bacteria into membrane-bound phagosomes. Further evidence that TLR4 signaling is both necessary and sufficient for phagocytosis by epithelial cells was found as cultured enterocytes were able to internalize LPS-coated but not uncoated latex particles, and MD2/TLR4-transfected HEK-293 cells acquired the capacity to internalize E. coli, whereas non-transfected HEK-293 and HEK-293 transfected with dominant negative TLR4 bearing a P712H mutation did not. Strikingly, the internalization of Gram-negative bacteria into enterocytes in vivo and the translocation of bacteria across the intestinal epithelium to mesenteric lymph nodes were significantly greater in wild-type mice as compared with mice with mutations in TLR4 (27). These data suggest a novel mechanism by which bacterial translocation occurs, and suggest a critical role for TLR4 in the phagocytosis of bacteria by enterocytes in this process.
The work reviewed above indicates that activation of TLR4 within the intestine is deleterious to the host, through effects on intestinal barrier injury, repair, and bacterial translocation. The overriding concept that enterocyte TLR4 activation has negative effects on intestinal homeostasis is supported by work demonstrating that TLR4 plays an important role in protecting the host from the development of chemical-induced colonic inflammation through the maintenance of intestinal homeostasis and the production of cytoprotective factors (83–85). However, subsequent studies have demonstrated that TLR4 may play a permissive role in the development of spontaneous colonic inflammation (86), suggesting either that the net effects of TLR4 on intestinal inflammation are dependent on the specific disease process examined, the anatomic location of the disease process, or that the interaction with various downstream effectors influences the extent of intestinal inflammation that develops. It is noteworthy that the inflammation observed in NEC is predominantly localized to the small intestine as opposed to the colon (4,87), implying that the effects of TLR4 activation within small intestinal epithelial cells may lead to different effects than its role on the colonic epithelia. In support of this concept, it has been demonstrated previously that small intestinal enterocytes are more responsive to LPS than colonic enterocytes, due in part to differences in TLR4 expression and/or activity (88,89). Moreover, the increase in expression of TLR4 within the ileum that we have observed after exposure to hypoxia and endotoxin suggests that TLR4-dependent signaling within the small bowel mucosa may be increased after exposure to these stressors. The combined effects of the enhanced baseline sensitivity of the small intestine to LPS, and the upregulation of TLR4 expression in the intestine may partially explain the observed effects of enterocyte TLR4 in the induction of NEC. In support of this possibility, Caplan et al. have recently demonstrated that TLR4 expressing mice are more susceptible to the development of NEC in a model of formula feeding and cold asphyxia through a mechanism involving the enhanced interaction with luminal bacteria (33).
In addition to TLR4, other TLRs have been shown to play a role in the pathogenesis of intestinal inflammation, potentially via TLR-dependent signaling of the enterocytes themselves. For instance, both TLR2−/− and TLR9−/− mice were found recently to develop more severe intestinal inflammation compared with wild-type counterparts (90,91). Moreover, TLR5−/− mice have been found to develop spontaneous colitis (92) and the TLR5 ligand flagellin has been found to protect against enterocyte apoptosis (93). These findings indicate that TLR2, TLR5, and TLR9 may exert protective roles in the pathogenesis of intestinal inflammation, or indeed may provide support for the maintenance of intestinal homeostasis. Since TLR2, TLR5, and TLR9 share the downstream mediator MyD88, it is possible that these studies provide mechanistic insights into the protective role of MyD88 in the maintenance of intestinal homeostasis as identified by Medzhitov et al. (83). Once again, though the story is more complicated than appears on first glance, as activation of TLR3, the only TLR family member that does not require MyD88 to signal, with the specific ligand polyinosinic:polycytidylic acid (poly I:C) protected against the severity of DSS-induced colitis (94).
How do we reconcile the apparently contradictory roles of TLRs in the development of intestinal inflammation? It is possible that there may be cross talk between various TLR family members in the maintenance of intestinal inflammation, and the balance between intestinal homeostasis versus intestinal injury may be a reflection of the relative balance between TLRs and their associated signaling molecules. Alternatively, different TLRs within the intestine may be more or less susceptible to upregulation by different physiological stressors. We and others also have shown that intestinal mucosal TLR expression varies in different parts of the GI tract (SC Gribar and DJ Hackam, unpublished report) (95), which could explain in part the regional effects of TLR signaling on intestinal inflammation that is observed. It also may be possible that unique, epithelial-specific, intracellular signaling networks are activated by specific TLR ligation in enterocytes. Additional studies designed to delineate the precise interaction between the various enterocyte TLRs and their downstream receptors are required to resolve these possibilities.
Further insights regarding a potential role for TLR signaling in the pathogenesis of intestinal inflammation may be learned from studying genetic polymorphisms in humans with diseases of intestinal inflammation and sepsis. The TLR4 Asp299Gly mutation is known to render TLR4 hyporesponsive to endotoxin (99). This mutation has been associated with an increased incidence of inflammatory bowel disease (ulcerative colitis and Crohn’s disease) (100,101). Furthermore, pancolitis, the most severe manifestation of ulcerative colitis, is more common in patients with the TLR1 Arg80Thr polymorphism and the TLR2 Arg753Gly polymorphism (102). In patients with Crohn’s disease, the TLR1 Ser602Ile polymorphism is associated with a reduced risk of developing ileal disease (102). While no genetic polymorphisms have been associated with NEC, further study is necessary as the current observations were made on small cohorts of patients (103).
TLR-Dependent Signaling in the Pulmonary Epithelium: A Role in the Pathogenesis of Pulmonary Inflammation?
Pulmonary inflammatory diseases represent a broad spectrum of conditions that include allergic asthma, acute lung injury and acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary disease (COPD), and infectious pneumonia (96 has a recent review). Although these diseases traditionally have been considered to reflect the combined effects of activation of the adaptive immune system with the release of antibodies and mobilization of host immune cells, recent evidence has demonstrated an important role for the TLR family members of the innate immune system in their pathogenesis. And, in parallel with the mechanisms leading to the development of mucosal inflammation in the intestine, an emerging body of literature now provides evidence that epithelial TLR signaling plays a central role (15). Previous authors have shown that the pulmonary epithelium expresses a variety of TLRs (see Table 3), suggesting their role in the pathogenesis of pulmonary inflammation. In support of a role for TLR signaling in the pulmonary epithelium in the development of pulmonary inflammation, Noulin et al. have demonstrated that TLR4 and MyD88-dependent signaling are required for the bronchoconstriction, cytokine response, protein leak, and neutrophil recruitment observed in response to inhaled endotoxin (28). Furthermore, using MyD88-/-bone marrow chimeras, Noulin et al. demonstrated that both resident and hematopoietic cells are necessary for the mucosal inflammatory response to inhaled endotoxin (28). Hajjar et al. demonstrated that MyD88-deficient mice transplanted with bone marrow from MyD88-expressing mice showed reduced chemokine production compared with MyD88 expressing mice that were transplanted with MyD88-expressing bone marrow in a model of experimental Pseudomonas aerogeninosa pneumonia, indicating a requirement for resident pulmonary parenchymal cells in the response to experimental pneumonia. The local pulmonary cytokine response was predominately dependent on competent MyD88 signaling in bone-marrow derived cells, suggesting that collaboration between local parenchymal cells, including epithelial cells, and bone-marrow derived cells is required (29). In a model of bacterial pneumonia that utilizes inhaled LPS, the uptake of LPS was observed in bronchial epithelial cells and was associated with increased TLR2 and TLR4 expression in the bronchial epithelium (97). Similarly, in an equine model of recurrent airway obstruction associated with inhaled endotoxin-rich stable dust, increased epithelial expression of TLR4 was observed and was associated with increased IL-8 expression by the airway epithelium (98). Taken together, these reports provide supportive evidence for an important role for epithelial TLR signaling in the pathogenesis of mucosal inflammation in the pulmonary system.
Several groups have shown that airway epithelial cells (AEC) express TLRs and secrete cytokines in response to TLR activation. AECs have been shown to express TLR2 and TLR4 and release IL-8 in response to Streptococcus pneumoniae, lipoteichoic acid, and lipopolysaccharide (99,100). Furthermore, TLR9 activation in bronchial epithelial cells has been shown to potentiate IL-8 release from bronchial epithelial cells (101). Although it has been shown that multiple TLRs may signal in the airway epithelium (15), microarray analysis of the lung has revealed that TLR4 signaling accounts for 74% of the pulmonary response to experimental Klebsiella pneumoniae pneumonia by comparing the pulmonary response in wildtype mice to C3H/HeJ TLR4 mutant mice. The particular TLR4-dependent responses included genes that are involved in cytokine and chemokine induction, neutrophil activation and recruitment, growth factor receptors, and TLR adaptor molecules (102).
In addition to the evidence for TLR signaling in pulmonary epithelial cells in vitro, a variety of studies have shown that TLR activation may lead to the development of pulmonary inflammation in vivo. For instance, the TLR4 mutant strains C3H/HeJ and C57BL/10ScCr showed reduced clearance of pulmonary H. influenzae and E. coli (103,104). In an experimental Chlamydia pneumoniae pulmonary infection model, both TLR4 and TLR2 were found to be required for survival (105), while TLR4 and CD14 were found to play an important role in the response to respiratory syncytial virus (RSV) infection (106). In response to pulmonary Streptococcus pneumoniae infection, TLR2-deficient mice revealed only modestly reduced inflammatory response and unchanged bacterial clearance (107) compared with wild-type counterparts. TLR3-deficient mice developed a survival advantage compared with wild-type mice, as well as reduced expression of IL-6 in the bronchoalveolar fluid in a murine model of influenza A virus infection (108).
Additional evidence implicating a role for TLR signaling in the development of pulmonary inflammation may be found in studies examining the development of pulmonary inflammation in human patients with TLR polymorphisms. For instance, polymorphisms in TLR4 (Ala299Gly and Thr399Ile), which are known to lead to hyporesponsiveness to LPS (187), lead to a marked resistance to infection with Legionella pneumophila (188). These TLR4 mutations have been correlated with the development of severe RSV infection in infants (120). An inactivating polymorphism in TLR5 (TLR5392STOP) that encodes a stop codon in the ligand binding domain of TLR5 is associated with an increased susceptibility to infection with Legionella pneumophila causing Legionnaire’s disease (188). Taken in aggregate, the results of these in vitro and in vivo studies provide evidence for a role for TLR signaling in the pathogenesis of pulmonary inflammation. Additional studies are required utilizing pulmonary-specific TLR deletions to further delineate the relative contributions of pulmonary epithelial cell versus infiltrating leukocytes in the development of mucosal inflammation in the lung.
TLR-Dependent Signaling in the Uroepithelial Tract: A Role in the Pathogenesis of Urinary Tract Inflammation?
Akin to the gastrointestinal and pulmonary tracts, dysregulated epithelial signaling in the genitourinary system may lead to marked organ dysfunction. The expression of multiple TLRs within the urinary epithelium has now been established, suggesting the possibility that TLR signaling may regulate the interaction of the urinary epithelium with potential pathogens (see Table 3). TLR signaling within urinary tract epithelial cells leads to pro-inflammatory signaling in response to uropathogenic E. coli (UPEC) and LPS (109,110). Furthermore, modulation of uroepithelial inflammation may be mediated by sensitization of the uroepithelium through regulation of epithelial TLR expression in response to infection or injury. An increase in TLR4 expression in the urinary epithelium has been observed during systemic sepsis in a murine model of cecal ligation and puncture (111), and an increase in renal epithelial TLR2 and TLR4 expression has been observed in a murine model of local renal inflammation induced by ischemia (112). Further demonstrating a role for TLR activation in uroepithelial inflammation, TLR4 mutant C3H/Hej mice failed to clear uropathogenic E. coli (UPEC) and showed reduced inflammatory mediator production compared with wild-type controls (109). TLR4 mutant C3H/Hej mice were resistant to LPS-induced renal failure, had less renal neutrophilic infiltrate, and less renal cell apoptosis compared with wild-type controls (113). In addition to TLR4, other TLRs may participate in the development of uroepithelial inflammation. For instance, TLR5-deficient mice were found to be more susceptible to experimental UPEC urinary tract infection compared with wild-type counterparts (16), while mice with null mutations in TLR11, which is normally found to be strongly expressed in the bladder and kidney epithelium, developed markedly less severe kidney inflammation compared with wild-type counterparts (114). TLR2-deficient mice were protected from tubular injury and renal function deterioration in a model of kidney ischemia-reperfusion (115). The clinical significance of a role for TLR signaling in the pathogenesis of genitourinary inflammation is found in clinical studies in which the incidence of acute rejection after kidney transplantation is reduced in patients who received a graft heterozygous for either the TLR4 Asp299Gly or Thr399Ile polymorphism compared with grafts without these mutations (189), although conflicting results have been reported (190). Taken together, these studies suggest an important role for TLR signaling in the development of urinary tract inflammation in a variety of models.
Which cells are required for the development of TLR-induced inflammation in the urinary tract? Evidence suggests that both epithelial and non-epithelial cell types may play a role. For instance, when TLR4 mutant C3H/Hej mice were transplanted with wild-type hematopoietic cells, the mice were unable to mount the necessary response to UPEC (30). By contrast, in a model of cisplatin-induced renal injury, the development of inflammation was dependent on competent TLR4 signaling in resident renal parenchymal cells, as demonstrated in the study of TLR4−/− bone marrow chimeras (31). Additional work is required to define more accurately the relative roles of TLR signaling within the epithelium versus the leukocytes in the development of mucosal inflammation in the epithelial tract.
Peaceful Coexistence: Mechanisms Allowing Epithelial Cells to Interact with Bacteria without Initiating an Exaggerated Inflammatory Response
The information reviewed above highlights the important roles that TLRs play in the regulation of the inflammatory response at mucosal surfaces. However, it is well known that these mucosal surfaces are constantly bathed in bacteria, and yet appear to mount little, if any, inflammatory response. These observations lead to the question, “What controls the activation of TLRs during basal states, and what leads to their activation during inflammatory conditions?” While a complete answer to this question remains lacking, current evidence suggests that the regulation of TLR activity occurs through altering TLR or co-receptor expression, TLR localization, TLR polarity, or signaling intermediate or negative regulatory protein expression, as described below.
Regulation of epithelial TLR expression has been suggested as a mechanism for the regulation of epithelial cell responsiveness in the setting of commensal bacterial exposure and during disease states. For instance, low expression of TLR4, as has been observed in colonic biopsies from humans (75), has been suggested as a mechanism for colonocyte LPS hyporesponsiveness. Similar findings of low TLR4 expression have been observed in colonocyte cell lines (HT-29, SW480, colo205) and increased TLR4 expression after IFN-γ or TNF-α priming, as may occur during inflammatory states, has been shown to enable LPS responsiveness (88). Similarly, low expression of TLR2 has been implicated in bronchial epithelial cell hyporesponsiveness to Gram-positive bacteria (15).
Expression of TLR co-receptors in the epithelial cells also may play a role in epithelial TLR responsiveness. Hyporesponsiveness to LPS in colonic epithelial cell lines (Caco-2, T84, SW837, and HT-29) in the basal state is associated with low or absent expression of the TLR4 core-ceptor MD-2 (88,191) and priming of cultured colonocytes (HT-29) with IFN-γ or TNF-α enabled LPS responsiveness in a mechanism that involved increased MD-2 expression (192). Recently, we also have demonstrated a transient increase in the expression of the LPS co-receptor CD14 in enterocytes after exposure to LPS (193). In the pulmonary system, absent expression of the TLR2 coreceptor CD36 has been implicated in the hypore-sponsiveness of bronchial epithelial cell to Gram-positive bacteria (194).
Changes in the subcellular localization of TLRs also may play a role in their responsiveness. Dissimilar to plasma membrane localized TLR4 in macrophages, TLR4 has been shown to be localized predominately in the Golgi apparatus in enterocytes (174), and TLR4 activation in enterocytes has been shown to require intracellular recognition of LPS in the Golgi apparatus and recruitment of IRAK-1 and MyD88 to the Golgi apparatus (195). Furthermore, colonic HT-29 and colo205 cells express TLR4 predominately in cytoplasmic fractions and are hyporesponsive to LPS in basal states (89). Intracellular TLR redistribution has been suggested as a mechanism for flagellin tolerance, as prolonged flagellin exposure resulted in redistribution of TLR5 to an intracellular location in T84 colonocytes. Increased cell surface TLR expression also has been suggested as a mechanism of increased TLR sensitivity. Colonic SW480 cells are LPS-responsive and express TLR4 on the cell surface, where LPS internalization is not necessary for TLR4-LPS interaction (89). Also, increased TLR9 surface expression was noted in response to DNA from pathogenic bacteria in HT-29 colonocytes (196).
Epithelial cell polarity and differential localization of TLRs on the apical and basolateral cell surface also has been shown to play a role in TLR sensitivity as the apical surface of epithelial cells is more likely to encounter bacteria in the normal state, whereas the basolateral surface of epithelial cells may be more likely to encounter TLR ligands only in states of disease. In support of this concept, TLR4 and TLR2 are expressed at the apical pole of T84 cells and redistribute to a cytoplasmic compartment near the basal pole with activation (77). Furthermore, differential TLR9 signaling has been shown in colonic HCA-7 epithelial cells. Apical TLR9 activation leads to attenuation of activation of NF-κB pathways, whereas basolateral TLR9 activation leads to activation of NF-κB signaling (91). In the pulmonary system, TLR2 was located at the apical pole of airway epithelial cells, and increased surface expression was observed in response to bacteria, whereas TLR4 was noted predominately in a basolateral location (160). In a recent finding by Soong et al., TLR2 became enriched in lipid rafts on the apical surface after bacterial infection in airway epithelial cells, suggesting a role in the regulation of TLR sensitivity (197).
The regulation of signaling intermediate molecules also may affect TLR sensitivity. For instance, although fetal intestinal cells are known to be responsive to LPS, postnatal endotoxin hyporesponsiveness of enterocytes has been observed, and recently shown to be due to a decrease in the expression of the TLR4 signaling intermediate, IRAK1 (198). Negative regulatory molecules may play a role in regulating epithelial TLR signaling, including peroxisome proliferator-activated receptor-γ; the cytoplasmic zinc finger protein, A20; and the negative regulator of TLR signaling, IRAK-M, as has been reviewed recently (199). The relevance of these molecules to signaling within epithelial cells remains to be definitely demonstrated.
Therapeutic Manipulation of TLR Signaling in the Setting of Mucosal Inflammation
Given the importance of TLR signaling to the development of mucosal inflammation, it is understandable that a great deal of interest exists in the development of agents that can interfere with TLR-signaling pathways. Such an anti-inflammatory approach may have particular relevance in the case of epithelial inflammation, due to ready access of the gastrointestinal, pulmonary, and urinary mucosa through ingestion, inhalation, or instillation via catheter delivery methods. Considerable attention has been placed on developing agents that are capable of modulation of the TLR4-mediated response, in particular through manipulation of the lipid A moiety of LPS. Such lipid A mimetics, termed aminoalkyl glucosaminide phosphates (AGPs), have been demonstrated to reduce inflammation in experimental models of systemic sepsis induced by intravenous injection of Listeria monocytogenes (116), pulmonary infection after intranasal administration of influenza virus (116), murine models of colitis including DSS-induced colitis (117), and spontaneous colitis in multidrug resistance gene 1a-deficient mice (117). In parallel studies, soluble TLRs may reduce TLR signaling by binding to circulating ligands, rendering them unable to initiate pro-inflammatory signaling. Brandl et al. demonstrated that the synthetic molecule “LPS-Trap” was capable of blocking LPS-mediated macrophage activation in vitro by fusing MD-2 to the C-terminus of a soluble form of TLR4 (118). Iwami et al. cloned an alternatively spliced soluble murine TLR4 (smTLR4) that, when transfected into murine macrophages, was secreted and inhibited LPS-mediated macrophage NF-κB activation and TNF-α release in vitro (119). In addition, TLR-signaling intermediates have been targeted chemically to minimize the host inflammatory response. The synthetic peptide-mimetic compound ST2825 prevents MyD88 homodimer formation leading to an inhibition in MyD88-dependent signaling, and prevents TLR9-dependent inflammation in vivo (120). A cyclohexene derivative, TAK-242, prevents TLR4 activation and was found to reduce the cytokine response to endotoxemic shock in mice (121,122). The Vaccinia virus protein A52R also reduces the TLR-mediated response by interacting with IRAK2 and TRAF6, and was found to reduce the cytokine response in an animal model of infectious otitis media (123,124) and to increase survival in models of endotoxemia (125).
As the effects of TLR signaling in hematopoietic cells, as well as epithelial cells, are more clearly defined, manipulation of TLR signaling may play a larger role in the treatment of patients with diseases of inflammation, and, as evidence continues to mount suggesting a protective role for particular TLR signaling, directed and specific TLR activation may hold therapeutic promise.
Putting It All Together: A Model for the Role of the Epithelium in the Development of Mucosal Inflammation
Mucosal surfaces and the epithelial cells that line them are constantly exposed to potential pathogens. The evidence reviewed above suggests that the innate immune system, comprised of TLRs and their associated molecules, plays a pivotal role in the regulation of mucosal inflammation in response to invading pathogens. However, the very fact that these mucosal surfaces are bathed in potential pathogens as part of their daily existence and yet don’t develop inflammation under normal conditions raises an important scientific question: When does epithelial TLR signaling within mucosal surfaces become pathological? Or stated differently, when is the balance tipped between “physiological” signaling and “pathological” signaling in favor of a pathological response? A definitive answer to this important question not only is necessary to fully elucidate the steps required for the development of mucosal inflammatory diseases, but is central for the design of effective anti-inflammatory strategies.
Our current thinking in this area based upon our work and the work of others is shown in Figure 3, in which the intensity of TLR signaling within the epithelium varies depending upon the prevailing degree of systemic stress. Under basal conditions, epithelial-bacterial interactions that may occur via TLRs are likely to play roles in the regulation of processes that regulate barrier integrity, such as epithelial migration, proliferation, and apoptosis (Figure 3A). However, during states of systemic stress, such as hypoxia or remote infection, we submit that the extent of TLR signaling within the epithelium becomes exaggerated in response to PAMPS and DAMPS that are encountered. This “tips the balance” in favor of mucosal barrier disruption, and adversely affects mucosal repair while worsening mucosal injury (Figure 3B). The extent of inflammation that develops within the local microenvironment likely is compounded further by the contribution of TLR activation on leukocytes, and the release of proinflammatory molecules. Under conditions in which the balance of TLR signaling within the epithelium can be “tipped back” to a homeostatic state, mucosal inflammation may not develop. By contrast, when the extent of TLR signaling is persistent, we propose that a “feedforward” loop develops within the mucosa, resulting in persistent TLR signaling, cytokine release, and mucosal inflammation. The evaluation of the factors that maintain the degree of TLR signaling within the mucosa in the maintenance of homeostasis and the pathogenesis of disease is a topic of in tensive investigation.

A model of TLR-mediated inflammation in the mucosa. (A) During basal conditions, TLR signaling is required for mucosal homeostasis. (B) Under conditions of physiological stress, TLR signaling in epithelial cells becomes exaggerated in part through increased TLR expression. This leads to an impairment in epithelial function, increased injury, and decreased repair, resulting in mucosal inflammation.
Conclusions and Directions for Further Research
The importance of mucosal inflammation as a clinical problem is well accepted; however, the molecular and cellular signaling pathways that lead to its development remain incompletely understood. Although much attention has been placed on the role of the epithelium as a target in the mucosal inflammatory cascade, recent evidence has shed light upon the critical role that the epithelium itself, signaling in part through Toll-like receptors, may play in the initiation of a pro-inflammatory cascade in response to external stimuli. The field of mucosal inflammation research is likely to be advanced significantly through success in the following areas of study: 1) What are the relative roles of TLR signaling within the epithelium versus circulating leukocytes in the pathogenesis of mucosal inflammation? 2) What is the precise trigger for TLR signaling within the epithelium that adversely affects the host, and what are the essential roles played by mucosal TLRs in the maintenance of mucosal homeostasis? 3) Are there TLR-signaling molecular intermediates that differ between epithelial cells and leukocytes, and do such molecules confer epithelial-specific responses in the development of mucosal inflammation? 4) What regulates the interplay between the epithelium and the other cellular constituents of the mucosa, including neurons, endothelial cells, and endocrine cells during TLR activation? It is our belief that by addressing these important questions, one can be optimistic for the development of novel classes of anti-inflammatory strategies aimed specifically at the treatment of these devastating diseases of mucosal inflammation.
References
Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. (2006) The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn’s disease and ulcerative colitis patients. J. Med. Microbiol. 55:1141–9.
Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, Acero D, Garcia-Gil LJ. (2006) Abnormal microbiota composition in the ileocolonic mucosa of Crohn’s disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm. Bowel Dis. 12:1136–15.
Xavier RJ, Podolsky DK. (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–34.
Anand R, Leaphart CL, Mollen K, Hackam D. (2007) The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock 27:124–33.
Hippenstiel S, Opitz B, Schmeck B, Suttorp N. (2006) Lung epithelium as a sentinel and effector system in pneumonia—molecular mechanisms of pathogen recognition and signal transduction. Respir. Res. 7:97.
Holgate ST. (2007) Epithelium dysfunction in asthma. J. Allergy Clin. Immunol. 120:1233–44; quiz 1245–36.
Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B. (2000) Respiratory syncytial virus bronchiolitis in infancy is an important risk factor for asthma and allergy at age 7. Am. J. Respir. Crit. Care Med. 161:1501–7.
Bonventre JV, Zuk A. (2004) Ischemic acute renal failure: an inflammatory disease? Kidney Int. 66:480–5.
Parsons CL. (2007) The role of the urinary epithelium in the pathogenesis of interstitial cystitis/prostatitis/urethritis. Urology 69:9–16.
Haraoka M, et al. (1999) Neutrophil recruitment and resistance to urinary tract infection. J. Infect. Dis. 180:1220–9.
Lewis K, et al. (2008) Decreased epithelial barrier function evoked by exposure to metabolic stress and nonpathogenic E. coli is enhanced by TNF-alpha. Am. J. Physiol. 294:G669–78.
Cleret A, et al. (2007) Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J. Immunol. 178:7994–8001.
Frank JA, Wray CM, McAuley DF, Schwendener R, Matthay MA. (2006) Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am. J. Physiol. 291:L1191–8.
Rachmilewitz D, et al. (2004) Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 126:520–8.
Mayer AK, et al. (2007) Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 178:3134–42.
Andersen-Nissen E, et al. (2007) Cutting edge: Tlr5−/− mice are more susceptible to Escherichia coli urinary tract infection. J. Immunol. 178:4717–20.
Sappington P, et al. (2002) HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 123:790–802.
Jiang D, et al. (2005) Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 11:1173–9.
Boodoo S, Spannhake EW, Powell JD, Horton MR. (2006) Differential regulation of hyaluronaninduced IL-8 and IP-10 in airway epithelial cells. Am. J. Physiol. 291:L479–86.
Uematsu S, Akira S. (2008) Toll-Like receptors (TLRs) and their ligands. Handb. Exp. Pharmacol. 1–20.
Kawai T, Akira S. (2007) TLR signaling. Semin. Immunol. 19:24–32.
Kawai T, Akira S. (2006) TLR signaling. Cell Death Differ. 13:816–25.
Lee MS, Kim YJ. (2007) Signaling pathways downstream of pattern-recognition receptors and their cross talk. Ann. Rev. Biochem. 76:447–80.
Matzinger P. (1994) Tolerance, danger, and the extended family. Ann. Rev. Immunol. 12:991–1045.
Bianchi ME. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81:1–5.
Miyake K. (2007) Innate immune sensing of pathogens and danger signals by cell surface Toll-like receptors. Semin. Immunol. 19:3–10.
Neal MD, et al. (2006) Enterocyte TLR4 mediates phagocytosis and translocation of bacteria across the intestinal barrier. J. Immunol. 176:3070–9.
Noulin N, et al. (2005) Both hemopoietic and resident cells are required for MyD88-dependent pulmonary inflammatory response to inhaled endotoxin. J. Immunol. 175:6861–9.
Hajjar AM, et al. (2005) An essential role for nonbone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am. J. Respir. Cell. Mol. Biol. 33:470–5.
Schilling JD, Martin SM, Hung CS, Lorenz RG, Hultgren SJ. (2003) Toll-like receptor 4 on stromal and hematopoietic cells mediates innate resistance to uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 100:4203–8.
Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. (2008) TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J. Am. Soc. Nephrol. 19:923–32.
Leaphart CL, et al. (2007) A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 179:4808–20.
Jilling T, et al. (2006) The roles of bacteria and TLR4 in rat and murine models of necrotizing enterocolitis. J. Immunol. 177:3273–82.
Phipps S, Lam CE, Foster PS, Matthaei KI. (2007) The contribution of toll-like receptors to the pathogenesis of asthma. Immunol. Cell Biol. 85:463–70.
Akira S, Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. (1997) Ahuman homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–7.
Hoshino K, et al. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–52.
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–22.
Theiner G, et al. (2008) TLR9 cooperates with TLR4 to increase IL-12 release by murine dendritic cells. Mol. Immunol. 45:244–52
Pestka S, Krause CD, Walter MR. (2004) Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202:8–32.
Kawai T, et al. (2001) Lipopolysaccharide stimulates the MyD88-independent pathway and results in activation of IFN-regulatory factor 3 and the expression of a subset of lipopolysaccharideinducible genes. J. Immunol. 167:5887–94.
Horng T, Barton GM, Medzhitov R. (2001) TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2:835–41.
Fitzgerald KA, et al. (2001) Mal (MyD88-adapterlike) is required for Toll-like receptor-4 signal transduction. Nature 413:78–83.
Burns K, et al. (1998) MyD88, an adapter protein involved in interleukin-1 signaling. J. Biol. Chem. 273:12203–9.
Muzio M, Natoli G, Saccani S, Levrero M, Mantovani A. (1998) The human toll signaling pathway: divergence of nuclear factor kappaB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J. Exp. Med. 187:2097–101.
Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. (1997) MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity 7:837–47.
Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. (1996) TRAF6 is a signal transducer for interleukin-1. Nature 383:443–6.
Deng L, et al. (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–61.
Shibuya H, et al. (1996) TAB1: an activator of the TAK1 MAPKKK in TGF-beta signal transduction. Science 272:1179–82.
Takaesu G, et al. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell. 5:649–58.
Ishitani T, et al. (2003) Role of the TAB2-related protein TAB3 in IL-1 and TNF signaling. EMBO J. 22:6277–88.
Wang Q, Wang X, Hernandez A, Kim S, Evers B. (2001) Inhibition of the phosphatidylinositol 3-kinase pathway contributes to HT29 and Caco-2 intestinal cell differentiation. Gastroenterology 120:1381–92.
Shirakabe K, et al. (1997) TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J. Biol. Chem. 272:8141–4.
Moriguchi T, et al. (1996) Anovel kinase cascade mediated by mitogen-activated protein kinase kinase 6 and MKK3. J. Biol. Chem. 271:13675–9.
An H, et al. (2002) Involvement of ERK, p38 and NF-kappaB signal transduction in regulation of TLR2, TLR4 and TLR9 gene expression induced by lipopolysaccharide in mouse dendritic cells. Immunology 106:38–45.
Morse D, et al. (2003) Suppression of inflammatory cytokine production by carbon monoxide involves the JNK pathway and AP-1. J. Biol. Chem. 278:36993–8.
Zhang Y, et al. (2001) The regulatory effect of ERK1/2 signal pathway on production of TNFαlpha induced by LPS in mice Kupffer cells. Chin. J. Traumatol. 4:139–42.
Hayashi F, et al. (2001) The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–103.
Hemmi H, et al. (2002) Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat. Immunol. 3:196–200.
Hacker H, et al. (2000) Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor-associated factor (TRAF) 6. J. Exp. Med. 192:595–600.
Schnare M, Holt AC, Takeda K, Akira S, Medzhitov R. (2000) Recognition of CpG DNAis mediated by signaling pathways dependent on the adaptor protein MyD88. Curr. Biol. 10:1139–42.
Yamamoto M, et al. (2003) Role of adaptor TRIF in the MyD88-independent Toll-like receptor signaling pathway. Science 301:640–3.
Hoebe K, et al. (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424:743–8.
Sharma S, et al. (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–51.
Fitzgerald KA, et al. (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–6.
Nakaya T, et al. (2001) Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 283:1150–6.
Sato S, et al. (2003) Toll/IL-1 receptor domaincontaining adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 171:4304–10.
Meylan E, et al. (2004) RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 5:503–7.
Caplan MS, Kelly A, Hsueh W. (1992) Endotoxin and hypoxia-induced intestinal necrosis in rats: the role of platelet activating factor. Pediatr. Res. 31:428–34.
De Plaen IG, et al. (2002) Endotoxin, but not platelet-activating factor, activates nuclear factor-kappaB and increases IkappaBalpha and IkappaBbeta turnover in enterocytes. Immunology 106:577–83.
Anand RJ, Leaphart CL, Mollen KP, Hackam DJ. (2007) The role of the intestinal barrier in the pathogenesis of necrotizing enterocolitis. Shock 27:124–33.
Gardiner KR, et al. (1995) Significance of systemic endotoxaemia in inflammatory bowel disease. Gut 36:897–901.
Ruemmele FM, et al. (2002) Lipopolysaccharide modulation of normal enterocyte turnover by toll-like receptors is mediated by endogenously produced tumour necrosis factor alpha. Gut 51:842–8.
Otte J, Cario E, Podolsky D. (2004) Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology 126:1054–70.
Cario E, Podolsky DK. (2000) Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 68:7010–7.
Funda DP, et al. (2001) CD14 Is Expressed and Released as Soluble CD14 by Human Intestinal Epithelial Cells In Vitro: Lipopolysaccharide Activation of Epithelial Cells Revisited. Infect. Immun. 69:3772–81.
Cario E, et al. (2002) Commensal-associated molecular patterns induce selective toll-like receptor-trafficking from apical membrane to cytoplasmic compartments in polarized intestinal epithelium. Am. J. Pathol. 160:165–73.
Szebeni B, et al. (2008) Increased expression of Toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin. Exp. Immunol. 151:34–41.
Gomariz RP, et al. (2005) Time-course expression of Toll-like receptors 2 and 4 in inflammatory bowel disease and homeostatic effect of VIP. J. Leukoc. Biol. 78:491–502.
Ortega-Cava CF, et al. (2003) Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J. Immunol. 170:3977–85.
Szebeni B, et al. (2007) Increased mucosal expression of Toll-like receptor (TLR)2 and TLR4 in coeliac disease. J. Pediatr. Gastroenterol. Nutr. 45:187–93.
Minino AM, Heron MP, Murphy SL, Kochanek KD. (2007) Deaths: final data for 2004. Natl. Vital Stat. Rep. 55:1–119.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. (2004) Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118:229–41.
Araki A, et al. (2005) MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. J. Gastroenterol. 40:16–23.
Fukata M, et al. (2005) Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G1055–65.
Rakoff-Nahoum S, Hao L, Medzhitov R. (2006) Role of toll-like receptors in spontaneous commensal-dependent colitis. Immunity 25:319–29.
Kosloske AM, Musemeche CA. (1989) Necrotizing enterocolitis of the neonate. Clin. Perinatol. 16:97–111.
Abreu MT, et al. (2001) Decreased expression of Toll-like receptor-4 and MD-2 correlates with intestinal epithelial cell protection against dysregulated proinflammatory gene expression in response to bacterial lipopolysaccharide. J. Immunol. 167:1609–16.
Suzuki M, Hisamatsu T, Podolsky DK. (2003) Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular Toll-like receptor 4-MD-2 complex. Infect. Immun. 71:3503–11.
Cario E, Gerken G, Podolsky DK. (2007) Toll-Like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology 132:1359–74.
Lee J, et al. (2006) Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell Biol. 8:1327–36.
Vijay-Kumar M, et al. (2007) Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest. 117:3909–21.
Vijay-Kumar M, et al. (2006) Flagellin suppresses epithelial apoptosis and limits disease during enteric infection. Am. J. Pathol. 169:1686–700.
Vijay-Kumar M, et al. (2007) Activation of tolllike receptor 3 protects against DSS-induced acute colitis. Inflamm. Bowel Dis. 13:856–64.
Ortega-Cava CF, et al. (2003) Strategic compartmentalization of Toll-like Receptor 4 in the mouse gut. J. Immunol. 170:3977–85.
Chaudhuri N, Whyte MK, Sabroe I. (2007) Reducing the toll of inflammatory lung disease. Chest 131:1550–6.
Saito T, Yamamoto T, Kazawa T, Gejyo H, Naito M. (2005) Expression of toll-like receptor 2 and 4 in lipopolysaccharide-induced lung injury in mouse. Cell Tissue Res. 321:75–88.
Berndt A, et al. (2007) Elevated amount of Toll-like receptor 4 mRNA in bronchial epithelial cells is associated with airway inflammation in horses with recurrent airway obstruction. Am. J. Physiol. Lung Cell Mol. Physiol. 292:L936–43.
Schmeck B, et al. (2006) Pneumococci induced TLR- and Rac1-dependent NF-kappaB-recruitment to the IL-8 promoter in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 290: L730–7.
Armstrong L, et al. (2004) Expression of functional toll-like receptor-2 and -4 on alveolar epithelial cells. Am. J. Respir. Cell. Mol. Biol. 31:241–5.
Parilla NW, Hughes VS, Lierl KM, Wong HR, Page K. (2006) CpG DNA modulates interleukin 1beta-induced interleukin-8 expression in human bronchial epithelial (16HBE14o-) cells. Resp. Res. 7:84.
Schurr JR, et al. (2005) Central role of toll-like receptor 4 signaling and host defense in experimental pneumonia caused by Gram-negative bacteria. Infect. Immun. 73:532–45.
Wang X, et al. (2002) Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J. Immunol. 168:810–5.
Lee JS, et al. (2005) TLR-4 pathway mediates the inflammatory response but not bacterial elimination in E. coli pneumonia. Am. J. Physiol. Lung Cell Mol. Physiol. 289: L731–8.
Rodriguez N, et al. (2006) Differential involvement of TLR2 and TLR4 in host survival during pulmonary infection with Chlamydia pneumoniae. Eur. J. Immunol. 36:1145–55.
Kurt-Jones EA, et al. (2000) Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 1:398–401.
Knapp S, et al. (2004) Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J. Immunol. 172:3132–8.
Le Goffic R, et al. (2006) Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2:e53.
Chassin C, et al. (2006) Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J. Immunol. 177:4773–84.
Tsuboi N, et al. (2002) Roles of toll-like receptors in C-C chemokine production by renal tubular epithelial cells. J. Immunol. 169:2026–33.
El-Achkar TM, et al. (2006) Sepsis induces changes in the expression and distribution of Toll-like receptor 4 in the rat kidney. Am. J. Physiol. Renal. Physiol. 290: F1034–43.
Wolfs TG, et al. (2002) In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J. Immunol. 168:1286–93.
Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. (2004) Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J. Immunol. 172:2629–35.
Zhang D, et al. (2004) Atoll-like receptor that prevents infection by uropathogenic bacteria. Science 303:1522–6.
Leemans JC, et al. (2005) Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J. Clin. Invest. 115:2894–903.
Cluff CW, et al. (2005) Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect. Immun. 73:3044–52.
Fort MM, et al. (2005) A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 174:6416–23.
Brandl K, Gluck T, Hartmann P, Salzberger B, Falk W. (2005) A designed TLR4/MD-2 complex to capture LPS. J. Endotoxin Res. 11:197–206.
Iwami KI, et al. (2000) Cutting edge: naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 165:6682–6686.
Loiarro M, et al. (2007) Pivotal Advance: Inhibition of MyD88 dimerization and recruitment of IRAK1 and IRAK4 by a novel peptidomimetic compound. J. Leukoc. Biol. 82:801–10.
Ii M, et al. (2006) A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 69:1288–95.
Yamada M, et al. (2005) Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl) cyclohex-1-ene-1-carboxylate. J. Med. Chem. 48:7457–67.
Harte MT, et al. (2003) The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 197:343–51.
McCoy SL, Kurtz SE, Macarthur CJ, Trune DR, Hefeneider SH. (2005) Identification of a peptide derived from vaccinia virus A52R protein that inhibits cytokine secretion in response to TLR-dependent signaling and reduces in vivo bacterial-induced inflammation. J. Immunol. 174:3006–14.
Tsung A, et al. (2007) A novel inhibitory peptide of Toll-like receptor signaling limits lipopolysaccharide-induced production of inflammatory mediators and enhances survival in mice. Shock 27:364–9.
Takeuchi O, et al. (2002) Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169:10–14.
Aliprantis AO, et al. (1999) Cell activation and apoptosis by bacterial lipoproteins through tolllike receptor-2. Science 285:736–9.
Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. (1999) Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 274:17406–9.
Takeuchi O, et al. (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 11:443–51.
Underhill DM, et al. (1999) The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401:811–5.
Ozinsky A, et al. (2000) The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 97:13766–71.
Vabulas RM, et al. (2002) HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J. Biol. Chem. 277:15107–12.
Asea A, et al. (2002) Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277:15028–34.
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–8.
Poltorak A, et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–8.
Kawasaki K, et al. (2000) Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharidemimetic signal transduction by Taxol. J. Biol. Chem. 275:2251–4.
Bulut Y, et al. (2002) Chlamydial heat shock protein 60 activates macrophages and endothelial cells through Toll-like receptor 4 and MD2 in a MyD88-dependent pathway. J. Immunol. 168:1435–40.
Ohashi K, Burkart V, Flohe S, Kolb H. (2000) Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 164:558–61.
Okamura Y, et al. (2001) The extra domain A of fibronectin activates Toll-like receptor 4. J. Biol. Chem. 276:10229–33.
Termeer C, et al. (2002) Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 195:99–111.
Johnson GB, Brunn GJ, Kodaira Y, Platt JL. (2002) Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J. Immunol. 168:5233–9.
Smiley ST, King JA, Hancock WW. (2001) Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 167:2887–94.
Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. (2002) Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U. S. A. 99:2281–6.
Takeuchi O, et al. (2001) Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13:933–40.
Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–31.
Heil F, et al. (2004) Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–9.
Jurk M, et al. (2006) Modulating responsiveness of human TLR7 and 8 to small molecule ligands with T-rich phosphorothiate oligodeoxynucleotides. Eur. J. Immunol. 36:1815–26.
Hemmi H, et al. (2000) AToll-like receptor recognizes bacterial DNA. Nature 408:740–5.
Yarovinsky F, et al. (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–9.
Guillot L, et al. (2002) Cutting edge: the immunostimulatory activity of the lung surfactant protein-A involves Toll-like receptor 4. J. Immunol. 168:5989–92.
Biragyn A, et al. (2002) Toll-like receptor 4-dependent activation of dendritic cells by beta-deensin 2. Science 298:1025–9.
Hoshino K, et al. (1999) Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162:3749–52.
Park JS, et al. (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279:7370–7.
Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. (2004) mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279:12542–50.
Ishii KJ, et al. (2001) Genomic DNA released by dying cells induces the maturation of APCs. J. Immunol. 167:2602–7.
Shi Y, Evans JE, Rock KL. (2003) Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425:516–21.
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. (1998) Afamily of human receptors structurally related to Drosophila Toll. Proc. Natl. Acad. Sci. U. S. A. 95:588–93.
Otte JM, Cario E, Podolsky DK. (2004) Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology 126:1054–70.
Melmed G, et al. (2003) Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host-microbial interactions in the gut J. Immunol. 170:1406–15.
Muir A, et al. (2004) Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell. Mol. Biol. 30:777–83.
Becker MN, Diamond G, Verghese MW, Randell SH. (2000) CD14-dependent lipopolysaccharide-induced beta-defensin-2 expression in human tracheobronchial epithelium. J. Biol. Chem. 275:29731–6.
Greene CM, et al. (2005) TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 174:1638–46.
Zhang Z, Louboutin JP, Weiner DJ, Goldberg JB, Wilson JM. (2005) Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect. Immun. 73:7151–60.
Naik S, Kelly EJ, Meijer L, Pettersson S, Sanderson IR. (2001) Absence of Toll-like receptor 4 ex plains endotoxin hyporesponsiveness in human intestinal epithelium. J. Pediatr. Gastroenterol. Nutr. 32:449–53.
Cario E, et al. (2000) Lipopolysaccharide activates distinct signaling pathways in intestinal epithelial cell lines expressing Toll-like receptors. J. Immunol. 164:966–72.
Ortega-Cava CF, et al. (2006) Epithelial toll-like receptor 5 is constitutively localized in the mouse cecum and exhibits distinctive down-regulation during experimental colitis. Clin. Vaccine Immunol. 13:132–8.
Droemann D, et al. (2003) Toll-like receptor 2 is expressed by alveolar epithelial cells type II and macrophages in the human lung. Histochem. Cell Biol. 119:103–8.
Hauber HP, et al. (2005) Toll-like receptors 4 and 2 expression in the bronchial mucosa of patients with cystic fibrosis. Can. Respir. J. 12:13–8.
Adamo R, Sokol S, Soong G, Gomez MI, Prince A. (2004) Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am. J. Respir. Cell. Mol. Biol. 30:627–34.
Shigeoka AA, et al. (2007) TLR2 is constitutively expressed within the kidney and participates in ischemic renal injury through both MyD88-dependent and -independent pathways. J. Immunol. 178:6252–8.
Backhed F, Soderhall M, Ekman P, Normark S, Richter-Dahlfors A. (2001) Induction of innate immune responses by Escherichia coli and purified lipopolysaccharide correlate with organ-and cell-specific expression of Toll-like receptors within the human urinary tract. Cell. Microbiol. 3:153–8.
Groskreutz DJ, et al. (2006) Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 176:1733–40.
Bambou JC, et al. (2004) In vitro and ex vivo activation of the TLR5 signaling pathway in intestinal epithelial cells by a commensal Escherichia coli strain. J. Biol. Chem. 279:42984–92.
Hornef MW, Frisan T, Vandewalle A, Normark S, Richter-Dahlfors A. (2002) Toll-like receptor 4 resides in the Golgi apparatus and colocalizes with internalized lipopolysaccharide in intestinal epithelial cells. J. Exp. Med. 195:559–70.
Guillot L, et al. (2004) Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J. Biol. Chem. 279:2712–8.
Samuelsson P, Hang L, Wullt B, Irjala H, Svanborg C. (2004) Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect. Immun. 72:3179–86.
Du X, Poltorak A, Wei Y, Beutler B. (2000) Three novel mammalian toll-like receptors: gene structure, expression, and evolution. Eur. Cytokine Netw. 11:362–71.
Pawar RD, et al. (2006) Toll-like receptor-7 modulates immune complex glomerulonephritis. J. Am. Soc. Nephrol. 17:141–9.
Droemann D, et al. (2005) Human lung cancer cells express functionally active Toll-like receptor 9. Respir. Res. 6:1.
Anders HJ, Schlondorff D. (2007) Toll-like receptors: emerging concepts in kidney disease. Curr. Opin. Nephrol. Hypertens. 16:177–83.
Rumio C, et al. (2004) Degranulation of paneth cells via toll-like receptor 9. Am. J. Pathol. 165:373–81.
Ewaschuk JB, et al. (2007) Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect. Immun. 75:2572–9.
Pedersen G, Andresen L, Matthiessen MW, Rask-Madsen J, Brynskov J. (2005) Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 141:298–306.
Anders HJ, et al. (2004) Activation of toll-like receptor-9 induces progression of renal disease in RL-Fas (lpr) mice. FASEB J. 18:534–6.
Chuang T, Ulevitch RJ. (2001) Identification of TLR10: a novel human Toll-like receptor preferentially expressed in immune cells. Biochim. Biophys. Acta. 1518:157–61.
Cambi A, Figdor CG. (2005). Levels of complexity in pathogen recognition by C-type lectins. Curr. Opin. Immunol. 17:345–51.
Arbour NC, et al. (2000) TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 25:187–91.
Hawn TR, et al. (2005). Toll-like receptor 4 polymorphisms are associated with resistance to Legionnaires’ disease. Proc. Natl. Acad. Sci. U. S. A. 102:2487–9.
Palmer SM, et al. (2006). Donor polymorphisms in Toll-like receptor-4 influence the development of rejection after renal transplantation. Clin. Transplant. 20:30–6.
Nogueira E, et al. (2007). Incidence of donor and recipient toll-like receptor-4 polymorphisms in kidney transplantation. Transplant. Proc. 39:412–4.
Lenoir C, et al. (2008). MD-2 controls bacterial lipopolysaccharide hyporesponsiveness in human intestinal epithelial cells. Life Sci. 82:519–28.
Abreu MT, et al. (2002). TLR4 and MD-2 expression is regulated by immune-mediated signals in human intestinal epithelial cells. J. Biol. Chem. 277:20431–7.
Mollen, KP, et al. (2008) Increased expression and internalization of the endotoxin coreceptor CD14 in enterocytes occur as an early event in the development of experimental necrotizing enterocolitis. J. Pediatr. Surg. 43:1175–81.
Mayer AK, et al. (2007). Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 178:3134–42.
Hornef MW, Normark BH, Vandewalle A, Normark S. (2003). Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J. Exp. Med. 198:1225–35.
Ewaschuk JB, et al. (2007) Surface expression of Toll-like receptor 9 is upregulated on intestinal epithelial cells in response to pathogenic bacterial DNA. Infect. Immun. 75:2572–9.
Muir A, et al. (2004). Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 30:777–83.
Lotz M, et al. (2006). Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J. Exp. Med. 203:973–84.
Shibolet O, Podolsky DK. (2007) TLRs in the Gut. IV. Negative regulation of Toll-like receptors and intestinal homeostasis: addition by subtraction. Am. J. Physiol. Gastrointest. Liver Physiol. 292:G1469–73.
Acknowledgments
DJH is supported by grant R01GM078238-01 from the National Institutes of Health and the State of Pennsylvania Tobacco Settlement Fund. SCG is supported in part by the Loan Repayment Program for Pediatric Research of the National Institutes of Health and a Resident Research Award from the American College of Surgeons. WMR is supported in part by the Loan Repayment Program for Pediatric Research of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Gribar, S.C., Richardson, W.M., Sodhi, C.P. et al. No Longer an Innocent Bystander: Epithelial Toll-Like Receptor Signaling in the Development of Mucosal Inflammation. Mol Med 14, 645–659 (2008). https://doi.org/10.2119/2008-00035.Gribar
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2008-00035.Gribar