
Abstract
While foreign pathogens and their products have long been known to activate the innate immune system, the recent recognition of a group of endogenous molecules that serve a similar function has provided a framework for understanding the overlap between the inflammatory responses activated by pathogens and injury. These endogenous molecules, termed alarmins, are normal cell constituents that can be released into the extracellular milieu during states of cellular stress or damage and subsequently activate the immune system. One nuclear protein, High mobility group box-1 (HMGB1), has received particular attention as fulfilling the functions of an alarmin by being involved in both infectious and non-infectious inflammatory conditions. Once released, HMGB1 signals through various receptors to activate immune cells involved in the immune process. Although initial studies demonstrated HMGB1 as a late mediator of sepsis, recent findings indicate HMGB1 to have an important role in models of non-infectious inflammation, such as autoimmunity, cancer, trauma, and ischemia reperfusion injury. Furthermore, in contrast to its pro-inflammatory functions, there is evidence that HMGB1 also has restorative effects leading to tissue repair and regeneration. The complex functions of HMGB1 as an archetypical alarmin are outlined here to review our current understanding of a molecule that holds the potential for treatment in many important human conditions.
Similar content being viewed by others

Introduction
Activation of the innate immune system is a necessary step in mounting an anti-microbial response to pathogens. Clinicians have long appreciated that infectious insults and tissue injury from sterile inflammatory states produce a similar inflammatory response. Recent advances in understanding the mechanisms of innate immune system activation have pointed to certain pattern recognition receptors, such as the family of Toll-like receptors, as a common pathway for immune recognition of both microbial invasion and tissue injury. By recognizing either pathogens or endogenous danger signals released upon cellular stress or damage, these pattern recognition receptors are capable of alerting the host of danger by activating the innate immune system. In this review, we will describe one such endogenous danger signal, High mobility group box-1 (HMGB1), and its role in the pathogene-sis of many disease states.
Danger Signaling
Pathogens have long been known to cause both local and systemic inflammation via activation of the immune system. Classically, descriptions of immune regulation arose from the idea that immune cells can discriminate self from non-self, and activate innate immunity when there is foreign invasion. This model, which stemmed from work done in the 1960s by Burnet and Medawar, has been modified through the years to account for new discoveries in immune cell function (1,2). Although the self/nonself model is suitable in explaining immune activation and inflammation that occurs as a consequence of clinical scenarios, such as invasion by foreign pathogens or transplant rejection, it fails to account for the inflammation which occurs in settings such as trauma or autoimmunity, which are void of non-self stimuli. In an attempt to better describe these phenomena, Matzinger has proposed the concept of danger signaling (3). In this model, initiation of the inflammatory response occurs in response to molecular patterns that are associated with both pathogens and some normal cellular components that are released by damaged cells during both infectious and sterile processes (4). The concept of cellular communication by “danger signals,” whether exogenous or endogenous, reconciled the paradox of immune activation in both infection and sterile injury.
In the danger model, immune activation is the result of recognition of molecular patterns by cellular receptors. Molecular elements from pathogens that elicit an immune response (i.e. lipopolysaccharide [LPS], bacterial DNA, viral RNA) are specific patterns termed Pathogen Associated Molecular Patterns (PAMPs). These PAMPs are generally invariant and it is their recognition by the immune system that triggers the inflammatory response (5,6). The cellular receptors that recognize these patterns are evolutionarily conserved and called Pattern Recognition Receptors (PRRs) (6).
Inflammatory responses following sterile injury closely mimic those seen during infection, with similar cytokine and chemokine production patterns (7,8). Several endogenous molecules that are released during both infectious and sterile inflammation, such as HMGB1, heat shock proteins (HSPs), S100s, and hyaluronan, have been implicated as possessing the capacity to trigger the immune system through PRRs, much like PAMPs (9–11). These signals are normal cell constituents and are released either passively by necrotic cells or actively secreted by stressed cells in response to cellular injury. While the term PAMP is restricted to patterns located on pathogens, these endogenous analogues are termed alarmins (12). The exogenous PAMPs and endogenous alarmins are subgroups of the larger category of danger signals termed Damage Associated Molecular Patterns (DAMPs) (13,14).
Alarmins are of particular interest because of their role in both infectious and sterile inflammation. They are present either locally or systemically in severe sepsis, burns, infection, arthritis, and cancer (10,15–17). Alarmins are found in a variety of organelles in all cell types studied and maintain functions in normal cellular homeostasis. They are found in the nucleus as transcription factors (HMGB1), in the cytoplasm as calcium regulators (S100s), in exosomes as chaperones (HSPs), or as components of the cell matrix (hyaluronan). Although diverse in their locations during homeostatic conditions, alarmins have many common functional characteristics. In addition to immune activation, an alarmin is released rapidly during necrosis, sequestered in apoptosis, has potential for active secretion by immune cells, and ultimately promotes homeostasis (13). Because alarmins are a diverse group of ubiquitous molecules implicated in nearly all inflammatory states, understanding and ultimately modulating their activity may allow us to control the inflammatory processes.
HMGB-1: Nuclear Protein
The high mobility group (HMG) nuclear proteins were discovered in 1973 in an effort to better define the specific regulators of gene expression (18). This group of non-histone, chromatin-associated proteins has since been characterized to be involved in DNA organization and regulation of transcription. This family of proteins shares structural characteristics which make them unique from other chromosomal proteins. These characteristics include transcripts with long AT-rich 3′ untranslated regions as well as carboxy-terminal regions which are highly negatively charged (19). HMG proteins are constitutively expressed in the nucleus of eukaryotic cells. Collectively, they share functional motifs that bind specific DNA structures and induce conformational changes without specificity for target sequences.
HMGB1 is a member of a subfamily of the HMG proteins. On average, HMGB1 is found at concentrations of 106 molecules per cell and non-specifically binds to the minor grooves in DNA (19). This binding is regulated by two 80 amino acid HMG-1 domains (or boxes), each structurally represented as three α-helices in a characteristic L-shaped fold (20). The nuclear localization of HMGB1 and its affinity for DNA is regulated through phosphorylation and acetylation, and has been found to have a dynamic relationship with chromatin (21). Like the other members of this protein family, HMGB1 plays an important role in DNA architecture and transcriptional regulation.
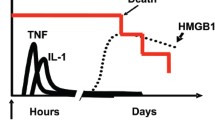
HMGB1 first was implicated as an important endogenous signaling molecule in 1999 when Wang et al. described the cytokine activity of HMGB1 by identifying it as a late mediator of endotoxin-related lethality in mice (10). This study reported increased serum levels of HMGB1 from 8 to 32 h after endotoxin exposure, attenuation of lethality with delayed administration of an HMGB1 neutralizing antibody, and lethality with direct administration of HMGB1 (10). This groundbreaking work sparked renewed interest in HMGB1 as an important component of the immune response.
The specific interactions and functions of the HMGB1 DNA binding domains have been described. As noted above, the structure of HMGB1 contains two separate “boxes,” the A- and B-boxes, each containing ∼80 amino acids in an L-shaped fold, along with an acidic C-terminal tail (20). These separate structural motifs appear to function differently when isolated from HMGB1. Several studies have identified the B-box domain as important for many of the proinflammatory properties of HMGB1 including cytokine release (22,23). In comparison, the A-box does not possess the pro-inflammatory properties of the B-box and instead competes with HMGB1 for binding sites leading to attenuation of the inflammatory cascade (24).
HMGB1 Release
HMGB1 is released passively during cellular necrosis by almost all cells which have a nucleus and signals neighboring cells of ongoing damage (25). However, HMGB1 also is secreted actively by immune cells such as monocytes, macrophages, and dendritic cells (10,16,26). Active secretion of HMGB1 is generally through non-traditional, leaderless pathways which are not routed through the endoplasmic reticulum or Golgi apparatus, similar to IL-1 (27). During normal cellular homeostasis the dynamic relationship of HMGB1 with the nucleus and cytoplasm heavily favors the nucleus. However, HMGB1 localizes in secretory lysosomes when hyper-acetylated on lysine residues and then is released upon appropriate signaling stimuli (21,28). When HMGB1 is not acetylated, it remains localized to the nucleus and is not secreted or released, such as during apoptosis (25). How the acetylation of HMGB1 is regulated is yet to be determined but is surmised to involve deacetylases in the nucleus (16,21,29). In other studies, specifically with TNFα stimulated macrophages, the secretion of HMGB1 is dependent upon phosphorylation (30). While these modifications are clearly important in the release of HMGB1, it currently is unclear how these specific modifications differ.
Stimuli for secretion of HMGB1 from immune cells are diverse and include PAMPs, cytokines, and certain states of cellular stress. Macrophages release HMGB1 starting approximately 8 h following exposure to LPS (10,31–33) but do not demonstrate the same response to CpG DNA (34). It is notable that release due to LPS is dependent, at least partially, on TNFα (31). HMGB1 also can be released in response to polyinosinic-polycytidylic acid (a model of viral infection) (33,35), while in vitro viral stimulation has demonstrated only passive release of HMGB1 due to cytokines or necrotic cells (36). In addition to these PAMPs that result in the release of HMGB1, endogenous molecules such as cytokines released during other states of injury can result in secretion of HMGB1. While first demonstrated with IFNγ, macrophages also release HMGB1 in response to stimulation with TNFα, IL-1, and oxidative stress (35,37–40). Interestingly, the PAMPs and cytokine stimuli for HMGB1 secretion from macrophages occurs through distinct pathways (35). For example, while LPS regulates HMGB1 release by hyper-acetylation, TNFα-induced secretion is mediated through phosphorylation, which directs it to the cytoplasm for release (30).
While it was thought initially that HMGB1 was released only from cells of the immune system, other cells have since been demonstrated to actively secrete alarmins. The first non-immune cell to be studied for active HMGB1 secretion was the pituicyte, which was found to release HMGB1 in response to IL-1 or TNFα stimulation (41). Enterocytes release HMGB1 following stimulation with a cytokine mixture (42). Hepatocytes also can release HMGB1 in response to hypoxic conditions or oxidative stress; this release appears to be mediated by calcium signaling changes within the cell (43).
The inhibition of HMGB1 also has been an important topic for those seeking to ameliorate injury by decreasing the level of HMGB1 release. A variety of pharmacologic agents have been studied for their potential to inhibit release of HMGB1; however, a full discussion of pharmacologic inhibition of HMGB1 is beyond the scope of this paper. It is worth noting, though, that HSP72, an endogenous molecule that has itself been indicted as an alarmin, has been demonstrated to inhibit HMGB1 release. Originally, it was demonstrated that brief heat shock resulted in decreased HMGB1 release from LPS-stimulated macrophages, but no specific pathway was identified (32). Recently, HSP72 overexpression has been shown to inhibit HMGB1 release from macrophages in response to LPS, TNFα, or oxidative stress (hydrogen peroxide). This inhibition appears to be due to intranuclear interactions between HSP72 and HMGB1 (44,45).
HMGB1 Receptors
As noted above, PRRs are a group of molecules that recognize the molecular patterns of DAMPs. These receptors may be activated by PAMPs, alarmins, or both, to activate the immune system. Several important receptors have been implicated in HMGB1 signaling, including the receptor for advanced glycation end products (RAGE) and members of the Toll-like family of receptors (TLRs). RAGE is a transmembrane protein expressed at low levels in normal tissues that is upregulated at sites where its ligands accumulate (46). The receptor first was identified to bind advanced glycation end products in diabetes, but has since been identified to bind other ligands and is involved in multiple inflammatory states (46,47). RAGE was also the first receptor demonstrated to bind HMGB1 (48). At the time, the consequences of HMGB1 interaction with RAGE were unknown, but it was discovered later that HMGB-1 signaling through RAGE promotes chemotaxis and the production of cytokines in a process that involves the activation of the transcription factor nuclear factor-κB (NF-κB) (49,50). Other RAGE-dependent effects of HMGB1 appear to involve the maturation (23,51–53) and migration (38,53–56) of immune cells as well as the upregulation of cell surface receptors (57–59).
In addition to RAGE, the Toll-like family of receptors has been demonstrated to be important in HMGB1 signaling. Members of the TLR family share many structural similarities, both extracellularly and intracellularly, but they differ from each other in ligand specificity, expression patterns, and, in some instances, the signaling pathways they activate. Generally, TLRs can recognize both DAMPs and PAMPs and hence are involved in immune response to both infection and injury. Members of the Toll family have specific ligands, and TLR4 might be the most well known and for its role in the bacterial endotoxin (LPS) recognition complex (60). TLR4, TLR2, and TLR9 have all been implicated as HMGB1 receptors. HMGB1 binding of TLR2 and TLR4 results in NF-κB upregulation (61–63), thus making it likely that HMGB1 stimulation of these receptors can lead to cytokine release. Interestingly, HMGB1-mediated TLR4 activation is different from that resulting from LPS stimulation (50,62). For example, when applied to cell cultures, HMGB1 activates both IKKα and IKKβ, whereas LPS only activates IKKβ. Additionally, the MAPK protein activation and cytokine production profiles differ between HMGB1- and LPS-treated cells.
There is much speculation that HMGB1 does not act alone in the triggering of receptor activation. Proof of this concept was provided by Tian et al. who demonstrated that HMGB1-DNA complexes activate TLR9 signaling (64). HMGB1 involvement in TLR9 activation appears to be mediated by HMGB1-DNA complexes rather than HMGB1 alone. While HMGB1-DNA complex stimulation of TLR9 is involved in maturation of immune cells and cytokine secretion (64,65), there is some evidence that these complexes may also suppress the immune response in some cell types (66). In addition to the HMGB1-DNA interactions that activate TLR9, HMGB1 interactions with other cytokines such as IL-1β, IFNγ, and TNFα lead to an increased pro-inflammatory response compared with HMGB1 stimulation alone (67). Ultimately, whether other interactions or modifications of HMGB1 are required for pattern recognition receptor binding or activation is uncertain.
Pro-Inflammatory Effects of HMGB1
As HMGB1 has multiple downstream signaling responses due to activation of different receptors, it also induces cell specific responses when it stimulates cells of the immune system (Figure 1). HMGB1 induces DC maturation as measured by the increased expression of many cell surface markers as well as the secretion of inflammatory cytokines (23,51–53). Monocytes stimulated with HMGB1 have an increased capacity for adhesion (38) and release numerous cytokines and inflammatory mediators (61,68–70). This effect is augmented when administered concomitantly with other cytokines (67,71). Neutrophil stimulation with HMGB1 increases the interaction of MAC-1 and RAGE, thus activating the adhesive and migratory function of these cells (56). Furthermore, HMGB1 stimulates the production of reactive oxygen species by neutrophils through a TLR4 dependent activation of NAD(P)H oxidase (72), as well as increases the activation of NF-κB which results in increased production and release of cytokines (50,62). T cells stimulated with HMGB1 release cytokines and appear to have increased proliferation, survival, and Th1 functional polarization (23,51).

HMGB1 is an endogenous nuclear protein that is released due to a variety of stimuli to activate proinflammatory responses in multiple cell types.
The response of endothelial cells to exogenous HMGB1 has been studied in relation to its possible pro-inflammatory role in vascular disease. Endothelial cells release TNFα, IL-8, and MCP-1, all of which enhance the local inflammatory environment (57,58). Additionally, HMGB1 stimulation appears to increase the expression of ICAM-1 and VCAM-1 on the surface of endothelial cells, thus increasing the adhesion of inflammatory cells (57,58). These effects appear to be at least partially mediated by RAGE, which also appears to be upregulated in HMGB1-stimulated endothelial cells (57,58).
Regenerative Effects of HMGB1
In contrast to their role in promoting inflammation, the ability of alarmins to promote tissue repair and regeneration is of increasing interest (73). Importantly, HMGB1 induces migration of stem cells toward inflamed regions to promote repair and regeneration (74). Furthermore, it results in increased mesangioblast and endothelial proliferation and migration to sites of inflammation and induces transit of these cells across the endothelial layer (75,76). Myoblasts stimulated by HMGB1 migrate toward damaged regions and stimulate repair (77,78). These effects on specific cell types are reflected in increased regeneration at the tissue level. In smooth muscle, HMGB1 induces proliferation and rapid changes in cellular architecture leading to cell migration (79,80). In skeletal muscle, HMGB1 promotes increased myogenesis and angiogenesis (49,76,77). Further confirmation of this is reflected in the finding that inhibition of HMGB1 signaling leads to decreased vessel density and tissue regeneration (77). Additionally, direct injection of exogenous HMGB1 to peri-infarcted car-diomyocytes in mice results in increased myocytes within the infarcted area and ultimately improved outcomes by both structural and functional measures (81). In an animal model of wound healing, topically applied HMGB1 accelerates the process in diabetic mice while inhibition of HMGB1 signaling slows it in normal mice, thus implicating functional HMGB1 signaling as an important component of diabetic wound healing (82). The ability of HMGB1 to stimulate angiogenesis has been demonstrated independently in studies demonstrating that exogenous HMGB1 stimulates endothelial proliferation and migration (75,83). Interestingly, many of these restorative effects are mediated through the same receptors (i.e. RAGE) that mediate the pro-inflammatory properties of the molecule (75,77–79,84). Taken together, the regenerative properties of HMGB1 represent a potentially important manner by which the functions of an alarmin can be manipulated to promote healing as opposed to injury.
HMGB1 and Disease
While early work on HMGB1 demonstrated its role as a late mediator of sepsis (10), more recently HMGB1 has been implicated as a putative danger signal involved in the pathogenesis of a variety of non-infectious inflammatory conditions including autoimmunity, cancer, trauma, and hemorrhagic shock, and ischemia-reperfusion injury. Furthermore, it has been studied in a number of organ systems including liver, heart, pancreas, brain, bone, and kidney. In addition to these studies highlighting the pro-inflammatory effects of HMGB1 in in vivo models of multiple diseases, there is emerging evidence to suggest that HMGB-1 can participate in tissue repair and remodeling as well as preconditioning; all of which are increasingly being recognized as important capacities of danger signals.
HMGB1 and Autoimmunity
Considerable evidence exists implicating extracellular HMGB1 in the pathogenesis of a variety of autoimmune diseases. Anti-HMGB1 antibodies are present in the serum of patients with rheumatoid arthritis and drug-induced systemic lupus erythematosus (85,86). Furthermore, HMGB1 has been found to be overexpressed in the extracellular milieu of synovial biopsy specimens in rheumatoid and experimental arthritis (87,88). When given via direct intra-articular injection, HMGB1 induces arthritis in mice, while treatment with HMGB1 antagonists ameliorates collagen-induced arthritis in both rats and mice (89,90). Increased extracellular HMGB1 also is detectable in the dermis and epidermis of skin lesions in patients with cutaneous lupus erythematosus and in biopsy specimens of the minor salivary glands of patients with Sjogren’s Syndrome (91,92).
HMGB1 and Cancer
HMGB1 was initially identified as a nuclear DNA-binding protein. As such, it plays a role in the transcription of several genes, some of which include those that have been implicated in cancer development including E-selectin, TNFα, insulin receptor, and BRCA (93–96). Furthermore, cancer cells that have undergone necrotic cell death can release HMGB1 into the local microenvironment. Extracellular HMGB1 can lead to chronic inflammatory/reparative responses that, in the setting of cancer, may lead to tumor cell survival, expansion, and metastases (97). Interestingly, numerous studies suggest that HMGB-1 plays a role in metastasis development, and thus links it to poor prognosis in a variety of cancers including prostate, breast, pancreas, and colon (98–106).
In contrast to its potential role in tumor growth and spread, it also has been shown that HMGB1-induced TLR4 signaling is required for effective responses to chemo-radiation in established tumors in animal models (107). These findings are validated further by the observation that node positive breast cancer patients carrying a loss of function TLR4 allele tend to relapse more quickly after treatment with chemo-radiation as compared to patients with wild-type TLR4 alleles (107,108). More recently, HMGB1 has been recognized as a tumor derived DAMP capable of recruiting and activating eosinophils. The role of tumor infiltrating eosinophils is still being elucidated as they can either limit tumor growth through destructive effector functions or promote tumor growth through immunoregulation and tissue repair/remodeling (109,110).
In addition to the evidence linking HMGB1 to established cancers, it has also been linked to the pathogenesis of premalignant conditions. Elevated serum levels of HMGB1 are present in mice with chemically induced colitis. Furthermore, neutralizing antibodies to HMGB1 decrease tumor incidence and size in colitis-associated cancer models (111).
The sum of these findings suggests that HMGB1 plays a role in tumor development, growth, and spread. Additionally, immune responses to established cancers prior to, as well as during, systemic treatment appear dependent upon HMGB1/TLR-4 mediated signaling cascades. Consequently, HMGB1 warrants further investigation as a possible therapeutic target in malignant processes.
HMGB1 and Trauma/Hemorrhagic Shock
End organ dysfunction in trauma and hemorrhagic shock results from systemic inflammatory responses (112). Barsness et al. provided indirect evidence that HMGB1 may be involved in the pathogenesis of end organ injury following hemorrhagic shock by linking TLR4 activation to the development of acute lung injury (113). Subsequently, it has been shown that pulmonary HMGB1 levels are increased as early as 4 h after the initiation of hemorrhagic shock (114). Furthermore, HMGB1 neutralizing antibodies ameliorate hemorrhage-induced acute lung injury (114) as well as gut barrier dysfunction and ultimately lead to improved survival (115). These findings are even more relevant given the clinical observation that circulating HMGB1 levels are elevated in trauma patients with hemorrhagic shock as compared with normal volunteers (115).
Additionally, HMGB1 has been shown to play a central role in the initiation and propagation of the inflammatory response following traumatic injury. In a mouse femur fracture model, administration of neutralizing HMGB1 antibodies results in decreased serum IL-6 and IL-10 levels. This blunted systemic response corresponds with decreased hepatic and gut barrier dysfunction as measured by serum transaminase levels and NF-κB activation, respectively. Interestingly, the contribution of HMGB1 to end organ injury in this model appears once again to be TLR4 dependent, as mutants are protected when compared with wild-types. No additional protection is seen when antibodies are administered to TLR4 mutants (116).
HMGB1 and Viral Illness
The role of HMGB1 in the pathogenesis of acute or chronic viral illnesses remains elusive. As mentioned, HMGB1 can escape extracellularly upon necrotic cell death. West Nile encephalitis and acute hepatitis are viral-induced pathologies associated with necrotic cell death either via direct cytotoxic effects of the virus itself or the inflammatory response to viral infection (36). In a recent study utilizing hepatitis B virus (HBV) transgenic mice injected with virus specific cytotoxic T lymphocytes (CTLs), HMGB1 translocates from the nucleus to the cytoplasm of hepatocytes surrounding CTL-containing necroinflammatory foci. Furthermore, treatment with HMGB1 inhibitors in this model significantly decreases the recruitment of inflammatory cells (117).
In addition to passive release from necrotic cells, HMGB1 can be secreted actively by inflammatory cells (monocytes, macrophages, dendritic cells, etc.). A number of viral illnesses including SARS and influenza result in the elevation of proinflammatory cytokines (TNF, IL-1, IL-6, types I and II interferon) that could be responsible for inducing release of HMGB1 from inflammatory cells, thus leading to the propagation of the immune response (36).
HMGB1 and Ischemia/Reperfusion Injury
A preponderance of evidence exists implicating HMGB1 in the pathogenesis of ischemia/reperfusion injury (IRI) in multiple organ systems including kidney, brain, heart, and liver. Much of the seminal work identifying extracellular HMGB1 as an alarmin that initiates the inflammatory response resulting from IRI was performed using a model of partial warm hepatic IRI in mice. In this model, tissue levels of HMGB1 are elevated as early as 1 h following reperfusion (118) and continue to increase for up to 24 h following the insult. Furthermore, in this same model, neutralizing antibodies to HMGB1 ameliorate the damage resulting from IRI in a TLR4-dependent manner.
The importance of HMGB1 signaling through TLR4 in hepatic IRI was further clarified in subsequent in vivo experiments by the generation of chimeric mice in which TLR4-derived bone marrow cells were shown to be largely responsible for the initiation and propagation of the inflammatory response. In this set of experiments, mice expressing TLR4 mutant bone marrow-derived cells were protected from liver damage as compared to those expressing TLR4 wild type bone marrow-derived cells. This occurred independently of the TLR4 phenotype expressed on hepatocytes (119). Subsequently, it was shown that increasing the number of hepatic dendritic cells worsened liver damage in TLR4 wildtype but not TLR4 mutant animals (59), thus suggesting that dendritic cells are involved in the recognition of and response to DAMPs (HMGB1 included) released after acute injury in this model. The molecular mechanisms of HMGB1/ TLR4-dependent IRI involve the production of reactive oxygen species in a process that appears to be dependent on calcium signaling via the calcium/ calmodulin-dependent kinases (CaMK) whose activation is ultimately involved in the release of HMGB1 (43).
While the significant body of literature described above elucidates the role of HMGB1 in hepatic IRI, similar work in other organ systems including the heart (120,121), kidney (122), and brain (123–125) has provided confirmatory evidence that HMGB1 is involved in the initiation of the inflammatory response following IRI.
HMGB1 and Preconditioning
A well characterized effect of pathogen-associated molecular patterns (PAMPs), such as LPS, is their ability to confer protection when administered in small doses prior to more significant insults in a process that has come to be known known as preconditioning. Therefore, alarmins ought to be able to precondition against later insults as well. Proof of principle of this concept has been demonstrated by HMGB1’s ability to provide protection when administered as a precondition agent in hepatic IRI (126). HMGB1 administration 1 h prior to the onset of injury results in a dose-dependent protection as evidenced by decreased circulating biochemical markers of liver damage as well as decreased serum TNFα and IL-6 levels. This preconditioning effect appears to be mediated through an inhibition of TLR4 signaling (126).
Conclusion
There is a growing body of literature surrounding the specific functions of HMGB1 on cells of the immune system as well as its role in important disease states. As the mechanisms promoting the release of HMGB1 and the signaling pathways it activates remain to be completely elucidated, evidence that suggests its potential as a therapeutic target/agent in various models of inflammation continues to accumulate. Importantly, human-subject studies which demonstrate its elevation in several disease states further indicate its importance. Interestingly, in addition to its pro-inflammatory roles, HMGB1 also appears to have several regenerative effects that lead to tissue repair. Understanding HMGB1 and its complex effects on the immune system may lead to the development of novel strategies to attenuate inflammation and/or promote tissue repair and regeneration in various clinical states.
References
Medawar P. (1964) Immunological Tolerance. In: Nobel Lectures, Physiology or Medicine 1942–1962. Elsevier Publishing Company, Amsterdam, The Netherlands.
Medawar PB, Woodruff MF. (1958) The induction of tolerance by skin homografts on newborn rats. Immunology. 1:27–35.
Matzinger P. (1994) Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045.
Matzinger P. (2002) The danger model: a renewed sense of self. Science. 296:301–305.
Medzhitov R, Janeway CA Jr. (2002) Decoding the patterns of self and nonself by the innate immune system. Science. 296:298–300.
Medzhitov R, Janeway CA Jr. (1997) Innate immunity: the virtues of a nonclonal system of recognition. Cell. 91:295–8.
DeMaria EJ, Pellicane JV, Lee RB. (1993) Hemorrhagic shock in endotoxin-resistant mice: improved survival unrelated to deficient production of tumor necrosis factor. J. Trauma. 35:720–4.
Mollen KP et al. (2006) Emerging paradigm: tolllike receptor 4-sentinel for the detection of tissue damage. Shock. 26:430–7.
Scheibner KA et al. (2006) Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J. Immunol. 177:1272–81.
Wang H et al. (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science. 285:248–51.
Roth J, Vogl T, Sorg C, Sunderkotter C. (2003) Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol. 24:155–8.
Oppenheim JJ, Yang D. (2005) Alarmins: chemotactic activators of immune responses. Curr. Opin. Immunol. 17:359–65.
Bianchi ME. (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81:1–5.
Harris HE, Raucci A. (2006) Alarmin(g) news about danger: workshop on innate danger signals and HMGB1. EMBO Reports. 7:774–8.
Foell D, Wittkowski H, Roth J. (2007) Mechanisms of disease: a ‘DAMP’ view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 3:382–90.
Lotze MT, Tracey KJ. (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 5:331–42.
Flohr AM et al. (2001) Variation of HMGB1 expression in breast cancer. AntiCancer Res. 21:3881–5.
Goodwin GH, Sanders C, Johns EW. (1973) A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. JBiochem. 38:14–9.
Bustin M. (1999) Regulation of DNA-dependent activities by the functional motifs of the high-mobility-group chromosomal proteins. Mol. Cell. Biol. 19:5237–46.
Weir HM et al. (1993) Structure of the HMG box motif in the B-domain of HMG1. EMBO J. 12:1311–9.
Bonaldi T et al. (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it toward secretion. EMBO J. 22:5551–60.
Li J et al. (2003) Structural basis for the proin-flammatory cytokine activity of high mobility group box 1. Mol. Med. 9:37–45.
Messmer D et al. (2004) High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J. Immunol. 173:307–13.
Yang H et al. (2004) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc. Natl. Acad. Sci. U. S. A. 101:296–301.
Scaffidi P, Misteli T, Bianchi ME. (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 418:191–5.
Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. (2000) HMG-1 as a mediator of acute lung inflammation. J. Immunol. 165:2950–4.
Gardella S et al. (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 3:995–1001.
Nickel W. (2003) The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur. J. Biochem. 270:2109–19.
Fu M, Wang C, Zhang X, Pestell RG. (2004) Acetylation of nuclear receptors in cellular growth and apoptosis. Biochem. Pharmacol. 68:1199–208.
Youn JH, Shin JS. (2006) Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J. Immunol. 177:7889–97.
Chen G et al. (2004) Bacterial endotoxin stimulates macrophages to release HMGB1 partly through. J. Leukoc. Biol. 76:994–1001.
Tang D et al. (2005) Heat shock response inhibits release of high mobility group box 1 protein induced by endotoxin in murine macrophages. Shock. 23:434–40.
Jiang W, Bell CW, Pisetsky DS. (2007) The relationship between apoptosis and high-mobility group protein 1 release from murine macrophages stimulated with lipopolysaccharide or polyinosinic-polycytidylic acid. J. Immunol. 178:6495–503.
Jiang W, Li J, Gallowitsch-Puerta M, Tracey KJ, Pisetsky DS. (2005) The effects of CpG DNA on HMGB1 release by murine macrophage cell lines. J. Leukoc. Biol. 78:930–6.
Jiang W, Pisetsky DS. (2006) The role of IFN-alpha and nitric oxide in the release of HMGB1 by RAW 264.7 cells stimulated with polyinosinic-polycytidylic acid or lipopolysaccharide. J. Immunol. 177:3337–43.
Wang H, Ward MF, Fan XG, Sama AE, Li W. (2006) Potential role of high mobility group box 1 in viral infectious diseases. Viral Immunol. 19:3–9.
Rendon-Mitchell B et al. (2003) IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J. Immunol. 170:3890–7.
Rouhiainen A et al. (2004) Regulation of monocyte migration by amphoterin (HMGB1). Blood. 104:1174–82.
Tang D et al. (2007) Hydrogen peroxide stimulates macrophages and monocytes to actively release HMGB1. J. Leukoc. Biol. 81:741–7.
Wahamaa H et al. (2007) HMGB1-secreting capacity of multiple cell lineages revealed by a novel HMGB1 ELISPOT assay. J. Leukoc. Biol. 81:129–36.
Wang H et al. (1999) Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 126:389–92.
Liu S et al. (2006) HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am. J. Physiol. Cell. Physiol. 290:C990–9.
Tsung A et al. (2007) HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 204:2913–23.
Tang D et al. (2007) The anti-inflammatory effects of heat shock protein 72 involve inhibition of high-mobility-group box 1 release and proin-flammatory function in macrophages. J. Immunol. 179:1236–44.
Tang D et al. (2007) Nuclear heat shock protein 72 as a negative regulator of oxidative stress (hydrogen peroxide)-induced HMGB1 cytoplasmic translocation and release. J. Immunol. 178:7376–84.
Chavakis T, Bierhaus A, Nawroth PP. (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect. 6:1219–25.
Stern D, Yan SD, Yan SF, Schmidt AM. (2002) Receptor for advanced glycation endproducts: a multiligand receptor magnifying cell stress in diverse pathologic settings. Adv. Drug Deliv. Rev. 54:1615–25.
Hori O et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270:25752–61.
Palumbo R et al. (2007) Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-kappaB activation. J. Cell. Biol. 179:33–40.
Park JS et al. (2003) Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am. J. Physiol. Cell. Physiol. 284:C870–9.
Dumitriu IE et al. (2005) Release of high mobility group box 1 by dendritic cells controls T cell activation via the receptor for advanced glycation end products. J. Immunol. 174:7506–15.
Telusma G et al. (2006) Dendritic cell activating peptides induce distinct cytokine profiles. Int. Immunol. 18:1563–73.
Yang D et al. (2007) High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 81:59–66.
Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P. (2005) Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur. J. Immunol. 35:2184–90.
Dumitriu IE, Bianchi ME, Bacci M, Manfredi AA, Rovere-Querini P. (2007) The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 81:84–91.
Orlova VV et al. (2007) Anovel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBOJ. 26:1129–39.
Fiuza C et al. (2003) Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 101:2652–60.
Treutiger CJ et al. (2003) High mobility group 1 B-box mediates activation of human endothelium. J. Intern. Med. 254:375–85.
Tsung A et al. (2007) Increasing numbers of hepatic dendritic cells promote HMGB1-mediated ischemia-reperfusion injury. J. Leukoc. Biol. 81:119–28.
Poltorak A et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282:2085–8.
Kokkola R et al. (2005) RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 61:1–9.
Park JS et al. (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J. Biol. Chem. 279:7370–7.
Park JS et al. (2006) High mobility group box 1 protein interacts with multiple Toll-like receptors. Am. J. Physiol. Cell. Physiol. 290:C917–24.
Tian J et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8:487–96.
Ivanov S et al. (2007) Anovel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 110:1970–81.
Popovic PJ et al. (2006) High mobility group B1 protein suppresses the human plasmacytoid dendritic cell response to TLR9 agonists. J. Immunol. 177:8701–7.
Sha Y, Zmijewski J, Xu Z, Abraham E. (2008) HMGB1 Develops Enhanced Proinflammatory Activity by Binding to Cytokines. J. Immunol. 180:2531–7.
Andersson U et al. (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 192:565–70.
Ren D, Sun R, Wang S. (2006) Role of inducible nitric oxide synthase expressed by alveolar macrophages in high mobility group box 1—induced acute lung injury. Inflamm. Res. 55:207–15.
Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. (2007) Pivotal advance: analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin). J. Leukoc. Biol. 81:49–58.
DeMarco RA, Fink MP, Lotze MT. (2005) Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol. Immunol. 42:433–44.
Fan J et al. (2007) Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J. Immunol. 178:6573–80.
Bianchi ME, Manfredi AA. (2007) High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol. Rev. 220:35–46.
Palumbo R, Bianchi ME. (2004) High mobility group box 1 protein, a cue for stem cell recruitment. Biochem. Pharmacol. 68:1165–70.
Mitola S et al. (2006) Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J. Immunol. 176:12–5.
Palumbo R et al. (2004) Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J. Cell. Biol. 164:441–9.
De MR et al. (2007) Multiple effects of high mobility group box protein 1 in skeletal muscle regeneration. Arterioscler. Thromb. Vasc. Biol. 27:2377–83.
Sorci G, Riuzzi F, Arcuri C, Giambanco I, Donato R. (2004) Amphoterin stimulates myogenesis and counteracts the antimyogenic factors basic fibroblast growth factor and S100B via RAGE binding. Mol. Cell. Biol. 24:4880–94.
Degryse B et al. (2001) The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell. Biol. 152:1197–206.
Porto A et al. (2006) Smooth muscle cells in human atherosclerotic plaques secrete and proliferate in response to high mobility group box 1 protein. FASEB J. 20:2565–6.
Limana F et al. (2005) Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C−kit+ cell proliferation and differentiation. Circ. Res. 97:e73–83.
Straino S et al. (2008) High-mobility group box 1 protein in human and murine skin: involvement in wound healing. J. Invest. Dermatol. 2008, Jan 31 [Epub ahead of print].
Schlueter C et al. (2005) Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am. J. Pathol. 166:1259–63.
Chavakis E et al. (2007) High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ. Res. 100:204–12.
Wittemann B, Neuer G, Michels H, Truckenbrodt H, Bautz FA. (1990) Autoantibodies to nonhistone chromosomal proteins HMG-1 and HMG-2 in sera of patients with juvenile rheumatoid arthritis. Arthritis Rheum. 33:1378–83.
Ayer LM, Rubin RL, Dixon GH, Fritzler MJ. (1994) Antibodies to HMG proteins in patients with drug-induced autoimmunity. Arthritis Rheum. 37:98–103.
Kokkola R et al. (2002) High mobility group box chromosomal protein 1: a novel proinflammatory mediator in synovitis. Arthritis Rheum. 46:2598–603.
Taniguchi N et al. (2003) High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 48:971–81.
Kokkola R et al. (2003) Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 48:2052–8.
Pullerits R et al. (2003) High mobility group box chromosomal protein 1, a DNA binding cytokine, induces arthritis. Arthritis Rheum. 48:1693–700.
Ek M, Popovic K, Harris HE, Naucler CS, Wahren-Herlenius M. (2006) Increased extracellular levels of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in minor salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 54:2289–94.
Popovic K et al. (2005) Increased expression of the novel proinflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis Rheum. 52:3639–45.
Fashena SJ, Reeves R, Ruddle NH. (1992) A poly(dA-dT) upstream activating sequence binds high-mobility group I protein and contributes to lymphotoxin (tumor necrosis factor-beta) gene regulation. Mol. Cell. Biol. 12:894–903.
Thanos D, Maniatis T. (1992) The high mobility group protein HMG I(Y) is required for NF-kappa B-dependent virus induction of the human IFN-beta gene. Cell. 71:777–89.
Baldassarre G et al. (2003) Negative regulation of BRCA1 gene expression by HMGA1 proteins accounts for the reduced BRCA1 protein levels in sporadic breast carcinoma. Mol. Cell. Biol. 23:2225–38.
Brunetti A, Manfioletti G, Chiefari E, Goldfine ID, Foti D. (2001) Transcriptional regulation of human insulin receptor gene by the high-mobility group protein HMGI(Y). FASEB J. 15:492–500.
Dong XE et al. (2007) High mobility group box I (HMGB1) release from tumor cells after treatment: implications for development of targeted chemoimmunotherapy. J. Immunother. 30:596–606.
Bussemakers MJ, van de Ven WJ, Debruyne FM, Schalken JA. (1991) Identification of high mobility group protein I(Y) as potential progression marker for prostate cancer by differential hybridization analysis. Cancer Res. 51:606–11.
Leman Es et al. (2003) Nuclear matrix localization of high mobility group protein I(Y) in a transgenic mouse model for prostate cancer. J. Cell. Biochem. 88:599–608.
Fedele M et al. (1996) Human colorectal carcinomas express high levels of high mobility group HMGI(Y) proteins. Cancer Res. 56:1896–901.
Nestl A et al. (2001) Gene expression patterns associated with the metastatic phenotype in rodent and human tumors. Cancer Res. 61:1569–77.
Tarbe N et al. (2001) Transcriptional profiling of cell lines derived from an orthotopic pancreatic tumor model reveals metastasis-associated genes. AntiCancer Res. 21:3221–8.
Ram TG, Reeves R, Hosick HL. (1993) Elevated high mobility group-I(Y) gene expression is associated with progressive transformation of mouse mammary epithelial cells. Cancer Res. 53:2655–60.
Dolde CE, Mukherjee M, Cho C, Resar LM. (2002) HMG-I/Y in human breast cancer cell lines. Breast Cancer Res. Treat. 71:181–91.
Takaha N, Hawkins AL, Griffin CA, Isaacs WB, Coffey DS. (2002) High mobility group protein I(Y): a candidate architectural protein for chromosomal rearrangements in prostate cancer cells. Cancer Res. 62:647–51.
Kuniyasu H, Chihara Y, Kondo H, Ohmori H, Ukai R. (2003) Amphoterin induction in prostatic stromal cells by androgen deprivation is associated with metastatic prostate cancer. Oncol. Rep. 10:1863–8.
Apetoh L et al. (2007) The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220:47–59.
Apetoh L et al. (2007) Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13:1050–9.
Lotfi R, Lee JJ, Lotze MT. (2007) Eosinophilic granulocytes and damage-associated molecular pattern molecules (DAMPs): role in the inflammatory response within tumors. J. Immunother. (1997). 30:16–28.
Cormier SA et al. (2006) Pivotal Advance: eosinophil infiltration of solid tumors is an early and persistent inflammatory host response. J. Leukoc. Biol. 79:1131–9.
Maeda S et al. (2007) Essential roles of high-mobility group box 1 in the development of murine colitis and colitis-associated cancer. Biochem. Biophys. Res. Commun. 360:394–400.
Abraham E, Carmody A, Shenkar R, Arcaroli J. (2000) Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 279:L1137–45.
Barsness KA et al. (2004) Hemorrhage-induced acute lung injury is TLR-4 dependent. Am. J. Physiol. Regul. Integr. Comp Physiol. 287:R592–9.
Kim JY et al. (2005) HMGB1 contributes to the development of acute lung injury after hemorrhage. Am. J. Physiol. Lung Cell Mol. Physiol. 288:L958–65.
Yang R et al. (2006) Anti-HMGB1 neutralizing antibody ameliorates gut barrier dysfunction and improves survival after hemorrhagic shock. Mol. Med. 12:105–14.
Levy RM et al. (2007) Systemic inflammation and remote organ injury following trauma require HMGB1. Am. J. Physiol. Regul. Integr. Comp Physiol. 293:R1538–44.
Sitia G, Iannacone M, Muller S, Bianchi ME, Guidotti LG. (2007) Treatment with HMGB1 inhibitors diminishes CTL-induced liver disease in HBV transgenic mice. J. Leukoc. Biol. 81:100–7.
Tsung A et al. (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201:1135–43.
Tsung A et al. (2005) Hepatic ischemia/ reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J. Immunol. 175:7661–8.
Goldstein RS et al. (2006) Elevated high-mobility group box 1 levels in patients with cerebral and myocardial ischemia. Shock. 25:571–4.
Oyama J et al. (2004) Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 109:784–9.
Wu H et al. (2007) TLR4 activation mediates kidney ischemia/reperfusion injury. J. Clin. Invest. 117:2847–59.
Faraco G et al. (2007) High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 103:590–603.
Kim JB, Lim CM, Yu YM, Lee JK. (2008) Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. J. Neurosci. Res. 86:1125–31
Kim JB et al. (2006) HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 26:6413–21.
Izuishi K et al. (2006) Cutting edge: high-mobility group box 1 preconditioning protects against liver ischemia-reperfusion injury. J. Immunol. 176:7154–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Klune, J.R., Dhupar, R., Cardinal, J. et al. HMGB1: Endogenous Danger Signaling. Mol Med 14, 476–484 (2008). https://doi.org/10.2119/2008-00034.Klune
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2008-00034.Klune