
Abstract
Receptor for advanced glycation end-products (RAGE) is known to be involved in microvascular complications in diabetes. RAGE is also profoundly associated with macrovascular complications in diabetes through regulation of atherogenesis, angiogenic response, vascular injury, and inflammatory response. The potential significance of RAGE in the pathogenesis of cardiovascular disease appears not to be confined solely to nondiabetic rather than diabetic conditions. Numerous truncated forms of RAGE have recently been described, and the C-terminally truncated soluble form of RAGE has received much attention. Soluble RAGE consists of several forms, including endogenous secretory RAGE (esRAGE), which is a spliced variant of RAGE, and a shedded form derived from cell-surface RAGE. These heterogeneous forms of soluble RAGE, which carry all of the extracellular domains but are devoid of the transmembrane and intracytoplasmic domains, bind ligands including AGEs and can antagonize RAGE signaling in vitro and in vivo. ELISA systems have been developed to measure plasma esRAGE and total soluble RAGE, and the pathophysiological roles of soluble RAGE have begun to be unveiled clinically. In this review, we summarize recent findings regarding pathophysiological roles in cardiovascular disease of RAGE and soluble RAGE and discuss their potential usefulness as therapeutic targets and biomarkers for the disease.
Similar content being viewed by others


Introduction
Receptor for advanced glycation end-products (receptor for AGEs, RAGE) is a multiligand cell-surface protein that was isolated from bovine lung in 1992 by Schmidt et al. and Neeper et al. (1,2). RAGE belongs to the immunoglobulin superfamily of cell surface molecules and has an extracellular region containing one V-type immunoglobulin domain and two C-type immunoglobulin domains (1,2) (Figure 1). The extracellular portion of the receptor is followed by a hydrophobic transmembrane-spanning domain and then by a highly charged, short cytoplasmic domain that is essential for intracellular RAGE signaling. RAGE was initially identified as a receptor for N-carboxymethyllysine (CML)-modified proteins (3), a major AGE in vivo (4). RAGE also interacts with other nonglycated peptide ligands, including S100/calgranulin (5), amphoterin (also termed high-mobility group box 1 protein, HMGB1) (6,7), amyloid fibrils (8), transthyretin (9), and a leukocyte integrin, Mac-1 (10). The common characteristics of these ligands are the presence of multiple β-sheets (10–12). RAGE is thought to interact with these ligands through their shared three-dimensional structure.

Numerous truncated forms of RAGE. There are three major spliced variants of RAGE: full length, N-terminally truncated, and C-terminally truncated. The C-terminally truncated form of RAGE is secreted from the cell and is named endogenously secreted RAGE (esRAGE). esRAGE has a V-domain, which is essential for binding with ligands, and is capable of competing with RAGE signaling as a decoy receptor. There are other forms of soluble RAGE (sRAGE) that are cleaved from cell-surface RAGE by matrix metalloproteinases. The ELISA assay for sRAGE measures all soluble forms including esRAGE in human plasma, while the ELISA for esRAGE measures only esRAGE, using polyclonal antibody raised against the unique C-terminus of the esRAGE sequence.
Ligand engagement of RAGE leads to prolonged inflammation, resulting in RAGE-dependent expression of proinflammatory mediators such as monocyte chemoattractant protein 1 and vascular cell adhesion molecule-1 (VCAM-1) (13,14). The engagement of RAGE has been reported to induce activation of the transcription factor nuclear factor-κB (NF-κB). However, the RAGE signaling pathway leading to NF-κB activation is largely unknown. RAGE has a short cytosolic portion that contains 43 amino acids (2). Adaptors and/or scaffold proteins that interact with the cytosolic tail of RAGE have just begun to be identified. The RAGE mutant lacking the 43-residue C-terminal tail fails to activate NF-κB, and expression of the mutant receptor results in a dominant negative effect against RAGE-mediated production of proinflammatory cytokines from macrophages (5,6). Thus, the cytosolic portion of RAGE appears to be critical in transducing the signal from the cell surface to downstream targets.
Rage Plays a Central Role in Atherosclerosis and Cardiovascular Diseases
RAGE and Atherosclerosis
In human diabetic atherosclerotic plaques, RAGE was demonstrated to be up-regulated and its expression colocalized with inflammatory markers such as cyclooxygenase 2 and matrix metalloproteinases, particularly in macrophages at the vulnerable regions of atherosclerotic plaques (15,16). These findings suggest potential roles of RAGE in human diabetic vascular disease. This hypothesis has been extensively examined in a murine model of accelerated atherosclerosis. In apoE-null mice, induction of diabetes by streptozotocin for six weeks was found to be associated with a significant increase in the atherosclerotic lesion area at the aortic sinus compared with nondiabetic apoE-null mice (17). This diabetes-associated atherosclerotic lesion has been found to exhibit increased accumulation of AGEs and S100/calgranulins and enhanced expression of RAGE (18). Daily treatment of the mice with genetically engineered murine soluble RAGE (sRAGE) suppressed diabetes-associated acceleration of lesion area and complexity, an effect that was independent of glycemic and lipid profiles (17). Similarly, sRAGE was found to prevent progression of atherosclerosis in apoE-null mice with insulin-resistant type 2 diabetes (db/db background). Vascular inflammatory phenotypes, such as overexpression of VCAM-1, tissue factor, and matrix metalloproteinase in the mice, were also prevented by administration of sRAGE (19). Bucciarelli et al. (20) found that administration of sRAGE stabilized the atherosclerotic lesion area and complexity at an advanced phase in diabetic apoE-null mice, and suppressed inflammatory markers such as expression of cyclooxy-genase 2, VCAM-1, JE-monocyte chemoattractant protein 1, matrix metalloproteinase 9 activity, tissue factor, and phosphorylation of p38 MAP kinase. The role of RAGE in progression of atherosclerosis may be independent of the presence of diabetes, because sRAGE administration was also found to significantly stabilize atherosclerotic lesion area and complexity in nondiabetic apoE-null mice (20).
RAGE and Impaired Angiogenic Response
In diabetes, progressive vasodegeneration in microvascular beds is the major underlying factor in initiation and progression of vascular complications (21–23). Recent observations in diabetic patients and diabetic animals also indicated that an increase in cardiovascular events and severity in diabetes can be associated with impairment in angiogenic response or development of new collateral vessels in response to local ischemia and/or inflammation (24–27). The AGE-RAGE axis has been found to be involved in impaired angiogenic responses in diabetes. Goova et al. (28) demonstrated that blockade of RAGE by sRAGE restores effective wound healing in diabetic mice. Tamarat et al. (29) demonstrated that blockade of AGE formation by aminoguanidine improves ischemia-induced angiogenesis in diabetic mice. Recently, ablation of galectin-3, a receptor for AGEs, has been shown to abolish the AGE-mediated increase in retinal ischemia and restore the neovascular response to that seen in controls (27).
To directly examine the role of RAGE in impairment of angiogenic response in diabetes, we compared angiogenic response in diabetic RAGE-null or wildtype mice (30). We found that angiogenic response determined by matrigel-plaque assay containing vascular endothelial growth factor (VEGF) in streptozotocintreated diabetic mice was significantly decreased compared with that of nondiabetic mice. In sharp contrast, in RAGEnull mice the angiogenic response of diabetic mice was not significantly different from that of nondiabetic mice (Figure 2). The impairment of angiogenic response in diabetes was associated with RAGE-mediated decreases in proliferation and increases in cell death (Figure 3A). The changes in cellular function were associated with activation of NF-κB in vascular cells. In this model, expression of VEGF was found to be decreased in diabetic mice (Figure 3B). The decrease in expression of VEGF in diabetes, however, was also observed in RAGE-null mice (Figure 3B). This result was in good agreement with an in vitro finding that a decrease in VEGF secretion in smooth muscle cells cultured on glycated type 1 collagen was again independent of the presence of RAGE (Figure 3C). These results suggest that the RAGE-mediated decrease in angiogenic response in diabetes is not necessarily explained by regulation of proangiogenic factors such as VEGF.

Decrease in angiogenic response in diabetes is dependent on the presence of RAGE. Angiogenic response in control and streptozotocin-induced diabetic RAGE +/+ or RAGE −/− mice was evaluated through the matrigel assay. VEGF (300 ng/mL) was suspended in matrigel, and the matrigel plugs were implanted subcutaneously into the abdominal midline of 13-week-old mice. The plugs were removed two weeks later, and processed for histochemical analysis. Bars represent 50 µm. Summary of the quantitative analyses is shown in the right panel. Data are shown as mean ± SD. *P < 0.05 vs. control, ANOVA with multiple comparison (Scheffe type). Modified from Figure 2 in (30).

(A) Decrease in PCNA-positive cells and increase in TUNEL-positive cells in diabetes are dependent on the presence of RAGE. PCNA- and TUNEL-positive cells were determined by immunohistochemical analysis. Arrowhead indicates TUNEL-positive cells. Bars represent 100 µm. Modified from (30). (B) VEGF mRNA expression was suppressed in diabetic mice independent of the presence of RAGE. Total RNA was isolated from the matrigel, and the expression of the mRNA was quantitated by real-time RT-PCR. Column represents mean ± SD. *P< 0.05 vs. control, Student t-test. Modified from reference (30). (C) VEGF mRNA expression in vascular smooth muscle cell (SMC) is suppressed in a culture on AGE-collagen independent of the presence of RAGE. Quiescent RAGE +/+ or RAGE −/− mouse SMCs were cultured on native or AGE-fibrillar collagen for 24 h with or without 100 µM deferoxamine. Total RNA was isolated, and mRNA for VEGF was determined by real-time quantitative RT-PCR. Cont, deferoxamine (−); DFO: deferoxamine (+). *P< 0.05 vs. native collagen, Student t-test.
RAGE and Neointimal Expansion after Arterial Injury
In addition to the acceleration of atherosclerosis and impairment of collateral new vessel formation, diabetic subjects have demonstrated accelerated responses to arterial injury (i.e.; therapeutic angioplasty) (31,32), which is associated with an increase in neointimal formation, enhanced vascular smooth muscle cell proliferation and migration, and production of extracellular matrix. The augmented response to arterial injury in diabetes has been shown to be associated with RAGE, because administration of sRAGE caused decreased neointimal expansion in hyperglycemic fatty Zucker rats (33). In nondiabetic RAGE-null mice, Sakaguchi et al. (34) showed that RAGE also plays an important role in neointimal expansion induced by arterial injury. Even in nondiabetic mice, RAGE transcripts in smooth muscle cells were found to be increased by arterial endothelial denudation, which was associated with induction of S100 transcripts and CML-modified AGE adducts (34).
RAGE Modifies Functions of Inflammatory Cells
It is now well recognized that mononuclear cells, such as lymphocytes and monocytes/macrophages, play fundamental roles in the progression of atherosclerosis (35). Through RAGE, AGEs, S100A12, and HMGB1 can bind to mononuclear phagocytes and modify their functions, including cell activation, chemotaxisis, transendothelial migration, and expression of key inflammatory mediators (5,36–38). Inhibition of RAGE has been found to markedly decrease infiltration of immune and inflammatory cells, such as CD4+ T cells, leading to suppression of autoimmune encephalitis (39), autoimmune diabetes (40), and allograft rejection (41). HMGB1, an essential component of DNA-containing immune complexes that stimulated cytokine production in plasmacytoid dendritic cells and B cells, was also found to involve RAGE and toll-like receptor 9, which is implicated in the pathogenesis of systemic autoimmune diseases (42).
RAGE also functions as an endothelial adhesion receptor promoting leukocyte recruitment (10). In an animal model of thioglycollate-induced acute peritonitis in both nondiabetic and diabetic mice, leukocyte recruitment was significantly impaired in RAGE-deficient mice compared with wild-type mice. In addition, RAGE-dependent leukocyte adhesion to endothelial cells was mediated by a direct interaction of RAGE with the β2-integrin Mac-1, and the RAGE-Mac-1 interaction was augmented by the proinflammatory S100-protein. Orlova et al. (43) recently defined a novel function of HMGB1 in promoting Mac-1-dependent neutrophil recruitment, which requires the presence of RAGE on neutrophils but not on endothelial cells. HMGB1 was also found to enhance the interaction between Mac-1 and RAGE. Consistently, HMGB1 stimulated in a RAGE-dependent manner activation of Mac-1, Mac-1-mediated adhesive and migratory functions, and activation of NF-κB of neutrophils. Taken together, these findings demonstrate that RAGE plays a pivotal role as a proinflammatory molecule acting at the sides of both inflammatory and endothelial cells, which may underlie part of the mechanism in progression of atherosclerosis.
sRAGE, the C-Terminally Truncated Form of Rage
Numerous truncated forms of RAGE have recently been described (44–48) (Figure 1). Two major spliced variants of RAGE mRNA, N-terminal and C-terminal truncated forms, have been most extensively characterized (45). The N-truncated isoform of RAGE mRNA codes for a 303-amino-acid protein lacking the N-terminal signal sequence and the first V-like extracellular domain. The N-truncated form is incapable of binding with AGEs, because the V-domain is critical for binding of the ligand (1). The N-truncated form of RAGE appears to be expressed on the cell surface, similarly to full-length RAGE, although its biological roles remain to be elucidated (49). This form of RAGE has been suggested to be involved in angiogenic regulation in a fashion independent of the classical RAGE signaling pathway (49).
The C-terminal truncated form of RAGE lacks the exon 10 sequences encoding the transmembrane and intracytoplasmic domains (45). This spliced variant mRNA of RAGE encodes a protein consisting of 347 amino acids with a 22-amino-acid signal sequence, and is released from cells. This C-truncated form is now known to be present in human circulation and is named endogenous secretory RAGE (esRAGE) (45). esRAGE was found to neutralize the effects of AGEs on endothelial cells in culture (45). Adenoviral overexpression of esRAGE in vivo in mice reverses diabetic impairment of vascular dysfunction (30) (Figure 4). Thus, the decoy function of esRAGE may involve a feedback mechanism by which esRAGE prevents the activation of RAGE signaling. It has also been suggested that some sRAGE isoforms that could act as decoy receptors may be cleaved proteolytically from the native RAGE expressed on the cell surface (50), suggesting heterogeneity of the origin and nature of sRAGE. This proteolytic generation of sRAGE was initially described in mice (51). Thus, the molecular heterogeneity of the diverse types of sRAGE in human plasma could exert significant protective effects against RAGEmediated toxicity. However, the endogenous action of sRAGE may not be confined to a decoy function against RAGE signaling. In an HMGB1-induced arthritis model, for example, sRAGE was found to interact with Mac-1 and to act as an important proinflammatory and chemotactic molecule (52). Further analyses are warranted to understand more about the endogenous activity of sRAGE.

Adenovirus-mediated overexpression of esRAGE restores diabetes impairment of angiogenic response. Control or streptozotocin-induced diabetic C57BL/6J mice were used to examine the effect of esRAGE overexpression in vivo. VEGF (300 ng/mL) and either Ad-esRAGE or Ad-LacZ were suspended in matrigel (5 × 108 pfu/mL), and the matrigel plugs were implanted subcutaneously into the abdominal midline of 13-week-old mice. The plugs were removed two weeks later and processed for histochemical analysis. _Bars represent 50 µm. Summary of the quantitative analyses (n = 5 for each group) is shown in the right panel. Data are shown as mean ± SD. *P < 0.05 vs. Ad-Lac Z infected control group, **P < 0.05 vs. Ad-Lac Z infected group, ANOVA with multiple comparison (Scheffe type). Modified from (30).
sRAGE and esRAGE as Potential Biomarkers for Cardiovascular and Metabolic Diseases
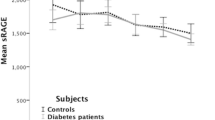
Because sRAGE and esRAGE may be involved in feedback regulation of the toxic effects of RAGE-mediated signaling, recent clinical studies have focused on the potential significance of circulating sRAGE and esRAGE in a variety of pathophysiological conditions. Table 1 summarizes currently available findings. First, Falcone et al. (53) reported that total sRAGE levels are significantly lower in patients with angiographically proven coronary artery disease (CAD) than in age-matched healthy controls. The association between circulating sRAGE and angiographic observations was shown to be dose dependent, with individuals in the lowest quartile of sRAGE exhibiting the highest risk for CAD. Importantly, this cohort consisted of a nondiabetic population, suggesting that the potential significance of sRAGE is not confined to diabetes. Falcone et al. also showed that the association between sRAGE and the risk of CAD was independent of other classical risk factors. The same research group also showed that patients with Alzheimer disease have lower levels of sRAGE in plasma than patients with vascular dementia and controls, suggesting a role for the RAGE axis in Alzheimer disease as well (54). After development of an ELISA system to specifically measure human esRAGE (55), we measured plasma esRAGE and cross-sectionally examined its association with atherosclerosis in 203 type 2 diabetic and 134 nondiabetic age- and sex-matched subjects (56). esRAGE levels were inversely correlated with carotid and femoral atherosclerosis, measured as intimal-medial thickness (IMT) by arterial ultrasound. Stepwise regression analyses revealed that plasma esRAGE was an independent factor associated with carotid IMT, and was the third strongest factor, following age and systolic blood pressure (56). Importantly however, when nondiabetic and diabetic groups were separately analyzed, inverse correlation between plasma esRAGE level and IMT was significant in the nondiabetic population only, suggesting potential importance of esRAGE in nondiabetic conditions (Figure 5). Another study also reported no association of plasma esRAGE with IMT in diabetes in 110 white type 2 diabetic subjects (57). Another Japanese research group found an in-verse correlation between plasma esRAGE and carotid atherosclerosis in type 1 (58) and type 2 diabetic subjects (59). The association between plasma sRAGE and atherosclerosis must be confirmed in larger studies, particularly in diabetic subjects, but recent findings suggest that plasma esRAGE and sRAGE may be markers for atherosclerosis and cardiovascular disease.

Plasma esRAGE level was inversely correlated with carotid and femoral atherosclerosis in a nondiabetic population but not in a diabetic population. Atherosclerosis was determined as intimal-medial thickness measured by arterial ultrasound. N = 337 including 203 type 2 diabetic patients. For the carotid artery, plasma esRAGE level exhibited a significant inverse correlation with atherosclerosis in nondiabetic subjects (r = −0.245, P = .008, n = 134), but not in diabetic subjects (r = 0.007, P = .923, n = 203). Similarly for the femoral artery, plasma esRAGE level exhibited a significant inverse correlation with atherosclerosis in nondiabetic subjects (r = −0,278, P= .003, n = 134), but not in diabetic subjects (r = −0.048, P = .493, n = 203).
Several metabolic components well established as risk factors for cardiovascular diseases have also been shown to be associated with altered plasma sRAGE and esRAGE levels. We have shown that plasma esRAGE levels are decreased in subjects with metabolic syndrome and are inversely correlated with several components of metabolic syndrome including body mass index, blood pressures, insulin resistance index, fasting plasma glucose, serum triglyceride, and lower HDL-cholesterol levels (56). The majorities of these correlations remained significant even when the nondiabetic or type 2 diabetic subpopulation was extracted for analyses. An inverse correlation between esRAGE (and sRAGE) and body mass index was also found for control subjects (60), those with type 1 diabetes (61), and those with end-stage renal disease (ESRD) (62). Patients with hypertension have been found to have lower plasma sRAGE and esRAGE levels (56,63).
The findings regarding plasma levels of sRAGE in diabetes are quite confusing. We and other groups have found that plasma esRAGE level is significantly lower in type 1 and type 2 diabetic patients than in nondiabetic controls (56,58). Plasma sRAGE levels have also been shown to be decreased in diabetic subjects (64,65), although conflicting findings have also been reported for type 1 (66) and type 2 diabetes (67,68). We examined plasma sRAGE levels with a different ELISA system that uses esRAGE as a standard protein and different sets of antibodies against the whole RAGE molecule (69). In our hands, type 2 diabetic subjects without overt nephropathy exhibited significantly (P < 0.001, Student t-test) lower plasma sRAGE levels (0.60 ± 0.28 ng/mL) than nondiabetic controls (0.77 ± 0.34 ng/mL). Of note, when diabetic subjects alone were extracted for analyses, a direct association was not observed between plasma soluble RAGE (both sRAGE and esRAGE) levels and the status of glycemic control (i.e., glycated HbA1c) (56,57,61,64,70). These complex findings in diabetic subjects suggest that levels of plasma soluble forms of RAGE are not determined simply by status of glycemic control, and that even plasma esRAGE and sRAGE levels may be under the control of distinct mechanisms.
Another important component that can affect plasma sRAGE is the presence of chronic kidney disease (CKD), which may explain controversial findings of plasma sRAGE in diabetes. It has been shown that in peripheral monocytes from subjects with varying severities of CKD, RAGE expression is closely associated with worsening of CKD and is strongly correlated with plasma levels of pentosidine, a marker for AGEs (71). Circulating sRAGE levels have been shown to be increased in patients with decreased renal function, particularly those with ESRD (57,67,72). As shown in Figure 6, plasma esRAGE levels in type 2 diabetic subjects without CKD are lower than those in nondiabetic controls, but levels gradually increased in accordance with progression of CKD. Plasma sRAGE levels in diabetic subjects without CKD were also significantly lower than those of nondiabetic controls (Figure 6). Thus, plasma sRAGE and esRAGE are markedly affected by the presence of CKD, an association that might make the previous findings regarding comparison between nondiabetic and diabetic subjects quite controversial. It remains to be determined whether the increase in plasma esRAGE in CKD is caused by decreased renal function alone or whether esRAGE levels are up-regulated to protect against toxic effects of the RAGE ligands. Successful kidney transplantation resulted in significant decreases in plasma sRAGE (73), indicating that the kidneys play a role in sRAGE removal.

Plasma esRAGE and sRAGE levels are influenced by the presence of chronic kidney disease (CKD) in patients with type 2 diabetes. CKD categories are based on glomerular filtration rate (GFR) estimated by the MDRD equation; I, GFR ≥ 90; II, GFR 60–89; III, GFR 30–59; IV, 15–29; V, GFR < 15 mL/min).
We recently reported an observational cohort study in which we longitudinally evaluated the effect of plasma esRAGE on cardiovascular mortality in patients with ESRD (62). Patients with ESRD have been reported to have a substantially increased rate of cardiovascular mortality. The cohort in our study included 206 ESRD subjects who had been treated by regular hemodialysis for more than three months. The median follow-up period of the subjects was 111 months. At the end of follow-up, 132 patients were confirmed to be alive on hemodialysis and 74 to have died; 34 deaths were due to fatal cardiovascular events. Even though the plasma esRAGE levels at baseline were higher in ESRD subjects than in those without kidney disease, the subjects in the lowest tertile of plasma esRAGE levels exhibited significantly higher cardiovascular mortality, but not noncardiovascular mortality (Figure 7). Importantly, even in the subpopulation of nondiabetic subjects alone, low circulating esRAGE level was a predictor of cardiovascular mortality, independent of the other classical risk factors. Our findings thus suggest that low circulating esRAGE level is a predictor for atherosclerosis and cardiovascular events in patients with ESRD. Because this study was designed to survey predictors for cardiovascular mortality in the population of ESRD patients, the number of fatal events was relatively small and statistical power may not have been high enough to detect important cardiovascular risk factors. Thus demonstration of the role of plasma esRAGE as a biomarker of cardiovascular mortality will require further large-scale prospective studies or nested case-control studies with required numbers of subjects calculated by power analysis.

Low plasma esRAGE level is a predictor of cardiovascular mortality in patients with ESRD. Cumulative mortalities in subjects with the lowest, middle, and highest tertiles of plasma esRAGE levels were estimated by Kaplan Meier analysis and the logrank test. Cardiovascular mortality in the lowest tertile of plasma esRAGE levels was significantly higher both in all subjects (A: P = .0028, n = 206) and in nondiabetic ESRD subjects (B: P = .0130, n = 171). In contrast, noncardiovascular mortality was not statistically different in all subjects (C: P = .89, n = 206) and in nondiabetic subjects (P = .91, n = 171). Modified from (62).
It is not yet known how esRAGE is involved in cardiovascular mortality. In our ESRD cohort, neither plasma pentosidine nor carboxymethyl lysine level predicted cardiovascular mortality. Moreover, the inverse correlation between low circulating esRAGE level and cardiovascular mortality was not dependent on plasma AGEs levels. Thus, the protective effect of esRAGE against car-diovascular mortality may not be entirely dependent on neutralization of toxic AGEs. Other endogenous ligands for RAGE, such as S100A12, may also be involved in the function of esRAGE. The plasma level of S100A12 has been shown to be increased in diabetes and inversely correlated with serum sRAGE level (64,74).
Do sRAGE and esRAGE Differ in Pathophysiological Significance?
It is unclear whether the pathophysiological significances of circulating esRAGE and sRAGE are distinct in different clinical settings. esRAGE appears to make up less than half of the total sRAGE in human plasma. In our analyses, the plasma esRAGE level in Japanese healthy subjects was found to be 0.25 ± 0.11 ng/mL (56), whereas the mean plasma sRAGE level in white healthy controls has been reported to be 1.3 ng/mL (53). We and others have shown that the plasma esRAGE level is decreased in diabetes (56,58). In contrast to esRAGE, findings regarding circulating sRAGE levels in both type 1 and type 2 diabetic patients are quite confusing and have been reported to be both increased (66-68) and decreased (64, 65). Humpert et al. also showed that sRAGE but not esRAGE is associated with albuminuria in patients with type 2 diabetes (57). We recently described a head-to-head comparison of plasma esRAGE and sRAGE levels, using esRAGE as a standard protein and different sets of antibodies, and showed that plasma esRAGE level was about two-fold less than that of plasma sRAGE (69). In this analysis, esRAGE and sRAGE levels were positively correlated, with a stronger correlation in healthy subjects than in type 1 diabetic patients. Because regulatory mechanisms are not understood for alternative splicing to generate esRAGE and for proteolytic shedding of cell-surface RAGE to generate sRAGE, the possibility of distinct roles for these forms of RAGE in certain disease conditions requires further examination.
Soluble Forms of Rage as Therapeutic Targets?
As described earlier, the potential usefulness of the soluble forms of RAGE for prevention and treatment of inflammatory diseases has been demonstrated in many animal models. Blockade of RAGE by administration of genetically engineered sRAGE successfully prevented the development of micro-(75,76) and macrovascular complications in diabetes (17,18,20). We have also shown that adenoviral overexpression of esRAGE successfully restored the impaired angiogenic response in diabetic mice (30). Sakaguchi et al. found that administration of sRAGE markedly suppressed neointimal formation following arterial injury in nondiabetic mice (34). Soluble RAGE has also been shown to effectively prevent the development of diabetes (40), protect against tumor growth and metastasis (7), improve the outcome of colitis (5), restore impaired wound healing (28), and suppress Alzheimer disease-like conditions (77). These effects of soluble RAGE in animal models could be explained by its decoy function, which inhibits RAGE interaction with its proinflammatory ligands and might be applicable to human diseases as well.
Further application of soluble forms of RAGE to the treatment of human diseases will require answers to several questions. Most importantly, limited findings are available regarding the mechanisms of regulation of circulating esRAGE and sRAGE in humans. A tissue microarray technique using a wide variety of normal adult human organ and tissue sample materials obtained from surgical and autopsy specimens revealed that esRAGE was widely distributed in tissues, including vascular endothelium, monocyte/macrophages, pneumocytes, and several endocrine organs (78). It is not yet known, however, from which organs or tissues plasma sRAGE and esRAGE originate. Circulating AGEs may be involved in regulation of the secretion or production of soluble RAGE, because AGEs are known to up-regulate RAGE expression in vitro (79). esRAGE may be simultaneously up-regulated by AGEs and act as a negative feedback loop to compensate for the damaging effects of AGEs. We and others have found positive correlations between plasma sRAGE or esRAGE and AGEs (60–62,67). As shown in Figure 8, correlation between plasma esRAGE and pentosidine was modest in all subjects (r = 0.616, P < 0.001), and was also statistically significant in only two subpopulations, hemodialysis and nonhemodialysis subjects. Plasma esRAGE was also significantly correlated with plasma CML (P = .492, P < 0.001), but the significant correlation was lost in the two sub-poulations of hemodialysis and nonhemodialysis subjects. AGEs-mediated regulation of soluble RAGE is further supported by the findings that the suppression of sRAGE expression in diabetic rat kidney was reversed by alage-brium blockade of AGEs accumulation (80). Other inflammatory mediators, such as S100, tumor necrosis factor α, and C-reactive protein, are potential candidates for regulation of the plasma level of sRAGE in humans (64,79,81). Moreover, Geroldi et al. (82) showed that high serum sRAGE is associated with extreme longevity, suggesting that understanding the intrinsic regulation of RAGE and soluble RAGE is important for longevity/antiaging strategies. Further understanding of the regulation of soluble RAGE will be helpful in identifying potential targets for its therapeutic application.

Correlation between plasma esRAGE and AGEs, pentosidine (A) and carboxymethyllysine (CML) (B). In all subjects, correlation between plasma esRAGE and both pentosidine (r = 0.616) and CML (r = 0.492) were significant (P< 0.001). Plasma pentosidine level was also significantly correlated with plasma esRAGE level in the two subpopulation of nonhemodialysis (open circle: r = 0.160, P= .005, n = 308) and hemodialysis subjects (closed circle: r = 0.216, P= .002, n = 206). In contrast, plasma CML level did not show significant correlation with plasma esRAGE level in the two subgroups of nonhemodialysis (open circle: r = 0.105, ns, n = 293) and hemodialysis subjects (closed circle: r = −0.030, ns, n = 204). Spearman’s rank correlation test was used for analyses.
Also important is investigation as to whether currently available pharmacological agents can regulate plasma sRAGE or esRAGE. Forbes et al. (83) showed that inhibition of angiotensin-converting enzyme (ACE) in rats increased renal expression of sRAGE, and that this effect was associated with decreases in expression of renal full-length RAGE protein. These investigators also showed that plasma sRAGE levels were significantly increased by inhibition of ACE in both diabetic rats and human subjects with type 1 diabetes. One attractive theory arising from these finings is that the protective effect of ACE inhibition against progression of renal dysfunction is mediated through regulation of RAGE versus soluble RAGE production. Other potential agents that may affect circulating soluble RAGE include the thiazolidinediones (84) and statins (85,86), both of which are known to modulate the AGEs-RAGE system in culture. Very recently, a randomized, open-label, parallel group study was performed with 64 participants randomized to receive add-on therapy with either rosiglitazone or sulfonylurea to examine the effect on plasma soluble RAGE (87). At six months, both rosiglitazone and sulfonylurea resulted in a significant reduction in HbA1c, fasting glucose, and AGE. However, significant increases in total sRAGE and esRAGE were seen only in the rosiglitazone group. Thus, thiazolidinedione is a promising candidate for increasing circulating levels of esRAGE and sRAGE. Future long-term prospective studies are warranted to evaluate whether modulation of circulating sRAGE can help to prevent atherosclerosis and cardiovascular disease.
Taken altogether, the findings discussed here indicate that the RAGE system plays a pivotal role in initiation and progression of atherosclerosis through regulation of vascular and inflammatory cells. sRAGE and esRAGE could serve as novel biomarkers for estimation of the risk of progression of atherosclerotic disorders. Further examination of the molecular mechanisms underlying RAGE and esRAGE regulation will provide important insights into potential targets for the prevention and treatment of cardiovascular diseases.
References
Schmidt AM, Vianna M, Gerlach M, et al. (1992) Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J. Biol. Chem. 267:14987–97.
Neeper M, Schmidt AM, Brett J, et al. (1992) Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 267:14998–15004.
Kislinger T, Fu C, Huber B, et al. (1999) N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274:31740–9.
Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW. (1995) N epsilon-(carboxymethyl)lysine is a dominant advanced glycation end product (AGE) antigen in tissue proteins. Biochemistry 34:10872–8.
Hofmann MA, Drury S, Fu C, et al. (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901.
Hori O, Brett J, Slattery T, et al. (1995) The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 270:25752–61.
Taguchi A, Blood DC, del Toro G, et al. (2000) Blockade of RAGE-amphoterin signaling suppresses tumor growth and metastases. Nature 405:354–60.
Yan SD, Chen X, Fu J, et al. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382:685–91.
Sousa MM, Yan SD, Stern D, Saraiva MJ. (2000) Interaction of the receptor for advanced glycation end products (RAGE) with transthyretin triggers nuclear transcription factor kB (NF-kB) activation. Lab. Invest. 80:1101–10.
Chavakis T, Bierhaus A, Al-Fakhri N, et al. (2003) The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J. Exp. Med. 198:1507–15.
Krieger M, Stern DM. (2001) Series introduction: multiligand receptors and human disease. J. Clin. Invest. 108:645–7.
Schmidt AM, Yan SD, Yan SF, Stern DM. (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Invest. 108:949–55.
Schmidt AM, Hori O, Chen JX, et al. (1995) Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice: a potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest. 96:1395–1403.
Basta G, Lazzerini G, Massaro M, et al. (2002) Advanced glycation end products activate endothelium through signal-transduction receptor RAGE: a mechanism for amplification of inflammatory responses. Circulation 105:816–22.
Cipollone F, Iezzi A, Fazia M, et al. (2003) The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation 108:1070–7.
Burke AP, Kolodgie FD, Zieske A, et al. (2004) Morphologic findings of coronary atherosclerotic plaques in diabetics: a postmortem study. Arterioscler. Thromb. Vasc. Biol. 24:1266–71.
Park L, Raman KG, Lee KJ, et al. (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation end-products. Nat. Med. 4:1025–031.
Kislinger T, Tanji N, Wendt T, et al. (2001) Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 21:905–10.
Wendt T, Harja E, Bucciarelli L, et al. (2006) RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 185:70–7.
Bucciarelli LG, Wendt T, Qu W, et al. (2002) RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 106:2827–35.
Mizutani M, Kern TS, Lorenzi M. (1996) Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Invest. 97:2883–90.
Stitt A, Gardiner TA, Anderson NL, et al. (2002) The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 51:2826–32.
Hammes HP, Du X, Edelstein D, et al. (2003) Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 9:294–9.
Abaci A, Oguzhan A, Kahraman S, et al. (1999) Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation 99:2239–42.
Rivard A, Silver M, Chen D, et al. (1999) Rescue of diabetes-related impairment of angiogenesis by intramuscular gene therapy with adeno-VEGF. Am. J. Pathol. 154:355–63.
Waltenberger J. (2001) Impaired collateral vessel development in diabetes: potential cellular mechanisms and therapeutic implications. Cardiovasc. Res. 49:554–60.
Stitt AW, McGoldrick C, Rice-McCaldin A, et al. (2005) Impaired retinal angiogenesis in diabetes: role of advanced glycation end products and galectin-3. Diabetes 54:785–94.
Goova MT, Li J, Kislinger T, et al. (2001) Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 159:513–25.
Tamarat R, Silvestre JS, Huijberts M, et al. (2003) Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc. Natl. Acad. Sci. USA 100:8555–60.
Shoji T, Koyama H, Morioka T, et al. (2006) Receptor for advanced glycation end products is involved in impaired angiogenic response in diabetes. Diabetes 55:2245–55.
Rozenman Y, Sapoznikov D, Mosseri M, et al. (1997) Long-term angiographic follow-up of coronary balloon angioplasty in patients with diabetes mellitus: a clue to the explanation of the results of the BARI study: Balloon Angioplasty Revascularization Investigation. J. Am. Coll. Cardiol. 30:1420–5.
Abizaid A, Kornowski R, Mintz GS, et al. (1998) The influence of diabetes mellitus on acute and late clinical outcomes following coronary stent implantation. J. Am. Coll. Cardiol. 32:584–9.
Zhou Z, Wang K, Penn MS, et al. (2003) Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 107:2238–43.
Sakaguchi T, Yan SF, Yan SD, et al. (2003) Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Invest. 111:959–72.
Ross R. (1999) Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–26.
Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, Stern D. (1993) Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J. Clin. Invest. 91:2155–68.
Shanmugam N, Kim YS, Lanting L, Natarajan R. (2003) Regulation of cyclooxygenase-2 expression in monocytes by ligation of the receptor for advanced glycation end products. J. Biol. Chem. 278:34834–44.
Rouhiainen A, Kuja-Panula J, Wilkman E, et al. (2004) Regulation of monocyte migration by amphoterin (HMGB1). Blood 104:1174–82.
Yan SS, Wu ZY, Zhang HP, et al. (2003) Suppression of experimental autoimmune encephalo-myelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat. Med. 9:287–93.
Chen Y, Yan SD, Colgan J, et al. (2004) Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J. Immunol. 173:1399–1405.
Moser B, Szabolcs MJ, Ankersmit HJ, et al. (2007) Blockade of RAGE suppresses alloimmune reactions in vitro and delays allograft rejection in murine heart transplantation. Am. J. Transplant. 7:293–302.
Tian J, Avalos AM, Mao SY, et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8:487–96.
Orlova VV, Choi EY, Xie C, et al. (2007) Anovel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. Embo. J. 26:1129–39.
Malherbe P, Richards JG, Gaillard H, et al. (1999) cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and characterization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res. Mol. Brain Res. 71:159–70.
Yonekura H, Yamamoto Y, Sakurai S, et al. (2003) Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 370:1097–109.
Schlueter C, Hauke S, Flohr AM, Rogalla P, Bullerdiek J. (2003) Tissue-specific expression patterns of the RAGE receptor and its soluble forms: a result of regulated alternative splicing? Biochim. Biophys. Acta. 1630:1–6.
Park IH, Yeon SI, Youn JH, et al. (2004) Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 40:1203–11.
Ding Q, Keller JN. (2005) Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci. Lett. 373:67–72.
Bierhaus A, Humpert PM, Morcos M, et al. (2005) Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 83:876–86.
Hudson BI, Harja E, Moser B, Schmidt AM. (2005) Soluble levels of receptor for advanced glycation endproducts (sRAGE) and coronary artery disease: the next C-reactive protein? Arterioscler. Thromb. Vasc. Biol. 25:879–82.
Hanford LE, Enghild JJ, Valnickova Z, et al. (2004) Purification and characterization of mouse soluble receptor for advanced glycation end products (sRAGE). J. Biol. Chem. 279:50019–24.
Pullerits R, Brisslert M, Jonsson IM, Tarkowski A. (2006) Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via beta2 integrin Mac-1. Arthritis Rheum. 54:3898–907.
Falcone C, Emanuele E, D’Angelo A, et al. (2005) Plasma levels of soluble receptor for advanced glycation end products and coronary artery disease in nondiabetic men. Arterioscler. Thromb. Vasc. Biol. 25:1032–7.
Emanuele E, D’Angelo A, Tomaino C, et al. (2005) Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch. Neurol. 62:1734–6.
Sakurai S, Yamamoto Y, Tamei H, et al. (2006) Development of an ELISA for esRAGE and its application to type 1 diabetic patients. Diabetes Res. Clin. Pract. 73:158–65.
Koyama H, Shoji T, Yokoyama H, et al. (2005) Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 25:2587–93.
Humpert PM, Djuric Z, Kopf S, et al. (2007) Soluble RAGE but not endogenous secretory RAGE is associated with albuminuria in patients with type 2 diabetes. Cardiovasc. Diabetol. 6:9.
Katakami N, Matsuhisa M, Kaneto H, et al. (2005) Decreased endogenous secretory advanced glycation end product receptor in type 1 diabetic patients: its possible association with diabetic vascular complications. Diabetes Care 28:2716–21.
Katakami N, Matsuhisa M, Kaneto H, Yamasaki Y. (2007) Serum endogenous secretory RAGE levels are inversely associated with carotid IMT in type 2 diabetic patients. Atherosclerosis 190:22–3.
Yamagishi S, Adachi H, Nakamura K, et al. (2006) Positive association between serum levels of advanced glycation end products and the soluble form of receptor for advanced glycation end products in nondiabetic subjects. Metabolism 55:1227–31.
Miura J, Yamamoto Y, Osawa M, et al. (2007) Endogenous secretory receptor for advanced glycation endproducts levels are correlated with serum pentosidine and CML in patients with type 1 diabetes. Arterioscler. Thromb. Vasc. Biol. 27:253–4.
Koyama H, Shoji T, Fukumoto S, et al. (2007) Low circulating endogenous secretory receptor for AGEs predicts cardiovascular mortality in patients with end-stage renal disease. Arterioscler. Thromb. Vasc. Biol. 27:147–53.
Geroldi D, Falcone C, Emanuele E, et al. (2005) Decreased plasma levels of soluble receptor for advanced glycation end-products in patients with essential hypertension. J. Hypertens. 23:1725–9.
Basta G, Sironi AM, Lazzerini G, et al. (2006) Circulating soluble receptor for advanced glycation end products is inversely associated with glycemic control and S100A12 protein. J. Clin. Endocrinol. Metab. 91:4628–34.
Devangelio E, Santilli F, Formoso G, et al. (2007) Soluble RAGE in type 2 diabetes: association with oxidative stress. Free Radic. Biol. Med. 43:511–8.
Challier M, Jacqueminet S, Benabdesselam O, Grimaldi A, Beaudeux JL. (2005) Increased serum concentrations of soluble receptor for advanced glycation endproducts in patients with type 1 diabetes. Clin. Chem. 51:1749–50.
Tan KC, Shiu SW, Chow WS, Leng L, Bucala R, Betteridge DJ. (2006) Association between serum levels of soluble receptor for advanced glycation end products and circulating advanced glycation end products in type 2 diabetes. Diabetologia 49:2756–62.
Nakamura K, Yamagishi SI, Adachi H, et al. (2007) Elevation of soluble form of receptor for advanced glycation end products (sRAGE) in diabetic subjects with coronary artery disease. Diabetes Metab. Res. Rev. 23:368–71.
Yamamoto Y, Miura J, Sakurai S, et al. (2007) Assaying soluble forms of receptor for advanced glycation end products. Arterioscler. Thromb. Vasc. Biol. 27:e33–4.
Humpert PM, Kopf S, Djuric Z, et al. (2006) Plasma sRAGE is independently associated with urinary albumin excretion in type 2 diabetes. Diabetes Care 29:1111–3.
Hou FF, Ren H, Owen WF Jr, et al. (2004) Enhanced expression of receptor for advanced glycation end products in chronic kidney disease. J. Am. Soc. Nephrol. 15:1889–96.
Kalousova M, Hodkova M, Kazderova M, et al. (2006) Soluble receptor for advanced glycation end products in patients with decreased renal function. Am. J. Kidney Dis. 47:406–11.
Kalousova M, Bartosova K, Zima T, Skibova J, Teplan V, Viklicky O. (2007) Pregnancy-associated plasma protein a and soluble receptor for advanced glycation end products after kidney transplantation. Kidney Blood Press. Res. 30:31–7.
Kosaki A, Hasegawa T, Kimura T, et al. (2004) Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 89:5423–8.
Wendt TM, Tanji N, Guo J, et al. (2003) RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 162:1123–37.
Bierhaus A, Haslbeck KM, Humpert PM, et al. (2004) Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Invest. 114:1741–51.
Arancio O, Zhang HP, Chen X, et al. (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. Embo. J. 23:4096–105.
Cheng C, Tsuneyama K, Kominami R, et al. (2005) Expression profiling of endogenous secretory receptor for advanced glycation end products in human organs. Mod. Pathol. 18:1385–96.
Tanaka N, Yonekura H, Yamagishi S, Fujimori H, Yamamoto Y, Yamamoto H. (2000) The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 275:25781–90.
Coughlan MT, Thallas-Bonke V, Pete J, et al. (2007) Combination therapy with the advanced glycation end product cross-link breaker, alage-brium, and angiotensin converting enzyme inhibitors in diabetes: synergy or redundancy? Endocrinology 148:886–95.
Zhong Y, Li SH, Liu SM, et al. (2006) C-Reactive protein upregulates receptor for advanced glycation end products expression in human endothelial cells. Hypertension 48:504–11.
Geroldi D, Falcone C, Minoretti P, Emanuele E, Arra M, D’Angelo A. (2006) High levels of soluble receptor for advanced glycation end products may be a marker of extreme longevity in humans. J. Am. Geriatr. Soc. 54:1149–50.
Forbes JM, Thorpe SR, Thallas-Bonke V, et al. (2005) Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J. Am. Soc. Nephrol. 16:2363–72.
Marx N, Walcher D, Ivanova N, et al. (2004) Thiazolidinediones reduce endothelial expression of receptors for advanced glycation end products. Diabetes 53:2662–8.
Okamoto T, Yamagishi S, Inagaki Y, et al. (2002) Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J. 16:1928–30.
Cuccurullo C, Iezzi A, Fazia ML, et al. (2006) Suppression of RAGE as a basis of simvastatin-dependent plaque stabilization in type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 26:2716–23.
Tan KC, Chow WS, Tso AW, et al. (2007) Thiazolidinedione increases serum soluble receptor for advanced glycation end-products in type 2 diabetes. Diabetologia 50:1819–25.
Kalousova M, Jachymova M, Mestek O, et al. (2007) Receptor for advanced glycation end products: soluble form and gene polymorphisms in chronic haemodialysis patients. Nephrol. Dial. Transplant. 22:2020–6.
Nakamura K, Yamagishi S, Adachi H, et al. (2007) Serum levels of sRAGE, the soluble form of receptor for advanced glycation end products, are associated with inflammatory markers in patients with type 2 diabetes. Mol. Med. 13:185–9.
Nakamura K, Yamagishi SI, Adachi H, et al. (2007) Circulating advanced glycation end products (AGEs) and soluble form of receptor for AGEs (sRAGE) are independent determinants of serum monocyte chemoattractant protein-1 (MCP-1) levels in patients with type 2 diabetes. Diabetes Metab. Res. Rev. Aug 10; [Epub ahead of print].
Acknowledgments
The authors thank all colleagues at the Osaka City University Graduate School of Medicine and Kanazawa University Graduate School of Medical Science for their unflagging support of our projects. We apologize to all colleagues whose work we could not cite other than indirectly through other publications, because of space limitations. This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (17590946 to H.K and H.Y.) and a grant from the Osaka Kidney Foundation (OKF06-007 to H.K.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Koyama, H., Yamamoto, H. & Nishizawa, Y. RAGE and Soluble RAGE: Potential Therapeutic Targets for Cardiovascular Diseases. Mol Med 13, 625–635 (2007). https://doi.org/10.2119/2007-00087.Koyama
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2007-00087.Koyama