
Abstract
Whole body exposure of wild type control littermates and A2A adenosine receptor (A2AR) gene deleted mice to low oxygen containing inspired gas mixture allowed the investigation of the mechanism that controls inflammatory liver damage and protects the liver using a mouse model of T cell-mediated viral and autoimmune hepatitis. We tested the hypothesis that the inflammatory tissue damage-associated hypoxia and extracellular adenosine ➔ A2AR signaling plays an important role in the physiological anti-inflammatory mechanism that limits liver damage during fulminant hepatitis. After induction of T cell-mediated hepatitis, mice were kept in modular chambers either under normoxic (21% oxygen) or hypoxic (10% oxygen) conditions for 8 h. It was shown that the whole body exposure to hypoxic atmosphere caused tissue hypoxia in healthy animals as evidenced by a decrease in the arterial blood oxygen tension and increase of the plasma adenosine concentration (P < 0.05). This “hypoxic” treatment resulted in significantly reduced hepatocellular damage and attenuated levels of serum cytokines in mice with acute liver inflammation. The anti-inflammatory effects of hypoxia were not observed in the absence of A2AR in studies of A2AR gene-deficient mice or when A2AR have been pharmacologically antagonized with synthetic antagonist. The presented data demonstrate that total body hypoxia-triggered pathway provides protection in acute hepatitis and that hypoxia (upstream) and A2AR (downstream) function in the same immunosuppressive and liver tissue-protecting pathway.
Similar content being viewed by others

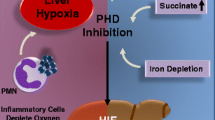

Introduction
Acute or chronic hepatitis due to viral infections or autoimmune hepatitis affects millions of patients and results in high long-term morbidity (1–4). It is believed that acute or chronic hepatitis in large degree both are caused by the over-active immune cells, rather than by the hepatitis virus itself. Indeed, for example, hepatitis B virus-specific T cell responses contribute not only to viral clearance and clinical recovery, but also to immune-mediated acute and chronic necro-inflammatory liver disease (5,6). Hence, the most prominent mechanism of liver injury in viral hepatitis seems to be CD8+ T cell-mediated, although non-virus-specific T cells also may be recruited to the liver and further aggravate the liver damage without contributing to virus control. In addition to T cells, neutrophilic granulocytes, mononuclear cells, and platelets accumulate in the liver area and can aggravate injury further. In autoimmune hepatitis, liver disease is triggered by yet to be identified antigens. The mechanism of autoimmunity appears to be similar to viral hepatitis i.e. acting through non-specific T cells which aggravate and enhance liver injury (reviewed in 7;8). It is important to uncover the mechanisms that regulate immune responses in the liver and that protect liver tissues from excessive collateral damage. Here, we focused on studies of the relationship between tissue hypoxia and the extracellular adenosine-mediated immunosuppression (9–11). It was suggested that the extracellular adenosine levels in the inflamed area was sufficiently high to trigger signaling by A2A and/or A2B adenosine receptors (A2AR and A2BR) and to suppress immune attack by surrounding cells, including activated T cells. This chain of events culminates in inhibition of overactive immune cells in a delayed negative feedback blockade due to the well-established immunosuppression by A2 adenosine receptors (11).
To date, four different and widely distributed adenosine receptors have been described: A1, A2A, A2B, and A3 (11). The high affinity A1 receptor and low affinity A3 receptor are Gi proteincoupled. The cAMP-elevating Gs protein-coupled A2 receptors are subdivided into high affinity A2AR and low affinity A2BR. Adenosine receptors are known to be immunosuppressive (11–14). CD8+ T cells, including antitumor CD8+ T cells and human T cells, predominantly express A2AR and A2BR but not A3 receptors. The cAMP-elevating signaling through A2AR or A2BR in T cells results in inhibition of T cell receptor-triggered activation of T cells and of many effector functions including proliferation, expansion, and secretion by T cells of cytokines such as IFN-γ and TNF-α (11,15).
We examined a tissue-protective effect of hypoxia via A2AR in a well-documented model of autoimmune and viral hepatitis which is triggered by i.v. injection of concanavalin A (Con A). Con A-induced liver injury has been studied extensively as an immunological model of acute hepatitis. Initially, it was described as a T cell-dependent liver injury model because the liver damage could not be induced in scid and nude mice (16). It was followed by the findings of proinflammatory cytokines (IFN-γ and TNF-α) and Kupffer cells as essential components of Con A-induced hepatitis (17–22). In addition to T cells, NKT cells also were found to be indispensable and IL-4 produced from NKT cells plays an important role in the induction of liver damage (23,24). We previously reported that A2AR regulates Con A-induced inflammatory responses in the liver as shown by exacerbation of liver damage in A2AR-deficient mice (25).
Here we utilized whole body exposure of mice to 10% oxygen containing inspired gas mixtures, which can predictably decrease local tissue oxygen tension, and demonstrate the hypoxia-driven immunosuppressive and liver tissue-protecting mechanism. Subsequent experiments using an A2AR antagonist and A2AR-deficient mice showed an important role of A2AR in the mechanism of hypoxia-driven hepatoprotection. It is concluded that this hypoxia ➙ A2AR mediated anti-inflammatory pathway is non-redundant in liver tissue protection.
Materials and Methods
Animals
Animal experiments were conducted in accordance to the “U.S. Government Principles For The Utilization And Care Of Vertebrate Animals Used in Testing, Research And Training” (u]http://oacu.od.nih.gov/regs/USGovtPrncpl.htm) and have been approved by the National Institutes of Health (NIH) Animal Care and Use Committee. Mice were maintained under specific pathogen-free conditions at the NIH animal care facilities. All mice used were on a C57BL/6 genetic background. The A2AR−/− mice were backcrossed at least ten times and all phenotypic comparisons were conducted by comparing A2AR−/− and their A2AR+/+ littermates. The breeding strategy was based on homozygous inter-breeding and all mice used in the experiments presented were nine- to twelve-week-old age-matched females. Hepatitis was induced by the tail vein i.v. injection of Con A (11.5 mg/kg; Sigma Chemicals, St. Louis, MO, USA); sham animals received vehicle only.
Pre-Treatment by A2-Receptor Antagonist
Adenosine A2 receptor antagonist ZM241385 (Tocris Cookson, Ballwin, MO, USA) was injected subcutaneously at a dose of 10 mg/kg body weight 1.5 h prior and again 1.5 h after Con A injection. Control animals received the same volume of the vehicle solution only. In these pharmacological studies with ZM241385, we have confirmed that the initial in vivo levels of ZM241385 in serum are sufficiently high to antagonize the adenosine A2 receptor-induced cAMP accumulation in immune cells (data not shown).
Blood Analyses
Blood samples were collected from the retro-orbital vein to determine serum activities of alanine transaminase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH), as well as the concentrations of IL-4, IFN-γ, and TNF-α. The choice of time points was based on results of preliminary and published experiments (25). Serum chemistries were performed with an automated analyzer (Synchron LX20 Clinical System, Beckman Coulter, Brea, CA, USA) and cytokines were quantified by ELISA according to the manufacturer’s instructions (R&D systems, Minneapolis, MN, USA). Lactate concentrations also were determined by photometry according to the manufacturer’s instruction (Biomedical Research Service Center, University at Buffalo, Buffalo, NY, USA).
Control of Fraction of Inspired Oxygen (Oxygenation) and Partial Arterial OxygenPressure (Art pO2)
After Con A or sham injections, mice were placed in air-tight modular incubation chambers (Billups-Rothenberg, San Diego, CA, USA) and atmosphere was controlled by a constant gas flow (1.5 L/min) having 21% or 10% O2. In representative experiments, arterial blood was drawn from the carotid artery of anesthetized mice breathing either 10% or 21% oxygen at a flow rate of 1.5 L/min. Blood was sampled immediately into heparinized glass capillaries for analysis of blood oxygen pressure (Rapidlab 248; Chiron Diagnostics, Essex, UK).
Plasma Concentrations of Purines
Arterial blood samples were drawn from the carotid in mice breathing 10% oxygen or 21% oxygen atmosphere for 1.5 h. Due to a short half life of adenosine, the syringes were pre-filled with ice-cold physiological saline containing dipyridamole 2 × 10−4 M, EHNA (erythro-9-(2-hydroxy-3-nonyl)adenine) 2 × 10−5 M, EDTA-Na 2 × 10−2 M, EGTA 2 × 10−2 M, and DL-α-glycerophosphate 2 × 10−2 M to prevent degradation or additional formation of plasma adenosine by inhibition of ecto-nucleotidases and non-specific phosphatases. The samples were processed and plasma concentrations of adenosine and inosine determined by high-performance liquid chromatography (HPLC) as described previously (26,27).
Statistics
Data were evaluated for statistical significance by ANOVA (two-way) for the detection of intra- and intergroup differences and by t-test for comparisons of only two different conditions, respectively. Data are presented as mean values ± standard error of the mean (SEM) with a level of significance being less than 0.05. Statistically significant differences between survival rates were evaluated by Chi-square testing with a level of significance below 0.05. All statistical analyses were performed by SPSS 10.0 program (SPSS Inc., Chicago, IL, USA).
Results
The Role of A2AR in Con A-Induced Liver Inflammation
Treatment of C57BL/6 wild type mice with Con A (11.5 mg/kg) resulted in the induction of strong liver damage as is reflected by elevated levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH). These increases have been dependent as a function of time after injection of Con A (data not shown) having reached peak values at 8 h after injection of Con A. This time point (8 h) after induction of liver-localized and T cell-triggered immune response has been selected for subsequent experiments and liver damage (i.e. ALT/AST/LDH measurements).
It was found that Con A-induced levels of ALT, AST, as well as LDH were significantly higher in animals that were pre-treated by the adenosine A2 receptor antagonist ZM241385 (Figure 1). There was also an increase of the lactate concentration, which was significant as compared with the Con A-injected mice without ZM241385 (Table 1). IFN-γ concentrations increased significantly from undetectable low values determined prior to Con A induction/or sham treated mice (data not shown) to mean values of more than 1100 or 1200 pg/mL, in the absence and presence of ZM241385, respectively (Table 2).

Effects of Con A injection on serum liver enzyme activities in the absence and presence of A2 receptor antagonist ZM241385. Serum activities (I.U.) of alanine aminotransferase (ALT, Figure 1A), aspartate aminotransferase (AST, Figure 1B), and lactate dehydrogenase (LDH, Figure 1C) were determined 8 h after Con A (11.5 mg/kg i.v.) in the presence and absence of ZM241385 (10 mg/kg s.c. × 2). Sham treated animals had low and physiological liver enzyme activities (I.U.): Con A Sham: ALT 100 ± 10, AST 110 ± 10, LDH 150 ± 60; ZM241385 Sham: ALT 110 ± 15, AST 100 ± 10, LDH 160 ± 50 (Means ± SEM, n = 4 per group, paired t-test; + P < 0.05, *P < 0.01).
Effects of Hypoxia on Arterial Oxygen Tension and Plasma Purines
Breathing of an hypoxic gas mixture with a preset concentration of 10% oxygen and 90% nitrogen resulted in a reduction of the arterial oxygen tension (paO2) to mean values of approximately 50 mmHg in spontaneously breathing mice. In contrast, breathing of normal compressed air (21% oxygen) increased paO2 to normal values of > 75 mmHg after 1.5 h (Table 3). An inverse relation was observed for plasma concentrations of adenosine which were significantly higher (P < 0.05) during hypoxia as compared with normal oxygen concentration. This was also observed for the direct metabolite of adenosine, for example, inosine, which was also elevated by hypoxia in both vehicle and Con A-treated groups to a comparable degree (Table 3).
The Whole Body Hypoxia Exposure Protects Liver Tissue from Inflammatory Damage
Serum activities of ALT, AST, and LDH were reduced significantly (P < 0.0001) when the animals were subjected to 10% oxygen instead of 21% in the control group (Figure 2) for 8 h after Con A injection. The same reduction was observed for lactate levels (Table 1). At the same time point (8 h), serum concentrations of IFN-γ were decreased significantly in mice exposed to hypoxia and TNF-α concentrations were dropped by 80%, close to the detection limits of the assay (Table 4). The reduction of cytokine levels was also apparent in serum samples collected 1.5 h after Con A injection. Especially, induction of IL-4 by Con A injection was strongly impaired by treatment with hypoxia (Table 4). These results indicate that whole body hypoxia suppresses cytokines production and protects the liver from inflammatory tissue damage.

Hypoxia (10% O2) reduces serum liver enzyme activities after Con A injection. As compared with the release of liver enzymes after Con A challenge in normo-oxygenated atmosphere (filled dark bars), hypoxia for 8 h after Con A injection (bright gray bars) results in lower liver enzyme activities of ALT, AST, and LDH. Sham 10%: ALT 110 ± 10, AST 110 ± 10, LDH 160 ± 84 (Means ± SEM, n = 9–11 per group, paired t-test; + P < 0.05; **P < 0.001).
Hypoxia Provides Hepatoprotection Through A2AR-Dependent Mechanism
Although hypoxia can strongly diminish Con A-induced liver damage, pharmacological antagonism of A2AR by ZM241385 in Con A-injected mice under hypoxia (10% O2) significantly increased serum activities of ALT, AST, and LDH (Figure 3) and concentrations of lactate (Table 1). Instead of ZM241385-treated wild type A2AR+/+ animals, when the A2AR−/− (“knock-out”) mice were subjected to the same conditions, almost the same high liver enzyme activities were detected as the ZM241385-treated group. In addition, concentrations of IFN-γ were more than doubled when A2 receptors were antagonized pharmacologically or when animals were genetically deficient for A2AR (Table 2). Moreover, while the majority of hypoxia-treated mice survived the Con A challenge, pharmacological antagonism at the A2AR was associated with a significant increase of mortality between 6–8 h after Con A injection (Figure 4), indicating that the action at the A2AR is required for better survival after Con A-induced liver inflammation. In addition, confirmative control experiments with sham mice not having received Con A showed that housing in the modular chambers at hypoxic or normal oxygenation did not induce mortality under both experimental conditions (10% and 21% O2, n = 10; observation period up to 48 h). Taken together, hypoxia protects from inflammatory liver damage via A2AR-dependent mechanism.

Effects of pharmacological antagonism (ZM241385) or genetic “knockout” of A2AR gene on the hepatoprotective effects mediated by hypoxia. Mice of A2AR “wild type” (A2AR+/+) either without (left black bar) or with ZM241385 (bright gray bar) and homozygotic A2AR “knock-out” mice (A2AR−/−) (right dark gray bar) were subjected to Con A (11.5 mg/kg i.v.) injection and placed into hypoxic chambers (10% oxygen) for 8 h thereafter. Means ± SEM, n = 11–13 per group; paired t-test versus WT without ZM241385; + P < 0.05, *P < 0.01.

Effects of antagonism at the A2AR site on the mortality after Con A induced hepatitis under hypoxia. To verify the clinical effects of the antagonism of the hypoxia- > adenosine- > hepatoprotection pathway, mice were injected by Con A (11.5 mg/kg) and placed into 10% oxygen atmosphere for 8 h maximally. A2AR+/+ wild type mice treated with ZM241385 revealed a significantly higher mortality with death mostly between 6–8 h after Con A injection. Differences between survival rates were tested by Chi-square test, P < 0.05.
Discussion
The data shown in this report strongly support the newly emerging model that it is the tissue inflammation accompanying local tissue hypoxia that may represent the initial event in recruiting the tissue-protecting immunosuppressive mechanism.
It is shown that tissue hypoxia is associated with the accumulation of extracellular adenosine (28), which then triggers A2AR and A2BR leading to the accumulation of immunosuppressive intracellular cAMP in activated immune cells (29). Cyclic AMP, in turn, inhibits signaling pathways required for synthesis and secretion of pro-inflammatory and cytotoxic mediators by immune cells, terminates immune cells’ effector functions, and thereby protects remaining healthy tissues from continuous immune damage (reviewed in 9,30).
This conclusion has implications for the regulation of immune response in general even though reported experiments have been performed in a model of acute liver inflammation. The pathogenesis of viral and autoimmune hepatitis is explained by activities of T-lymphocytes, tissue macrophages, and granulocytes which enhance tissue damage and impair the local microcirculation and blood supply by releasing inflammatory mediators, for example, IFN-γ and TNF-α. These processes mimicked in an acute model of Con A-induced liver injury are mediated by a T cell-initiated cascade of pro-inflammatory processes, which have been validated as reproducing the liver damage resembling autoimmune and viral hepatitis in detailed studies using different genetically altered mice, recombinant decoy cytokine receptors, and monoclonal antibodies (17–21). These considerations led to the choice of this model and to testing the potentially tissue-protective role of hypoxia and endogenous adenosine in a standardized model of Con A-induced T cell activation and T cell-mediated tissue inflammation.
When animals were subjected to hypoxia of 10% instead of 21% oxygen for 8 h, Con A-induced hepatocellular damage was attenuated significantly as evidenced by the lower enzyme activities of the cytosolic enzyme ALT which has high organ specificity to the liver and also by lower levels of AST derived from the cell cytosol and mitochondria (31). Similarly, the decrease was also seen in the activity of LDH in the blood. This result was expected as LDH levels were shown to reflect the degree of liver cell damage (32). In addition, serum lactate concentrations were also reduced to the baseline levels of the sham-treated group when Con A-challenged mice were subjected to hypoxia. The measurements of lactate concentration are considered to be one of the most reliable biochemical parameters to determine the severity and outcome of acute liver failure and reflect the severity of liver injury (33). Hyperlactatemia develops under clinical conditions in approximately 80% of patients suffering from fulminant liver failure, for example, due to hepatitis-induced enhancement of hepatic and extrahepatic anaerobic glycolytic activity with higher lactate generation together with a reduced hepatocyte capacity and function to clear lactate, altogether resulting in a net hepatic lactate production (34,35). In control, the hepatocellular integrity in the absence of Con A (sham) was not affected by hypoxia.
In addition to the suppression of liver damage, we found that serum cytokine levels were decreased significantly in mice subjected to hypoxia (Table 4). This result suggests that hypoxia can impair the initiation of inflammatory responses which eventually will lead to extensive liver damage. The decrease of cytokine levels such as IFN-γ, TNF-α, and IL-4 may, at least in part, explain the attenuation of liver damage by hypoxia, because these cytokines are shown to be essential in the induction of liver damage by Con A. Indeed, the importance of IFN-γ and TNF-α has been shown by the inhibition of Con A-induced liver damage after neutralization by anti-cytokine antibodies and in gene-deficient mice (17–21). Furthermore, early IL-4 production from NKT cells is demonstrated to be essential in the induction of liver injury (23,24). Therefore, the impairment of early IL-4 production by hypoxia may suggest inhibition of NKT cells activation by hypoxia-A2AR pathway as an additional inhibitory mechanism of Con A-induced liver injury. In correspondence, inhibition of NKT cells by an A2AR agonist is reported recently (36).
As a major prerequisite to estimate the role of hypoxia and endogenous adenosine under inflammation, we confirmed first that exposure of the animals to 10% oxygen, with or without Con A treatment, resulted in decreased arterial blood pO2 values as compared with exposure to 21% oxygen. Hypoxic exposure (1.5 h) was paralleled by a significant increase of plasma concentrations of the purine nucleotide adenosine and its metabolites inosine and hypoxanthine in vehicle- and Con A-treated mice to the same degree, indicating that this increase was due to hypoxia treatment.
Hypoxia is associated with i) decrease in intracellular ATP; ii) increase in intracellular AMP; iii) inhibition of adenosine kinase; iv) accumulation of intracellular adenosine; and v) subsequent transport or diffusion of intracellular adenosine and accumulation of adenosine in extracellular space. The accumulation of extracellular adenosine from intracellular sources may be triggered by local tissue hypoxia that follows the excessive collateral immune damage to endothelial cells and microcirculation with ensuing interruption of normal blood and oxygen supply (9,29). A recently described generation of extracellular adenosine by hypoxia-triggered ATPase/ADPase CD39 and 5′-ectonucleotidase CD73 represents another important pathway (37–39). The CD39/ecto-nucleoside triphosphate diphosphohydrolase-type-1 (ENTPD1) is now recognized to be the key cell surface ecto-nucleotidase (39,40).
The conditions of reduced oxygen delivery result in increased extracellular tissue concentrations of adenosine after hypoxic challenge in heart muscle (41) and hippocampus (42). We confirmed the elevation of adenosine concentration in the blood from healthy mice subjected to 10% oxygen for 1.5 h as compared with animals treated with 21% oxygen (Table 3). Observations of increased adenosine in the blood also were correlated with the proportional increase of its metabolite inosine. Because the increase of extracellular adenosine levels was observed in mice subjected to 10% oxygen in the absence of Con A (no liver damage according to liver enzyme assays) as well as after Con A injection, we conclude that the increase of adenosine is resulted from hypoxia even in the absence of tissue damage.
Because whole body hypoxia rendered mice resistant to the induction of liver injury, it had to be tested whether hypoxia-driven hepatoprotection is mediated by the hypoxia ➙ extracellular adenosine ➙ A2AR pathway or by yet unknown hypoxia ➙ molecule “X” pathway. We hypothesized that if the anti-inflammatory mechanism triggered by hypoxia is dependent on the action of extracellular adenosine, then the antagonism of the A2AR or genetic deletion of A2AR in mice should inactivate the hypoxia-driven tissue protection. This, in turn, was expected to potentiate inflammatory liver damage even under hypoxic conditions (9) because the hypoxia-induced accumulation of extracellular adenosine will not be transmitted further by A2AR and, therefore, liver tissues will be damaged continuously by inflammatory effectors (25).
Here we show that the hepatoprotective effects of hypoxia during acute inflammation are abolished by pharmacological antagonism in mice treated with the selective antagonist ZM241385 as evidenced by significantly (P < 0.01) elevated liver enzyme activities, lactate, and IFN-γ concentration. In agreement with these measurements, we also observed significantly higher mortality under hypoxia when the A2AR-mediated protection was antagonized (mortality: 10% oxygen Con A = 5.5% versus Con A + ZM = 33%, P < 0.05). The lower survival rate emphasizes the importance of tissue hypoxia in control of liver tissue inflammation via adenosine and A2AR. Importantly, the genetic evidence for the crucial role of the A2AR in hypoxia-triggered hepatoprotection was provided by demonstrations of the aggravation of liver damage and inflammation even during hypoxia in mice with genetically deleted A2AR (Figure 3). This indicates that the hypoxia ➙ A2AR pathway is non-redundant and is critical in limiting liver inflammation as it is in lung inflammation (43). Moreover, in contrast to our previous report testing hypoxia to control lung inflammation as initiated by bacterial toxin LPS (43) which activates immune cells of innate immunity, here we provide evidence that Con A-induced T cell-dependent inflammatory liver damage is also under the control of hypoxia ➙ A2AR pathway.
In conclusion, here we demonstrated that both hypoxia and A2AR are important in the downregulation of liver inflammation and that hypoxia and A2AR belong to the same non-redundant anti-inflammatory pathway. This could be an important and constitutive mechanism to control hepatic inflammation, because the liver is hypoxic even under normo-oxygenated conditions.
References
Poynard T, Yuen MF, Ratziu V, Lai CL. (2003) Viral hepatitis C. Lancet. 362:2095–100.
Lai CL, Ratziu V, Yuen MF, Poynard T. (2003) Viral hepatitis B. Lancet. 362:2089–94.
Davis GL, Albright JE, Cook SF, Rosenberg DM. (2003) Projecting future complications of chronic hepatitis C in the United States. Liver Transplant. 9:331–8.
Wong JB. (2006) Hepatitis C: cost of illness and considerations for the economic evaluation of antiviral therapies. Pharmacoeconomics. 24:661–72.
Chisari FV, Ferrari C. (1995) Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 13:29–60.
Huang CF, Lin SS, Ho YC, Chen FL, Yang CC. (2006) The immune response induced by hepatitis B virus principal antigens. Cell. Mol. Immunol. 3:97–106.
Herkel J, Schuchmann M, Tiegs G, and Lohse AW. (2005) Immune-mediated liver injury. J. Hepatol. 42:920–3.
Rehermann B, Nascimbeni M. (2005) Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 5:215–29.
Sitkovsky MV et al. (2004) Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A(2A) receptors. Annu. Rev. Immunol. 22:657–82.
Sitkovsky MV, Ohta A. (2005) The ‘danger’ sensors that STOP the immune response: the A2 adenosine receptors? Trends Immunol. 26:299–304.
Sitkovsky M, Lukashev D. (2005) Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nat. Rev. Immunol. 5:712–21.
Montesinos MC, Desai A, Delano D, Chen JF, Fink JS, Jacobson MA, Cronstein BN. (2003) Adenosine A2A or A3 receptors are required for inhibition of inflammation by methotrexate and its analog MX-68. Arthritis Rheum. 48:240–7.
Lappas CM, Sullivan GW, Linden J. (2005) Adenosine A2A agonists in development for the treatment of inflammation. Expert Opin. Invest. Drugs. 14:797–806.
Linden J. (2005) Adenosine in tissue protection and tissue regeneration. Mol. Pharmacol. 67:1385–7.
Lappas CM, Rieger JM, Linden J. (2005) A2A adenosine receptor induction inhibits IFN-gamma production in murine CD4+ T cells. J. Immunol. 174:1073–80.
Tiegs G, Hentschel J, Wendel A. (1992) A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90:196–203.
Mizuhara H et al. (1994) T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J. Exp. Med. 179:1529–37.
Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. (1995) Concanavalin A-induced T-cell-mediated hepatic injury in mice: the role of tumor necrosis factor. Hepatology. 21:190–8.
Mizuhara H et al. (1996) Critical involvement of interferon gamma in the pathogenesis of T-cell activation-associated hepatitis and regulatory mechanisms of interleukin-6 for the manifestations of hepatitis. Hepatology. 23:1608–15.
Kusters S, Gantner F, Kunstle G, Tiegs G. (1996) Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 111:462–71.
Kusters S et al. (1997) In vivo evidence for a functional role of both tumor necrosis factor (TNF) receptors and transmembrane TNF in experimental hepatitis. Eur. J. Immunol. 27:2870–5.
Schumann J, Wolf D, Pahl A, Brune K, Papadopoulos T, van Rooijen N, Tiegs G. (2000) Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 157:1671–83.
Toyabe S et al. (1997) Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J. Immunol. 159:1537–42.
Kaneko Y et al. (2000) Augmentation of V alpha 14 NKT cell-mediated cytotoxicity by inter-leukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J. Exp. Med. 191:105–14.
Ohta A, Sitkovsky M. (2001) Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 414:916–20.
Hagemeier E, Kemper K, Boos KS, Schlimme E. (1983) On-line high-performance liquid affinity chromatography-high-performance liquid chromatography analysis of monomeric ribonucleoside compounds in biological fluids. J. Chromatogr. 282:663–9.
Thiel M, Holzer K, Kreimeier U, Mortiz S, Peter K, Messmer K. (1997) Effects of adenosine on the functions of circulating polymorphonuclear leukocytes during hyperdynamic endotoxemia. Infect. Immun. 65:2136–44.
Decking UK, Schlieper G, Kroll K, Schrader J. (1997) Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 81:154–64.
Sitkovsky MV. (2003) Use of the A(2A) adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem. Pharmacol. 65:493–501.
Linden J. (2001) Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 41:775–87.
Aranda-Michel J, Sherman KE. (1998) Tests of the liver: use and misuse. Gastroenterologist. 6:34–43.
Kuroki I, Miyazaki T, Mizukami I, Matsumoto N, Matsumoto I. (2004) Effect of sodium nitroprusside on ischemia-reperfusion injuries of the rat liver. Hepatogastroenterology. 51:1404–7.
Macquillan GC, Seyam MS, Nightingale P, Neuberger JM, Murphy N. (2005) Blood lactate but not serum phosphate levels can predict patient outcome in fulminant hepatic failure. Liver Transplant. 11:1073–9.
Murphy ND, Kodakat SK, Wendon JA, Jooste CA, Muiesan P, Rela M, Heaton ND. (2001) Liver and intestinal lactate metabolism in patients with acute hepatic failure undergoing liver transplantation. Crit. Care Med. 29:2111–8.
Bihari D, Gimson AE, Lindridge J, Williams R. (1985) Lactic acidosis in fulminant hepatic failure. Some aspects of pathogenesis and prognosis. J. Hepatol. 1:405–16.
Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. (2006) Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J. Exp. Med. 203:2639–48.
Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. (2004) Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 104:3986–92.
Kaczmarek E et al. (1996) Identification and characterization of CD39/vascular ATP diphospho-hydrolase. J. Biol. Chem. 271:33116–22.
Robson SC, Wu Y, Sun X, Knosalla C, Dwyer K, Enjyoji K. (2005) Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin. Thromb. Hemost. 31:217–33.
Kaczmarek E et al. (2005) Modulation of endothelial cell migration by extracellular nucleotides: involvement of focal adhesion kinase and phosphatidylinositol 3-kinase-mediated pathways. Thromb. Haemost. 93:735–42.
Cohen MV, Walsh RS, Goto M, Downey JM. (1995) Hypoxia preconditions rabbit myocardium via adenosine and catecholamine release. J. Mol. Cell Cardiol. 27:1527–34.
Zhang WL, Lu GW. (1999) Changes of adenosine and its A(1) receptor in hypoxic preconditioning. Biol. Signals Recept. 8:275–80.
Thiel M et al. (2005) Oxygenation inhibits the physiological tissue-protecting mechanism and thereby exacerbates acute inflammatory lung injury. PLoS Biol. 3:e174.
Acknowledgments
The authors are very thankful to Dr. William Paul and Dr. Patrick Smith (NIH, NIAID, Bethesda, MD, USA) for their support and to Pearl Powers, Dr. Gregory Scako, and Thomas Anderson (National Institutes of Health, Clinical Center, Bethesda, MD, USA) for their advice and help for quantification of liver enzymes. We do also very much acknowledge the continuous support of Prof. Dr. Klaus Peter, Dr. Martina Choukèr (Ludwig-Maximilians-University of Munich, Germany), and Heidi Unkle (Rockville, MD, USA) during this study. The NIH John E. Fogarty research grant was awarded to AC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Choukèr, A., Thiel, M., Lukashev, D. et al. Critical Role of Hypoxia and A2A Adenosine Receptors in Liver Tissue-Protecting Physiological Anti-Inflammatory Pathway. Mol Med 14, 116–123 (2008). https://doi.org/10.2119/2007-00075.Chouker
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2007-00075.Chouker