
Abstract
Prions are composed solely of the disease-causing prion protein (PrPSc) that is formed from the cellular isoform PrPC by a post-translational process. Here we report that short phosphorothioate DNA (PS-DNA) oligonucleotides diminished the levels of both PrPC and PrPSc in prion-infected neuroblastoma (ScN2a) cells. The effect of PS-DNA on PrP levels was independent of the nucleotide sequence. The effective concentration (EC50) of PS-DNA required to achieve half-maximal diminution of PrPSc was ~70 nM, whereas the EC50 of PS-DNA for PrPC was more than 50-fold greater. This finding indicated that diminished levels of PrPSc after exposure to PS-DNA are unlikely to be due to decreased PrPC levels. Bioassays in transgenic mice demonstrated a substantial diminution in the prion infectivity after ScN2a cells were exposed to PS-DNAs. Whether PS-DNA will be useful in the treatment of prion disease in people or livestock remains to be established.
Similar content being viewed by others


Introduction
The neurodegenerative diseases include Alzheimer’s, Parkinson’s, and Huntington’s diseases as well as the frontotemporal dementias, amyotrophic lateral sclerosis, and the prion diseases. Not since the introduction of L-dopa for treatment of Parkinson’s disease (1) has a meaningful advance in the therapeutics for neurodegenerative diseases been recorded. Despite this drought, studies on the pathogenesis of the neurodegenerative diseases have been impressive.
The results of numerous studies have converged to argue that prions are composed solely of the disease-causing prion protein, designated PrPSc. A posttranslational process generates PrPSc from the cellular isoform PrPC (2). Recent studies of prions produced in cell-free systems and bioassayed in mammals or fungi have demonstrated that only a protein is necessary for prion infectivity (3,4). Animal models can faithfully reproduce human prion disease, making them an excellent system in which to develop new pharmacotherapeutics. Moreover, expression of chimeric human-mouse PrP transgenes permits the study of human prions in mice with incubation times of ~100 days (5).
Several approaches to the therapeutics of prion disease have been investigated, including diminishing the levels of PrPC (6–9), slowing the conversion of PrPC into PrPSc (10–13), and enhancing the degradation of PrPSc (14). Anti-PrP antibodies have been shown to diminish the formation of PrPSc in ScN2a cells (15,16) and in mice inoculated intraperitoneally with prions (17–19).
Of all the compounds studied, quinacrine seems to offer the most hope as an anti-prion therapeutic due to its long history of clinical use and its potency against PrPSc. The concentration of quinacrine required for half-maximal reduction (EC50) of PrPSc in cultured ScNa2 cells was ~300 nM (12). To identify compounds with increased efficacy over quinacrine, bis-acridine molecules were synthesized. Some of these compounds exhibited EC50 values 10-fold lower than quinacrine (20). Neither quinacrine nor the bis-acridines have been shown to be effective in attempts to prolong the incubation periods of mice inoculated intracerebrally (i.c.) with prions (21,22). Oral quinacrine is currently being evaluated in the treatment of sporadic and variant Creutzfeldt-Jakob disease (CJD). In addition to quinacrine, pentosan polysulfate is being evaluated in humans, but this drug must be administered intrathecally. Pentosan polysulfate infused intraventricularly into mice has been reported to prolong the incubation time (23).
In a quest to identify new lead compounds for the treatment of prion disease, we investigated oligonucleotides as potential pharmacotherapeutics. Phosphorothioate DNA (PS-DNA) oligonucleotides were reported to slow prion propagation when administered intraperitoneally (i.p.) for 20 days consecutively beginning immediately after inoculation of prions (24). To extend these findings, we exposed ScN2a cells to 22-mer, single-stranded PS-DNAs of various sequences. Phosphorothioate modification renders oligonucleotides resistant to nucleases while maintaining their charge and structure, by replacing an oxygen in the backbone phosphate with a sulfur atom (25). We found that PS-DNAs diminished the levels of both PrPC and PrPSc in ScN2a cells. A brief preliminary description of our studies was reported earlier (26) and an extensive study of PS-DNAs as inhibitors of PrPSc formation by others was published recently (27). Here we report that the EC50 for PrPSc was ~70 nM and the effect of PS-DNA on PrP levels was independent of the nucleotide sequence. Because the EC50 of PS-DNA for PrPC was much higher than that for PrPSc, the diminished levels of PrPSc after exposure to PS-DNA could not be due to decreased levels of PrPC. Bioassays in transgenic mice demonstrated a substantial diminution in the prion infectivity after ScN2a cells were exposed to PS-DNAs. The mechanism by which PS-DNA diminished the level of PrPSc in cultured cells remains unknown.
Materials and Methods
Oligonucleotides
All oligonucleotides were purchased from TriLink Biotechnologies (San Diego, CA, USA) after HPLC purification and verification using mass spectroscopy. The oligonucleotides synthesized for the present study had the following base sequences:
-
CpG 22-mer CpG-PS-DNA: TGACTGT
-
GAACGTTCGAGATGA
-
Scr 22-mer SCR-PS-DNA: CAGTGATA
-
GCTATGTGAGCTAG
-
6-mer PS-DNA: TGTGAG
-
12-mer PS-DNA: CAGTGATAGCTA
-
15-mer PS-DNA: TGCTCAACAGTATGA
-
18-mer PS-DNA: CAGTGATAGCTAT
-
GTGAG
-
44-mer PS-DNA: CAGTGATAGCTAT
-
GTGAGCTAGCAGTGATAGCTA
-
TGTGAGCTAG
Treatment of Cells with Oligonucleotides
N2a cells (28,29) were grown in 10-cm dishes in minimal essential medium (MEM) until attaining 90% to 95% confluence. Cells were trypsinized and diluted ten-fold into 60-mm plates containing 4 mL of Dulbecco’s modified Eagle’s medium (DMEM). On the following day, the cells were washed once with fresh DMEM and 2.5 mL of new medium was applied to the cells. Oligonucleotides were then added to the dish at various concentrations and incubated for variable periods of time. Cells were incubated with 1 µM of 6-mer, 12-mer, 15-mer, 22-mer, and 44-mer PS-DNA for 48 h. A range of concentrations (0.01 µM to 10 µM) of 22-mer PS-DNA was also incubated with the cells for 48 h (Figure 2). All incubations were performed at 37°C. Cells were harvested in 0.5 mL cold lysis buffer (10 mM Tris-HCl, pH 8; 100 mM NaCl; 0.5% NP-40; and 0.5% deoxycholate). Cell lysates were incubated for 3 min at 4°C and DNA aggregates were collected from the lysate using a sterile tip. Stock cultures of cells were maintained in MEM. The cells were harvested using 4 mL of 0.5% trypsin and plated in a 1:20 dilution, fed on day 4, and trypsinized again in a 1:3 dilution onto 10-cm plates on day 6. All media were supplemented with 10% fetal bovine serum, 2 mM Glutamax (Gibco BRL, Carlsbad, CA, USA), 100 units/mL penicillin, and 100 units/mL streptomycin in a humidified 37°C incubator with 5% CO2.
Primary Cell Cultures
Cerebellar granule neurons and hippocampal neurons were prepared from 6-day-old (P6) FVB and FVB/Prnp0/0 mice, using an established dissociation protocol, as described previously (30). Briefly, P6 mice were decapitated and their heads immediately placed into a Petri dish containing both ice-cold Hank’s BSS, Ca2+- and Mg2+ -free without Phenol Red (UCSF cell tissue facility, San Francisco, CA, USA), and a high concentration of penicillin/streptomycin (Gibco) to slow down metabolism and decrease contamination. The whole brain was moved into fresh, ice-cold Hank’s solution. Under a dissecting microscope (Nikon SMZ 1500), the cerebellum or hippocampus was removed and cleaned of meninges tissue to decrease glial cell contamination in the culture. The tissue was transferred to a sterile 15-ml (Falcon, BD Sciences; Bedford, MA, USA) conical tube, which was then spun at 200g in a Beckman GS-6 centrifuge for 5 min. The supernatant was carefully aspirated, mixed thoroughly with a solution containing 20 units of Papain (Worthington, Lakewood, NJ, USA) and 0.005% of DNase (Worthington), and incubated at 37°C for 20–45 min, depending on tissue volume. The tissue was mechanically dissociated using a 5-ml pipette followed by a 1000-µLpipette tip. Cells were then passed through a 40-µm cell strainer (Falcon), spun at 200g and resuspended in 3 mL of Neurobasal Media-A (Gibco) that contained 10% fetal bovine serum (Gibco) and B27 supplement (Gibco). The cells were counted and plated in fresh Neurobasal-Media A at a concentration of 1 × 105 cells/well in 96-well plates that had been coated with poly-D-lysine (250 µg/mL). One day postplating, 0.1% Arabinose-C (Sigma) was added to the Neurobasal Media-A to further limit glial cell growth. PS-DNAs were then added at the indicated final concentrations in Neurobasal media-A and incubated for up to 5 days.
Cell-Survival Assay
Cell survival was assayed using calcein AM, according to the manufacturer’s protocol (Molecular Probes, Eugene, OR, USA). Calcein AM was prepared in DMSO/Dulbecco’s phosphate-buffered saline (PBS; Gibco/Invitrogen). Briefly, after plating on black, 96-well plates (Thermo LabSystems, Vanta, Finland), cells were treated with the PS-DNAs at various concentrations for up to 5 days. After treatment, cells were washed three times in Ca2+ -and Mg2+ -free Dulbecco’s PBS (Gibco/Invitrogen) before incubation at 37°C for 30 min in calcein AM (1:10). The survival of the cells was read using a fluorescence microplate reader (Tecan, Research Triangle Park, NC, USA) using a filter with excitation at 490 ± 10 nm and emission at 530 ± 15 nm.
Long-term Treatment of Cells
ScN2a cells were prepared as described above for days 1 and 2. For each treatment, we prepared three sets of plates. On day three, 2 mL of DMEM was added to the cells. On day four, 4 mL of new medium containing the oligonucleotides was applied to the cells. On day 6, one set of plates was trypsinized and cells were transferred to new 60-mm plates as described to enable continuous treatment. Another set of plates was allowed to grow without the presence of oligonucleotides. The third set of plates was washed with 2 mL PBS, followed by harvesting of the cells in 0.5 mL cold lysis buffer, as described above.
Competitive Binding Experiments
For antibody binding experiments, N2a cells were pre-incubated with a panel of different recHuM Fabs at a concentration of 20 µg/mL. After 6 h, 4 µM of 22-mer PS-DNA was added to the cells and incubated for an additional 6 h.
For experiments investigating the lysosomal pathway, N2a and ScN2a cells were pre-incubated with 4 different lysosomal inhibitors: 100 nM bafilomycin A, 10 mM monesin, 100 µM chloroquine, or 30 mM ammonium chloride. All compounds were purchased from Sigma. After pre-incubation for 1 h, cells were exposed to 20 µM 22-mer PS-DNA for 6 h. For experiments investigating the proteasomal pathway, N2a and ScN2a cells were exposed to 20 µM of 22-mer PS-DNA for 6 h. Then three different proteasomal inhibitors were added to the cells and incubated for 1 h: 15 µM lactacystin, 15 µM MG132, and 150 µM ALLN.
After incubation, cells were washed and harvested as described above. ScN2a cells were digested with 20 µg/mL PK for 1 h at 37°C. Immunoblots were prepared and probed with the anti-PrP Fab D13 antibody.
Antibodies and Immunoblotting
Protein concentration in each sample was determined using the bicinchoninic acid (BCA) kit (Pierce, Rockford, Illinois, USA). Levels of PrPSc were measured by densitometry of the PK-resistant bands on Western blots. PK (20 µg) was added to 1 mg/mL cell lysate and incubated for 1 h at 37°C. Digestion was stopped by 2mM phenylmethylsulfonylfluoride (PMSF), 800 µLof acetone was added, and samples were centrifuged at 15,000g at 4°C for 1 h on a tabletop centrifuge. Pellets were resuspended in 40 µL × 2 Laemmli buffer and boiled for 6 min. A sample of 20 µLwas loaded on a 12% Pre-Cast SDS/PAGE gel (BioRad, Hercules, California, USA) and protease-resistant PrPSc was probed using Fab D13 (31,32). To quantify PrPC levels, 40 µg from each sample was loaded on the same gels before PK digestion and detected using Fab D13. To demonstrate that an equal amount of total protein was loaded from the different samples, we used the anti-mouse tubulin antibody, anti-mouse Thy1 antibody, anti-mouse Trk B-3 antibody, anti-mouse ATF-3 antibody, or anti-mouse actin antibody (Sigma, St. Louis, Missouri, USA). For N-CAM staining, we used the monoclonal mouse IgG2a antibody RDI-N-CAM13abm (Research Diagnostics, Inc., Flanders, New Jersey, USA). For Dpl, we used the polyclonal antibody E6977.
Northern blot assays were performed as described (33) with the exception of the amount of RNA loaded on the gel.
Bioassays in Mice
Bioassays were performed as described (15). Briefly, ScN2a cells were treated with 1 x M 22-mer PS-DNA for 8weeks. Subsequently, 10-cm confluent tissue culture plates were lysed with 1 mL of PBS free of Ca2+ and Mg2+. Thirty microlitres of the cell lysate were inoculated i.c. into Tg(MoPrP-A)4053/Prnp+/+ mice that overexpress wild-type mouse PrP (34) and in wild-type CD-1 Swiss mice at the age of 7 weeks. Mice were scored daily for onset of clinical signs of prion disease.
Results
We exposed ScN2a cells to 22-mers of single-stranded PS-DNAs of various sequences. All the 22-mer PS-DNAs were transfected into ScN2a cells via DOTAP (Roche, Indianapolis, IN, USA) at a concentration of 1 µM, and incubated for 48 h. Subsequent studies showed that the PS-DNA did not need to be transfected to observe a reduction in PrPSc. Initially two different PS-DNA sequences were tested: one containing a CpG motif (purine, purine, cytosine, guanine, pyrimidine, pyrimidine), which is known to elicit the innate immune response and cytokine secretion (35), and a second that is a scrambled version of the CpG-PS-DNA. An additional control was the methylated version of the CpG-PS-DNA. We found that the PS-DNA sequence carrying the CpG motif and the scrambled PS-DNA were equally potent in abolishing the PrPSc signal seen on Western blots after limited Proteinase K digestion (Figure 1). Interestingly, the methylated CpG motif also abolished the PrPSc signal even though it is not able to activate the innate immune response. Moreover, 22-mer PS-DNA composed of poly-C, poly-T, or poly-A/C had the same effect as poly-G/C (data not shown). From these results, we concluded that the sequence of PS-DNA is irrelevant in the process that leads to a diminution in PrPSc levels in ScN2a cells. Because the PS-DNA did not need to be transfected for a diminution in the level of PrPSc to be observed, it seems likely that PS-DNA interacts with PrPSc at the cell surface.

Effect of different PS-DNA sequences and PS-DNA transfection on PrPSc in ScN2a cells. Cells were exposed to different oligonucleotides for 48 h. CpG, PS-DNA that contains the CpG motif as described in Methods; Scr, scrambled sequence of the CpG oligonucleotide; Met, methylated version of CpG oligonucleotide: C, control ScN2a cells without PS-DNA. In all transfected cells, DOTAP was added while the non-transfected cells were incubated with PS-DNA only. Cells were either undigested (-PK) or subjected to PK digestion at 37°C for 1 h (+PK), and all immunoblots were stained with the D13 anti-PrP antibody.

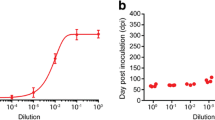
Effect of the concentration of PS-DNA on PrP in ScN2a and N2a cells. Increasing concentrations of 22-mer PS-DNA are plotted against the levels of PrPC in N2a cells (A) and PrPSc in ScN2a cells after PK digestion (B). The EC50 of PS-DNA for PrPC was ~5 µM, whereas that for PrPSc was ~70 nM. These results are scans of Western blots and are the average of three independent experiments.
Next, we incubated PS-DNAs of variable lengths with ScN2a cells. PS-DNAs composed of 6-mers, 12-mers, and 15-mers had no effect on the levels of PrPSc in ScN2a cells. In contrast, PS-DNAs composed of 18-mers, 22-mers, and 44-mers abolished PrPSc (data not shown). The mechanism by which PrPSc levels are lowered by 18- to 44-mer PS-DNAs remains to be established. Whether longer PS-DNAs adopt an active conformation that shorter PS-DNAs are unable to achieve is unknown.
It is noteworthy that double-stranded PS-DNA had the same effect as the single-stranded PS-DNA described above. In contrast to the phosphorothioate-modified DNA, unmodified, single-stranded phosphorodiester DNA as well as PS-RNA were unable to reduce PrPSc levels at the concentrations and lengths for which PS-DNA were highly effective.
The levels of PrPC and PrPSc in N2a cells were determined as a function of the concentration of a 22-mer PS-DNA. The PS-DNA was incubated with the cells for 48 h at 37°C. The curve relating increasing PS-DNA concentrations to the level of PrPC in N2a cells demonstrates an exponential relationship (Figure 2a). The EC50 of PS-DNA for PrPC was just under 4 µM. The curve relating increasing PS-DNA concentrations to the level of PrPSc in ScN2a cells is also exponential (Figure 2b). The EC50 of PS-DNA for PrPSc was approximately 70 nM, around 50-fold lower than that found for PrPC.
Because PS-DNA diminished PrP levels in a concentration-dependent manner, we investigated the time dependence of this process. A decrease in PrPSc and PrPC levels was detected after a minimal incubation of 4 h with 1 µM 22-mer PS-DNA (data not shown). Longer incubation times resulted in a progressive decline in the levels of PrPC and PrPSc. After 48 h of exposure to PS-DNA at 1 µM, PrPSc in ScN2a cells was undetectable by immunoblotting.
To extend our observations in ScN2a cells, we performed studies on PS-DNA added to ScGT1 cells (36). In agreement with results from ScN2a cells, both PrPSc and PrPC levels were reduced by PS-DNA in ScGT1 cells (data not shown). This finding excluded the possibility of a cell-line dependent artifact.
After determining that the effect of PS-DNA is dependent on duration of exposure, the length and concentration of PS-DNA, we wished to examine its mechanism of action. We asked whether the effect of PS-DNA is mediated via specific PrP degradation, a general decrease in cellular proteins, or reduction of proteins that are anchored by a glycosylphosphatidyl inositol (GPI) moiety, similar to PrP (37). Levels of tubulin and neural cell-adhesion molecule (N-CAM), a GPI-anchored protein (38) that binds to PrP (39,40), were measured before and after incubation with PS-DNA. The levels of neither protein were altered by PS-DNA (Figure 3). Furthermore, actin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), thymus cell antigen 1 (Thy1), tyrosine kinase receptor (TrkB-3), activating transcription factor 3 (ATF-3), and doppel, a PrP paralogue, were unaffected by the presence of PS-DNA, suggesting that PS-DNA selectively modifies PrP. The same pattern and levels of numerous proteins in the presence or absence of PS-DNA were found from PVDF membranes stained with Ponceau-S and from Comassie-stained gels after SDS-PAGE (data not shown).

N-CAM, tubulin, and PrP levels in ScN2a and N2a cells after exposure to PS-DNA. ScN2a and N2a cells incubated for 2 weeks with various concentrations of scrambled 22-mer PS-DNA indicate decreased levels of PrPC and PrPSc but no change in the level of the other proteins. Similar results were seen for shorter incubation times (data not shown). Cell lysates without PK digestion (-PK) were probed with PrP antibody (D13), and antibodies against N-CAM and tubulin on the same immunoblot. This can be accomplished because N-CAM, tubulin, and PrP have different molecular weights (140 kDa, 49.5 kDa, and 19.6 to 37.4 kDa, respectively). C, control with no PS-DNA.
Cell-survival assays revealed no toxicity of PS-DNA to primary cells, even at concentrations as high as 5 mM (data not shown). To ensure that the apparent reduction of PrPC and PrPSc levels on immunoblots is not due to steric hindrance of primary antibody binding to PS-DNA, we used a panel of antibodies directed against many different epitopes of native PrP. Recombinant (rec) chimeric HuM Fabs denoted E123, E149, D13, D18, R72 and R1, that recognize PrP epitopes 29–37, 72–86, 95–105, 133–157, 152–163 and 226–231, respectively (41,42), detected similar levels of PrPC or PrPSc. The similar results obtained with this panel of recHuM Fabs argue that our findings with PS-DNA are not artifacts.
To determine whether PS-DNA altered the levels of PrPC and PrPSc at the transcriptional level, PrP mRNA levels were measured at different durations of incubation with 22-mer PS-DNA at 1 µM. We selected three time-points at which immunoblots revealed reduced levels of PrPSc in ScN2a cells. Northern blot analysis revealed no change in the levels of PrP mRNA after exposure of cells to PS-DNAs for 4, 15, or 48 h.
Next, we examined the potential role of the lysosomal and proteasomal degradation pathways in PS-DNA-mediated attenuation of PrP levels. ScN2a and N2a cells were pre-exposed to inhibitors of these pathways followed by incubation with PS-DNA. Chloroquine, bafilomycin Aand monensin (data not shown), which are known to inhibit lysosomal degradation of proteins (43,44), diminished the reduction of PrPC by PS-DNA. Studies with chloroquine also showed that this lysosomal inhibitor could reduce the effect of PS-DNA on PrPSc levels in ScN2a cells. Interestingly, pre-incubation with ammonium chloride, another lysosomal inhibitor, did not interfere with the ability of PS-DNA to remove PrPSc and to reduce PrPC levels.
To investigate whether proteasome inhibitors show an effect similar to that seen with lysosomal inhibitors, lactacystin, MG132, or ALLN (45) were added to ScN2a cells prior to exposure to PS-DNA. None of these three proteasome inhibitors altered the effect of PS-DNA on PrPSc levels (data not shown), suggesting that PS-DNA exerts its effects on PrP via the lysosome but not through the proteasome.
To explore further the mechanism of PrP reduction meditated by PS-DNA, N2a cells were pre-incubated with recHuM Fabs directed against a variety of PrP epitopes (31). Because some PrP antibodies can eliminate PrP in N2a and ScN2a cells (15,16,46), we chose an incubation time and antibody concentration that would not affect PrP levels but enable the antibody to compete with the PS-DNA binding. N2a cells were incubated for 6 h with 20 µg/mL of various recHuM Fabs (15,31,32,47,48) followed by exposure to 4 µM 22-mer PS-DNA for an additional 6 h. The R1, D13, and D18 recHuM Fabs that bind to PrPC with high affinities (Kds: 1.8 ± 0.8 nM, 3.5 ± 0.5 nM, and 1.5 ± 0.6 nM, respectively) were able to block the effect of PS-DNA (Figure 4). In contrast, the E149 and R72 recHuM Fabs did not block the effect of PS-DNA on PrPC in N2a cells. Why some recHuM Fabs and not others prevented the effect of PS-DNA is unclear.

PrP reduction in the presence of PS-DNA can be blocked by preincubation with anti-PrP Fabs. N2a cells were incubated with 20 mg/mL of the indicated Fab for 6 h followed by incubation with 4 µM 22-mer PS-DNA for 6 h. The following Fabs that recognize different epitopes of PrP were used: E149, R72, R1, D13 and D18 (15). C, control, no antibody.
To investigate whether PS-DNA permanently disrupted the ability of ScN2a cells to form PrPC and PrPSc, we monitored PrPC and PrPSc levels in cells after cessation of PS-DNA treatment. ScN2a cells were incubated for 2 weeks with 22-mer PS-DNA followed by a recovery period of 1 week in media without PS-DNA. In comparison to treated ScN2a cells (Figure 3), ScN2a cells 1 week after cessation of treatment showed newly synthesized PrPC but no evidence of nascent PrPSc formation (Figure 5). The same results were observed after 3 weeks of recovery (data not shown).

PrP levels before and after the removal of 22-mer PS-DNA from ScN2a cells. ScN2a cells were exposed to various concentrations of PS-DNA for 2 weeks. Thereafter, the cells were grown for another week in DNA-free media, then harvested and digested with PK to determine levels of PrPSc. PrPC levels returned to normal levels whereas PrPSc levels were not restored after a recovery period of 1 week. Fab D18 was used to detect PrPC and PrPSc.
Bioassays were performed to confirm the depletion of PrPSc from ScN2a cells by PS-DNA. The ScN2a cells were exposed to 1 µM of a 22-mer PS-DNA for 8 weeks. At the end of the experiment, untreated control, and PS-DNA-treated ScN2a cells were homogenized and inoculated i.c. into wild-type CD-1 and Tg(MoPrP-A)4053 mice. The untreated cells caused disease in the CD-1 mice in 148 days and in Tg4053 mice in 68 days (Table 1). In contrast, the PS-DNA-treated cells caused disease in five of six CD-1 mice with a mean of 305 days, indicating that the prion titer was reduced by a factor of more than 104 (28). Similar results were observed with the Tg4053 mice: PS-DNA-treated cells did not cause disease Tg4053 mice in more than 400 days. Like the bioassays in CD-1 mice, the data from Tg4053 mice indicate a reduction in prion titer of > 104.
Please note that supplementary information is available on the Molecular Medicine website (www.molmed.org).
Discussion
When ScN2a cells were treated with a 22-mer PS-DNA, the levels of both PrPC and PrPSc diminished without affecting other proteins in the cells. This phenomenon was not sequence dependent because scrambled PS-DNA was as effective as a PS-DNA with a CpG motif. The lowering of PrPC levels by PS-DNA was similar whether or not the cells were infected with prions and probably occurred at the cell surface because some recHuM Fabs prevented this reduction of PrPC (Figure 4) (15). The effect of PS-DNA on PrPC levels contrasts with findings reported in a recent study (27).
While reduction of PrPC and PrPSc levels by the PS-DNA was not sequence-specific, it was dependent on the length of the oligonucleotide (Figure 2a). PS-DNAs composed of 6-mers, 12-mers, and 15-mers had no effect on the levels of PrPSc in ScN2a cells. In contrast, PS-DNAs composed of 18-mers, 22-mers, and 44-mers abolished PrPSc. Other investigators found a similar length-dependent relationship, where optimal reduction of PrPSc levels was achieved with PS-DNAs of 25–28 bases (27). They also found that the optimal size of PS-DNA for binding to recPrPs was 20 to 40 bases. The mechanism by which PrPSc levels are lowered by 18- to 44-mer PS-DNAs remains to be established. Whether longer PS-DNAs adopt an active conformation that shorter PS-DNAs are unable to achieve is unknown.
The reduction in PrPC and PrPSc levels was dependent upon the concentration of PS-DNAs. Because the EC50 for reduction of PrPC was ~50-fold greater than that for PrPSc, we concluded that the effect of PS-DNA on PrPSc did not result from a diminution in PrPC levels. Additionally, we found that PS-DNA did not act by decreasing the level of PrP mRNA. Some lysosomal inhibitors interfered with the effect of PS-DNA, but NH4Cl did not; furthermore, proteasome inhibitors had no effect on the lowering of PrPC and PrPSc levels by PS-DNA.
The removal of PrPSc in ScN2a cells by PS-DNA was dependent on the length of the oligonucleotide, dose, and incubation time. Treatment of ScN2a cells with 1 µM of the 22-mer PS-DNA for 48 h was sufficient to remove PrPSc in ScN2a cells based on Western immunoblotting. Treating ScN2a cells for 2–7 weeks with PS-DNA cured cells of prion infectivity as determined by immunoassay and bioassay (Table 1). Two weeks of incubation with 1 µM of 22-mer PS-DNA was sufficient to prevent any new PrPSc formation although newly synthesized PrPC was present (Figure 5).
The mechanism of PrPSc removal from ScN2a and ScGT-1 cells is unknown. Our studies suggest that PS-DNA binds to PrPSc and facilitates its degradation in lysosomes; however, the mechanism may be more complicated because the lysosomal inhibitor ammonium chloride failed to show an effect. Such a mechanism is reminiscent of branched polyamines that have been shown to enhance the clearance of PrPSc from ScN2a cells (14,49). Whether PS-DNA also inhibits the production of nascent PrPSc remains to be established.
Bioassays of ScN2a cells treated with 22-mer PS-DNA clearly show a reduction in prion titer by a factor of > 104 in CD-1 mice, and removed infectivity beyond detectable levels in Tg4053 mice on the FVB background. These findings in the two mouse lines suggest strain-specific differences in the susceptibility to prions. It is unclear whether PS-DNA can be used to treat patients dying of CJD. Other investigators have successfully extended the incubation period two- to three-fold by administering PS-DNAs to Tg(SHaPrP)7/Prnp0/0 mice inoculated with Syrian hamster prions (27). In these studies, prions were inoculated and PS-DNA administered simultaneously into the mice, either i.p. or subcutaneously. It is unknown if PS-DNA administered after prion infection has been established in the CNS will be effective. Anti-PrP antibodies administered after i.p. inoculation have been shown to prolong incubation times (17–19), but they are ineffective if the animals are inoculated i.c. with prions.
The lack of sequence specificity in the study reported here and in that of others (27) contrasts with studies of aptamers, in which sequence-specific oligonucleotides have been identified that bind tightly to PrP (50–53). In recent studies, the binding of thioaptamers is greatly enhanced by multiple rounds of selection against PrP (54). Whether PrP-specific PS-DNAs will prove to be more efficacious in treating prion disease than random sequences remains to be established.
In conclusion, it is unclear whether PS-DNAs can be used to treat prion diseases in humans effectively. Such molecules undoubtedly will require intravenous delivery and may even need intrathecal infusion. It is unknown whether the blood-brain barrier will allow PS-DNA to cross into the parenchyma and how PS-DNAs will distribute throughout the CNS. Furthermore, administration of DNA can elicit an immune response, which may be an additional impediment to the therapeutic use of PS-DNA. In spite of these potential problems, further investigation of PS-DNAs as pharmacotherapeutics for the treatment of prion diseases seems quite warranted.
References
Cotzias GC, Van Woert MH, Schiffer LM. (1967) Aromatic amino acids and modification of parkinsonism. N.Engl. J. Med. 276:374–9.
Prusiner SB. (2004) Prion Biology and Diseases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, p. 1050.
Legname G, Baskakov IV, Nguyen H-OB, et al. (2004) Synthetic mammalian prions. Science. 305:673–6.
Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. (2004) Conformational variations in an infectious protein determine prion strain differences. Nature. 428:323–8.
Korth C et al. (2003) Abbreviated incubation times for human prions in mice expressing a chimeric mouse—human prion protein transgene. Proc. Natl. Acad. Sci. U. S. A. 100:4784–9.
Prusiner SB et al. (1993) Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. U.S.A. 90:10608–12.
Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. (1994) High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol. Med. 1:19–30.
Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 302:871–4.
Safar JG et al. (2005) Prion clearance in bigenic mice. J. Gen. Virol. 86:2913–23.
Dohura K, Iwaki T, Caughey B. (2000) Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 74:4894–7.
Perrier V, Wallace AC, Kaneko K, Safar J, Prusiner SB, Cohen FE. (2000) Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc. Natl. Acad. Sci. U. S. A. 97:6073–8.
Korth C, May BCH, Cohen FE, Prusiner SB. (2001) Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. U. S. A 98:9836–41.
Kocisko DA, Baron GS, Rubenstein R, Chen J, Kuizon S, Caughey B. (2003) New inhibitors of scrapie-associated prion protein formation in a library of 2000 drugs and natural products. J. Virol. 77:10288–94.
Supattapone S, Nguyen H-OB, Cohen FE, Prusiner SB, Scott MR. (1999) Elimination of prions by branched polyamines and implications for therapeutics. Proc. Natl. Acad. Sci. U. S. A. 96:14529–34.
Peretz D et al. (2001) Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 412:739–43.
Enari M, Flechsig E, Weissmann C. (2001) Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc. Natl. Acad. Sci. U. S. A. 98:9295–9.
Heppner FL et al. (2001) Prevention of scrapie pathogenesis by transgenic expression of antiprion protein antibodies. Science. 294: 178–182.
Sigurdsson EM et al. (2003) Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci. Lett. 336:185–7.
White AR et al. (2003) Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 422:80–3.
May BCH et al. (2003) Potent inhibition of scrapie prion replication in cultured cells by bis-acridines. Proc. Natl. Acad. Sci. U. S. A. 100:3416–21.
Collins SJ, Lewis V, Brazier M, Hill AF, Fletcher A, Masters CL. (2002) Quinacrine does not prolong survival in a murine Creutzfeldt-Jakob disease model. Ann. Neurol. 52:503–6.
Barret A et al. (2003) Evaluation of quinacrine treatment for prion diseases. J. Virol. 77:8462–9.
Dohura K et al. (2004) Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J. Virol. 78:4999–5006.
Sethi S, Lipford G, Wagner H, Kretzschmar H. (2002) Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet. 360:229–30.
Stein CA, Cheng YC. (1993) Antisense oligonucleotides as therapeutic agents—is the bullet really magical? Science. 261:1004–12.
Prusiner SB, May BCH, Cohen FE. (2004) Therapeutic approaches to prion diseases. In: Prusiner SB (ed.) Prion Biology and Diseases. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp. 961–1014.
Kocisko DA et al. (2006) Potent antiscrapie activities of degenerate phosphorothioate oligonu-cleotides. Antimicrob. Agents Chemother. 50:1034–44.
Butler DA et al. (1988) Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J. Virol. 62:1558–64.
Bosque PJ, Prusiner SB. (2000) Cultured cell sublines highly susceptible to prion infection. J. Virol. 74:4377–86.
Kanaani J, Prusiner SB, Diacovo J, Baekkeskov S, Legname G. (2005) Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 95:1373–86.
Williamson RA et al. (1998) Mapping the prion protein using recombinant antibodies. J. Virol. 72:9413–8.
Peretz D et al. (1997) A conformational transition at the N-terminus of the prion protein features in formation of the scrapie isoform. J. Mol. Biol. 273:614–22.
Moore RC et al. (1999) Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J. Mol. Biol. 292:797–817.
Muramoto T, DeArmond SJ, Scott M, Telling GC, Cohen FE, Prusiner SB. (1997) Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an ahelix. Nat. Med. 3:750–5.
Krieg AM et al. (1995) CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 374:546–9.
Schätzl HM et al. (1997) A hypothalamic neuronal cell line persistently infected with scrapie prions exhibits apoptosis. J. Virol. 71:8821–31.
Stahl N, Borchelt DR, Hsiao K, Prusiner SB. (1987) Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 51:229–40.
He H-T, Finne J, Goridis C. (1987) Biosynthesis, membrane association, and release of N-CAM-120, a phosphatidylinositol-linked form of the neural cell adhesion molecule. J. Cell Biol. 105:2489–500.
Schmitt-Ulms G et al. (2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314:1209–25.
Santuccione A, Sytnyk V, Leshchyns’ka I, Schachner M. (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169: 41–54.
Matsunaga Y et al. (2001) Cryptic epitopes in N-terminally truncated prion protein are exposed in the full-length molecule: Dependence of conformation on pH. Proteins. 44:110–8.
Leclerc E et al. (2003) Conformation of PrPC on the cell surface as probed by antibodies. J. Mol. Biol. 326:475–83.
Stenseth K, Thyberg J. (1989) Monensin and chloroquine inhibit transfer to lysosomes of endocytosed macromolecules in cultured mouse peritoneal macrophages. Eur. J. Cell Biol. 49:326–33.
Furuchi T, Aikawa K, Arai H, Inoue K. (1993) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, blocks lysosomal cholesterol trafficking in macrophages. J. Biol. Chem. 268: 27345–8.
Yedidia Y, Horonchik L, Tzaban S, Yanai A, Taraboulos A. (2001) Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 20:5383–91.
Perrier V et al. (2004) Anti-PrPantibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J. Neurochem. 89:454–63.
Williamson RA et al. (1996) Circumventing tolerance to generate autologous monoclonal antibodies to the prion protein. Proc. Natl. Acad. Sci. U. S. A 93:7279–82.
Leclerc E et al. (2001) Immobilized prion protein undergoes spontaneous rearrangement to a conformation having features in common with the infectious form. EMBO J. 20:1547–54.
Supattapone S et al. (2001) Branched polyamines cure prion-infected neuroblastoma cells. J. Virol. 75:3453–61.
Proske D, Gilch S, Wopfner F, Schätzl HM, Winnacker EL, Famulok M. (2002) Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem. 3:717–25.
Adler V, Zeiler B, Kryukov V, Kascsak R, Rubenstein R, Grossman A. (2003) Small, highly structured RNAs participate in the conversion of human recombinant PrPSen to PrPResin vitro. J. Mol. Biol. 332:47–57.
Sayer NM, Cubin M, Rhie A, Bullock M, Tahiri-Alaoui A, James W. (2004) Structural determinants of conformationally selective, prion-binding aptamers. J. Biol. Chem. 279:13102–9.
Takemura K et al. (2006) DNAaptamers that bind to PrP(C) and not PrP(Sc) show sequence and structure specificity. Exp. Biol. Med. 231:204–14.
King D, Safar JG, Legname G, Prusiner SB. (In press) Thioaptamer interactions with prion proteins: sequence-specific and non-specific binding sites. J. Mol. Biol.
Acknowledgments
The authors thank Ana Serban, Rick Shefer, Patrick Bosque, Zoltan Kanyo, Gerold Schmitt-Ulms, and Diane Latawiec for helpful discussions. Special thanks to Hang Nguyen, Larry Steinman, Peter Hughes, and Christian Essrich for their excellent editorial contributions in improving this manuscript. We are very thankful to Anat Tiran, Lorna Malimovka, Véronique Perrier, Giuseppe Legname, Julie Vergara, Septima Hong, and Sam Barillas for technical assistance. This work was supported by grants from the National Institutes of Health as well as by a gift from the G Harold and Leila Y Mathers Charitable Foundation. MVK was supported by the Human Frontiers Science Program. WD is supported by NIH-NCI Grant #P20 CA091471. KG, MRS, DP, and SBP have financial interest in InPro Biotechnology, Inc.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Karpuj, M.V., Giles, K., Gelibter-Niv, S. et al. Phosphorothioate Oligonucleotides Reduce PrPSc Levels and Prion Infectivity in Cultured Cells. Mol Med 13, 190–198 (2007). https://doi.org/10.2119/2006-00073.Karpuj
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/2006-00073.Karpuj