
Abstract
Chloride binding by the hydrate phases of cementitious materials influences the rate of chloride ingress into these materials and, thus, the time at which chloride reaches the steel reinforcement in concrete structures. Chloride binding isotherms of individual hydrate phases would be required to model chloride ingress but are only scarcely available and partly conflicting. The present study by RILEM TC 283-CAM ‘Chloride transport in alkali-activated materials’ significantly extends the available database and resolves some of the apparent contradictions by determining the chloride binding isotherms of layered double hydroxides (LDH), including AFm phases (monosulfate, strätlingite, hydrotalcite, and meixnerite), and of alkali-activated slags (AAS) produced with four different activators (Na2SiO3, Na2O·1.87SiO2, Na2CO3, and Na2SO4), in NaOH/NaCl solutions at various liquid/solid ratios. Selected solids after chloride binding were analysed by X-ray diffraction, and thermodynamic modelling was applied to simulate the phase changes occurring during chloride binding by the AFm phases. The results of the present study show that the chloride binding isotherms of LDH/AFm phases depend strongly on the liquid/solid ratio during the experiments. This is attributed to kinetic restrictions, which are, however, currently poorly understood. Chloride binding by AAS pastes is only moderately influenced by the employed activator. A steep increase of the chloride binding by AAS occurs at free chloride concentrations above approx. 1.0 M, which is possibly related to chloride binding by the C–(N–)A–S–H gel in the AAS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Durable structures made from reinforced concrete elements require that the ingress of chlorides from the external environment into the concrete is impeded to avoid chloride-induced steel corrosion causing premature deterioration of the structures [1, 2]. The rate of ingress of chlorides into concrete is governed by (1) the pore structure of the material, (2) the moisture content within the concrete cover, (3) the ability of the material to bind Cl− ions, since chloride binding slows down the diffusion of Cl− ions, as well as (4) the chemistry of the pore solution [3, 4]. Chloride binding by conventional cements and concretes has been investigated extensively [3,4,5,6,7,8,9,10,11,12], and pertinent data is also accumulating for alkali-activated materials [13,14,15]. Though empirical data that links concrete mix designs, cement compositions, chloride binding capacities, and chloride diffusion coefficients are highly valuable, more versatile tools are needed, including computational models that can predict effective chloride diffusion coefficients of concretes based on their mix design and the resultant phase assemblage of their binder matrices [16,17,18]. Such models require knowledge of the chloride binding isotherms of the individual reaction products of cements and alkali-activated materials.
The secondary reaction products of conventional cements and alkali-activated slags (AAS) generally comprise layered double hydroxides (LDHs), which are a family of compounds with a structure consisting of a main layer that generally contains divalent cations (e.g., Ca2+, Mg2+, Zn2+) and trivalent cations (e.g., Al3+, Fe3+, Cr3+), and anions in the interlayer space between the main layers to balance their positive charge. The interlayer space also contains water molecules, the amount of which depends on the activity of water, i.e., the relative humidity. The anions in the interlayer can be exchanged; thus, LDHs are potentially able to bind or release significant amounts of Cl−, depending on the prevailing local aqueous environment.
In hydrated Portland cements and many blended cements, LDHs occur mainly as AFm phases, i.e., compounds in which the main layer contains Ca2+ and Al3+ (potentially substituted by Fe3+), and the anions in the interlayer are usually SO42−, OH−, CO32−, Cl− (after chloride ingress), or an aluminosilicate anion [19,20,21]. The general formula of iron-free AFm phases can be written as Ca2Al(OH)6·X·xH2O or 3CaO·Al2O3·CaX2·nH2O, where X represents a singly charged anion or half a doubly charged anion. In AAS binders with a significant fraction of MgO, LDHs with Mg2+ and Al3+ in the main layer are formed [18, 22, 23]. As their main-layer composition and structure are similar to those of the mineral hydrotalcite [approx. Mg6Al2(OH)16·CO3·4H2O], such phases are usually referred to as hydrotalcite-like compounds [24] or hydrotalcite-type phases, irrespective of the interlayer anions. A naturally occurring hydrotalcite-like compound with OH− as the interlayer anion is known and has been named meixnerite [25]. In AAS binders, LDH phases with a composition similar to meixnerite, i.e., with OH− as the predominant anion, occur and have been referred to as Mg–Al–OH LDH or MA-OH LDH [18, 23, 26]. A more detailed nomenclature scheme for synthetic LDH phases has been proposed and publicised as supplement to the International Mineralogical Association (IMA)-approved report on the nomenclature of the naturally occurring LDH phases (hydrotalcite supergroup) [27].
Despite the importance of chloride binding in LDH/AFm phases, chloride binding data for individual AFm and LDH phases applicable to modelling of chloride ingress into concrete are scarce. Hirao et al. [28] reported the chloride binding isotherm for monosulfate (4CaO·Al2O3·SO3·12H2O) in NaCl solutions. In the study of Ke et al. [29], chloride binding isotherms for strätlingite (2CaO·Al2O3·SiO2·7.5H2O) and a calcined hydrotalcite in NaOH/NaCl solutions with and without Na2CO3 were determined. Balonis et al. [21] conducted an extensive study of the thermodynamic stability of the Cl-containing AFm phases Friedel’s salt (3CaO·Al2O3·CaCl2·10H2O) and its solid solutions, and Kuzel’s salt (6CaO·2Al2O3·CaSO4·CaCl2·24H2O), in principle enabling the calculation of theoretical binding isotherms, but did not provide experimentally determined binding isotherms. Thermodynamic data for hydrotalcite-like compounds, including phases with OH−, CO32−, or Cl− in the interlayer, has been published recently [30].
The aim of the present study by RILEM TC 283-CAM was, thus, to extend the available database with a focus on AAS binders, i.e., for representative pore solutions with high concentrations of Na+. Nevertheless, the obtained data is also relevant for other cementitious systems. Four LDH/AFm phases (monosulfate, strätlingite, hydrotalcite, and meixnerite) and four AAS pastes with different activators (Na2SiO3, Na2O·1.87SiO2, Na2CO3, and Na2SO4), were produced and their chloride binding isotherms in NaOH/NaCl solutions with [Na+] = 1.5 M determined for different liquid/solid ratios. Selected solids after chloride binding were analysed by X-ray diffraction, and the phase changes during chloride binding by the AFm phases were simulated with thermodynamic modelling.
2 Materials and methods
2.1 Synthesis of LDH/AFm phases
Four different LDH/AFm phases were produced: monosulfate (nominally 3CaO·Al2O3·CaSO4·12H2O or 4CaO·Al2O3·SO3·12H2O), strätlingite (nominally 2CaO·Al2O3·SiO2·7.5H2O), hydrotalcite (nominally 4.67MgO·Al2O3·CO3·10H2O), and Mg–Al–OH LDH or meixnerite (approx. 4.67MgO·Al2O3·11H2O). The chemicals employed for the syntheses were commercially available compounds of analytical grade, except the sodium aluminate (NaAlO2) and the waterglass, which were of technical grade, and the tricalcium aluminate (3CaO·Al2O3), which was synthesized at the Cement Chemistry and Thermodynamics laboratory of Empa (Dübendorf, Switzerland). The protocols for the synthesis of the LDH/AFm phases, described below, were adopted from previous studies that established the thermodynamic properties or provided chloride binding isotherms of the respective phases.
Monosulfate was produced according to the method described by Matschei et al. [31]. A 1:1 molar mixture of 3CaO·Al2O3 and CaSO4 was suspended in water, the suspension aged at 85 °C for seven days and then filtered, and the filter residue washed (see Supplementary Text 1). Two batches of monosulfate were produced. The X-ray diffraction (XRD) pattern of batch 1 (Supplementary Fig. S1; for experimental conditions, see Sect. 2.3) exhibited major reflections of monosulfate 10.5-hydrate (4CaO·Al2O3·SO3·10.5H2O [32, 33]), and minor reflections of monosulfate 12-hydrate [4CaO·Al2O3·SO3·12H2O; Powder Diffraction File (PDF) 01-083-1289] and ettringite (6CaO·Al2O3·3SO3·32H2O; PDF 00-041-1451). The predominant occurrence of the 10.5-hydrate was caused by drying the material at 11% relative humidity (RH) for three weeks before characterisation and further experiments (see below). Batch 2 of monosulfate appeared to contain a somewhat higher fraction of ettringite, and the 12-hydrate was the dominating monosulfate (Supplementary Fig. S1).
Strätlingite was produced according to the method described by Matschei et al. [31]. Suspensions of Ca(OH)2 and NaAlO2 were mixed with a sodium silicate solution in stoichiometric amounts, the resulting suspension was stirred for four weeks at 20–25 °C and then filtered, and the filter residue was washed (see Supplementary Text 1). Two batches of strätlingite were produced. The XRD pattern of batch 1 (Supplementary Fig. S2) exhibited major reflections of strätlingite (PDF 01-080-1579), and minor reflections of monocarbonate (4CaO·Al2O3·CO3·11H2O; PDF 01-087-0493) and hydrogarnet (PDF 00-038-0368; pertaining to katoite with cell parameter a = 1.2358 Å) as the impurities in the material. The XRD pattern of the second batch (Supplementary Fig. S2) was very similar to that of batch 1, though batch 2 appeared to contain even less monocarbonate and hydrogarnet.
It is noted that the peaks of strätlingite in the XRD patterns were broad, and their intensities were much lower than the reflections of monosulfate, hydrotalcite, and meixnerite (Supplementary Fig. S4). This indicates a considerably lower degree of ordering, or lower ‘crystallinity’, of the strätlingite, compared to the other LDH/AFm phases. This observation is in accord with previous studies of synthetic strätlingite [29, 31], and it is likely also representative of the conditions in hydrated cement and AAS pastes, as indicated by the absence of noticeable reflections of strätlingite in XRD patterns of pastes for which thermodynamic modelling as well as spectroscopic and thermogravimetric data indicated the formation of significant amounts of this phase [34].
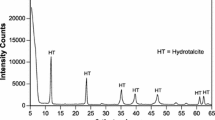
Hydrotalcite was produced in a way similar to the method described by Miyata [35, 36]. Solutions of MgCl2, AlCl3, NaOH and Na2CO3 were mixed in stoichiometric amounts, the pH of the resulting suspension was adjusted to pH = 10.0 ± 0.2 with NaOH solution, the suspension aged at ~ 45 °C for 20 min and filtered, and the filter residue washed; subsequently, the filter residue was dried at 80 °C, aged in water at 90–95 °C for four days, the solution filtered, and the obtained material washed (see Supplementary Text 1). The XRD pattern of the synthesized compound (Supplementary Fig. S3) showed it to be virtually pure hydrotalcite (PDF 01-070-2151), though minor reflections of an impurity or impurities at 9.4°, 16.2°, and 28.4° 2θ (d = 9.4, 5.5, and 3.1 Å) were detected, the latter possibly caused by boehmite [AlO(OH)].
A Mg–Al–OH LDH, in the following referred to as meixnerite, was produced according to the method described by Ke et al. [29]. Hydrotalcite was calcined at 500 °C for 3 h, the obtained material was suspended in 1 M NaOH solution, the suspension aged for ~ 24 h and then filtered, and the filter residue washed (see Supplementary Text 1). The XRD pattern of the synthesized compound was very similar to that of hydrotalcite, though the reflections of the latter were narrower (Supplementary Fig. S3). A peak of an impurity was found at 19.5° 2θ (d = 4.5 Å). The identification of the synthesized compound as meixnerite was possible through its differential thermogravimetry (DTG) curve, which differed from that of the hydrotalcite (Supplementary Fig. S5) and was in accord with previously published thermogravimetric curves of Mg–Al–OH LDH [29, 37,38,39]. In the attenuated total reflectance Fourier-transform infrared (ATR-FTIR) spectra of the hydrotalcite and the meixnerite, the relative intensity of the band of the carbonate anion at ~ 1365 cm−1 was considerably lower for the meixnerite (Supplementary Fig. S8), confirming that most of the carbonate had been lost during calcination of the hydrotalcite to produce meixnerite. Nevertheless, some of the carbonate had remained in the meixnerite, in line with previous studies that showed that decarbonation of hydrotalcite is usually not complete at 500 °C [36, 40].
Before characterisation and the chloride binding experiments, all synthesized materials were stored in argon-flushed desiccators over saturated LiCl solution (11% RH) for at least three weeks to allow them to assume a reproducible water content.
The DTG curves of all synthesized compounds are shown in Supplementary Fig. S5. The DTG curves were well in accord with previously published data for the respective compounds [29,30,31,32, 35,36,37,38,39,40,41] and with the presence of small amounts of ettringite and hydrogarnet as impurities in monosulfate and strätlingite, respectively, which were also identified by XRD. ATR-FTIR spectra of all synthesized compounds are presented in Supplementary Figs. S6–S8.
2.2 Alkali-activated slags
AAS pastes with four different activators were produced in two batches: batch 1 at the Bundesanstalt für Materialforschung und -prüfung (BAM), Berlin, and batch 2 at the Eidgenössische Technische Hochschule (ETH) Zürich. The ground granulated blast-furnace slag (GGBFS) used for batch 1 contained 43.8% CaO, 4.9% MgO, 12.0% Al2O3, and 33.4% SiO2 (full composition presented in Supplementary Table S1). The GGBFS used for batch 2 contained 42.1% CaO, 5.1% MgO, 11.4% Al2O3, and 35.0% SiO2 (full composition presented in Supplementary Table S1). Sodium silicate solutions for activation were produced from commercial waterglass, NaOH pellets (reagent grade) and deionised water. The Na2CO3 and Na2SO4 used as activators were of analytical grade.
In each batch, four AAS pastes with activators Na2SiO3, Na2O·1.87SiO2, Na2CO3, and Na2SO4, respectively, were produced with a water/binder (w/b) ratio of 0.40, where the b includes the activator (in the sodium silicate activators: solid fraction dissolved). The paste compositions, including the activator dosages are given in Table 1. After mixing, the pastes in batch 1 were cured sealed (wrapped in parafilm) at 23 °C for 6 months. The samples in batch 2 were cured under the same conditions for 90 days. After curing, the AAS pastes were manually ground, and the resulting powders were stored in argon- or nitrogen-flushed desiccators over saturated K2CO3 solution (43% RH) for 14 days to allow them to assume a reproducible water content.
The phase assemblages of AAS pastes such as those produced in the present study can be predicted by thermodynamic modelling [18, 23, 26]. For AAS based on a GGBFS with a composition (42.3% CaO, 5.2% MgO, 13.3% Al2O3, 32.3% SiO2) very similar to those of the GGBFS employed in the current study and similar activator compositions, the following phase assemblages are predicted [18]: AAS Na2SiO3 yields a sodium-substituted calcium aluminosilicate hydrate [C–(N–)A–S–H gel], strätlingite, Mg–Al–OH LDH, and hydrogarnet/katoite. AAS Na2O·1.87SiO2 yields C–(N–)A–S–H gel, strätlingite, and Mg–Al–OH LDH. AAS Na2CO3 yields C–(N–)A–S–H gel, monocarbonate, Mg–Al–OH LDH, and minor quantities of a sodium aluminosilicate gel (N–A–S–H gel) as well as calcite. AAS Na2SO4 yields C–(N–)A–S–H gel, monosulfate, ettringite, Mg–Al–OH LDH, and strätlingite. The composition of the C–(N–)A–S–H gels in the AAS is predicted to be in the range Ca/Si = 0.9–1.0, and Al/Si = 0.10–0.14.
XRD patterns of the AAS pastes after 28 or 48 days of curing are shown in Supplementary Fig. S9 (for experimental conditions, see Sect. 2.3). C–(N–)A–S–H gel was identified in all materials. Reflections of major secondary reaction products were Mg–Al–OH LDH for AAS Na2SiO3, monocarbonate for AAS Na2CO3, and ettringite for AAS Na2SO4. In addition, AAS Na2CO3 contained gaylussite [Na2Ca(CO3)2·5H2O], previously identified in sodium carbonate-activated slag also by Ke et al. [42], and a considerable amount of calcite. Calcite and vaterite were found also in all other AAS pastes, presumably caused by slight carbonation of the materials during storage and handling. In a 90-day cured sample of AAS Na2SO4, C–(N–)A–S–H gel and ettringite were the major reaction products, while smaller amounts of monosulfate and Mg–Al–OH LDH were also identified (Supplementary Fig. S10). Strätlingite was identified in none of the AAS pastes, but as discussed above (Sect. 2.1), strätlingite is difficult to detect by XRD, because of the generally low intensity and large width of its reflections. Thus, the experimentally determined phase assemblages were in reasonable agreement with the results of thermodynamic modelling.
2.3 X-ray diffraction
Powdered samples for XRD measurements were filled in sample holders by top loading. For the measurements of the LDH/AFm phases, the samples were covered with Kapton foil to prevent carbonation as well as uptake or release of water, except for two samples. These two exceptions were hydrotalcite and meixnerite after chloride binding, as in these cases the available amount of sample material was not sufficient to use a sample holder that could be covered with a stretched foil.
The XRD measurements were carried out with a Rigaku Ultima IV device using Cu Kα radiation (λ = 1.5419 Å) in Bragg–Brentano geometry. The parameters for the measurements were: cathode at 40 kV/40 mA, 0.5°/10 mm divergence slit, sampling interval 0.02° 2θ, and scanning speed 0.5° 2θ/min. The detector was a D/teX Ultra strip detector with 5° Soller slits. The scanning range was 5–75° 2θ for the LDH/AFm phases, and 5–65° 2θ for the AAS pastes.
2.4 Chloride binding isotherms
Chloride binding isotherms of LDH/AFm phases and AAS pastes were determined at BAM and at ETH to verify the reproducibility of the results. Batch 1 of the synthesized LDH/AFm phases and the AAS were used at BAM, and batch 2 of the LDH/AFm phases and AAS were used at ETH.
NaOH/NaCl solutions for the determination of chloride binding isotherms were prepared from NaOH, NaCl (analytical grade) and ultrapure water. [Na+] was kept at 1.5 M for all solutions to approximate the conditions in the pore solutions of alkali-activated materials (reviewed in Ref. [43]), while [Cl−] was varied as shown in Table 2.
Chloride binding isotherms were recorded by suspending appropriate amounts of solid samples (LDH/AFm phase or AAS paste) in NaOH/NaCl solution to yield specified liquid volume/solid mass (l/s) ratios. For the LDH/AFm phases, the employed l/s ratios were approx. 13, 26, 53, and 108 mL/g. For example, to obtain l/s = 26 mL/g, 0.75 g of LDH/AFm phase was suspended in 20 mL of NaOH/NaCl solution at BAM, and 0.47 g of LDH/AFm phase was suspended in 12.5 mL of NaOH/NaCl solution at ETH. For the AAS pastes, a l/s of 4 mL/g was always applied: 10 g of AAS paste in 40 mL solution at BAM; 5 g of AAS paste in 20 mL of solution at ETH. The bottles with the suspensions were tightly closed with a screw cap and sealed with Parafilm to prevent ingress of CO2, and then stored at 23 °C. During that period, the bottles with LDH/AFm phases were manually swung/panned three times per week. The bottles with AAS pastes were shaken for approx. 60 s each weekday.
After an equilibration time of 21 days, an aliquot of approx. 2 mL of the suspension was obtained and filtered through a 0.45-µm syringe filter (BAM), or the suspension was centrifuged at 4000 rpm and the supernatant filtered through a 0.22-µm syringe filter (ETH). The filtered solutions were then analysed for free chloride concentrations by potentiometric titration. The bound chloride, Qe, was calculated for each combination of material and solution from
where MCl is the molar mass of Cl, [Cl−]init is the initial chloride concentration in mol/L of the solution (cf. Table 2), [Cl−]final is the free chloride concentration in mol/L after equilibration for 21 days, Vsoln is the volume of solution used in the experiment, and msample is the initial mass of solid sample used in the experiment.
The chloride binding experiments at BAM were partly performed as duplicate experiments (monosulfate at l/s = 26 mL/g, and strätlingite at l/s = 26 and 53 mL/g) and partly as single experiments (monosulfate at l/s = 53 mL/g, strätlingite at l/s = 13 mL/g, hydrotalcite and meixnerite at l/s = 26 and 108 mL/g, and AAS pastes), due to the low yield of some of the syntheses and the limited availability of some of the starting materials. The experiments at ETH (monosulfate and strätlingite at l/s = 26 mL/g, and AAS pastes) were performed in duplicate.
Some of the LDH/AFm phase suspensions at BAM were centrifuged at 4000 rpm after the aliquot for chloride analysis had been obtained, and the solids recovered, washed with deionised water, and dried in argon-flushed desiccators over saturated LiCl solution (11% RH) for several weeks. The dried solids were then analysed by XRD (Sect. 2.3).
2.5 Thermodynamic modelling
Thermodynamic modelling was performed with the Gibbs Energy Minimization Software (GEMS) [44, 45], using the built-in Nagra/PSI thermodynamic database and the Cemdata18 database [46], which includes the data for Cl-containing AFm phases by Balonis et al. [21]. The Helgeson form of the extended Debye-Hückel equation (see, e.g., Ref. [26]) was employed to compute the activity coefficients of the ions in the solutions. Since modelling focused on the chloride binding in AFm phases during immersion in NaOH/NaCl solutions, the average ion size and the parameter for common short-range interactions of charged species in the extended Debye-Hückel equation were selected to be those for solutions dominated by sodium chloride (ȧ = 3.72 Å, bγ = 0.064 kg/mol). All thermodynamic modelling was done without access of air to simulate the experimental conditions during the chloride binding experiments. The reaction temperature was chosen to be 23 °C.
The formation of AFm phases was simulated by allowing the following amounts of the oxides to react: CaO, Al2O3, SO3, H2O in the ratios 2.25:1:0.83:10 to form monosulfate; and CaO, Al2O3, SiO2, H2O in ratios 1.34:1:0.84:5.6 to form strätlingite. These ratios had to be chosen to reduce the formation of by-products, mainly portlandite besides monosulfate, and C–(N–)A–S–H besides strätlingite. In the next step, NaCl, NaOH, and water were added to the bulk compositions in amounts representative of the solutions and l/s ratios in the chloride binding experiments (Sect. 2.4).
To simulate the formation of C–(N–)A–S–H besides strätlingite, both the ECSH I model [46] and the CNASH_ss model [46, 47] were included in the modelling. The rationale was that the C–S–H forming during the synthesis of strätlingite can be modelled with ECSH I, but the Na+ ions introduced by the NaOH/NaCl solutions interact with C–S–H to form C–(N–)A–S–H as modelled by the CNASH_ss model. Indeed, the computations yielded C–(N–)A–S–H as a major by-product in the systems with strätlingite and NaOH/NaCl, though in few cases C–S–H was predicted to form; in the following, these are always treated together and referred to as C–(N–)A–S–H.
Chloride binding by hydrotalcite and meixnerite were not modelled, as comparison of the results would have been possible only with partial chloride binding isotherms (Sect. 3.1), differentiation of hydrotalcite-like phases with different interlayer anions in mixtures is complex and potentially ambiguous (Sect. 3.1), and the presently available thermodynamic data [30] does not treat the partial exchange of the anions in the interlayer of these phases directly.
3 Results
3.1 Chloride binding by LDH/AFm phases
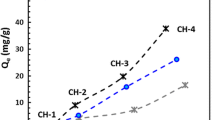
The chloride binding isotherms of the LDH/AFm phases are shown in Fig. 1. The isotherms for monosulfate and strätlingite at l/s ≈ 53 mL/g exhibited a continuous increase of bound chloride until a free Cl− concentration of approx. 0.5 M. At free [Cl−] above that value, the isotherms plateaued around the respective theoretical chloride binding capacities, i.e., the expected chloride binding when each SO42− ion in monosulfate is exchanged by two Cl− ions (119 mg/g), and each aluminosilicate anion in the interlayer of strätlingite is exchanged by a Cl− ion (87 mg/g).

Chloride binding isotherms of: A monosulfate (Ms); B strätlingite (Str); C hydrotalcite (Ht) and meixnerite (Mx). The approximate liquid volume/solid mass ratios (in mL/g; shown in parentheses) used for the experiments, and the laboratory where the data was obtained (BAM or ETH) are indicated in the legend. Empty symbols represent single experiments, and filled symbols represent duplicate experiments. Error bars represent one standard deviation in each direction for the duplicate experiments. Error bars/standard deviations for free Cl− are shown only for Ms, because in all other experiments, they were always smaller than the width of the symbols. For each LDH/AFm, the amount sorbed equivalent to one chloride ion per anion site is indicated by a dashed line, assuming an ideal stoichiometry of [0.5 × 4CaO·Al2O3·SO3·10.5H2O] per site for Ms, an ideal stoichiometry of [2CaO·Al2O3·SiO2·7.5H2O] per site for Str, an ideal stoichiometry of [0.5 × 4.67MgO·Al2O3·CO3·10H2O] per site for Ht, and an ideal stoichiometry of [0.5 × 4MgO·Al2O3·10H2O] per site for Mx
For lower l/s ratios (26 and 13 mL/g), both monosulfate and strätlingite exhibited a considerably reduced chloride binding. For monosulfate, chloride binding increased from 24 mg/g at the lowest free [Cl−] to 73 mg/g at free [Cl−] ≈ 0.7 M, and then increased only very slightly to approx. 82 mg/g, i.e., a value significantly lower than the theoretical chloride binding capacity of that phase. The chloride bound by strätlingite at l/s ≈ 26 mL/g increased slightly from approx. 26 mg/g to 34–41 mg/g at free [Cl−] ≈ 0.25 M, and then stayed approximately constant up to a free Cl− concentration of approx. 1.4 M. For strätlingite at l/s ≈ 13 mL/g, the evolution of bound chloride with free [Cl−] was similar to that obtained at l/s ≈ 26 mL/g, though Qe was generally slightly lower in the former case.
Only partial chloride binding isotherms could be recorded for hydrotalcite and meixnerite (Fig. 1C), due to the lower amounts of synthesized materials. Nevertheless, the measured data was in accordance with the observations made for monosulfate and strätlingite. At a high liquid volume/solid mass ratio (l/s ≈ 108 mL/g) and free [Cl−] = 1.15 M, meixnerite exhibited a chloride binding of 188 mg/g that was close to its theoretical binding capacity, i.e., the chloride binding expected when each OH− ion in the interlayer is exchanged by a Cl− ion (160 mg/g). At the lower l/s of 26 mL/g, however, both meixnerite and hydrotalcite exhibited much lower chloride binding, viz., 37–51 mg/g for meixnerite and 27–29 mg/g for hydrotalcite, for free Cl− concentrations in the range 0.25–1.0 M.
XRD patterns of the LDH/AFm phases as-synthesized and after chloride binding experiments at l/s = 26 mL/g and high initial Cl− concentrations (1.0 or 1.4 M) are shown in Fig. 2. The XRD patterns for the monosulfate samples after exposure to NaOH/NaCl solutions with initial [Cl−] of 1.0 and 1.4 M, respectively, were very similar. Upon exposure to chloride solutions, monosulfate was essentially absent, though a very minor reflection/shoulder at 9.9° 2θ (d = 8.9 Å) might indicate that trace quantities of monosulfate 12-hydrate had remained in the sample. The major phases in the chloride-exposed material were Kuzel’s salt (6CaO·2Al2O3·CaSO4·CaCl2·24H2O; PDF 00-019-0203) and Friedel’s salt (3CaO·Al2O3·CaCl2·10H2O; PDF 01-078-1219), i.e., a full transformation of monosulfate to Friedel’s salt had not occurred, in line with the fact that chloride binding for these samples at l/s = 26 mL/g was significantly below the theoretical chloride binding capacity of monosulfate. The amount of ettringite in the samples did not change significantly during chloride exposure.

X-ray diffraction patterns, as synthesized and after chloride binding experiments, of: A monosulfate (Ms); B strätlingite (Str); C hydrotalcite (Ht); D meixnerite (Mx). The liquid volume/solid mass ratios used for the chloride binding experiments were approx. 26 mL/g; the initial [Cl−] are indicated in the legends. Selected peaks are labelled: K, Kuzel’s salt; F, Friedel’s salt; Ett, ettringite; Ms-12, monosulfate 12-hydrate; Ms-10.5, monosulfate 10.5-hydrate; Str, strätlingite; Mc, monocarbonate; Hg, hydrogarnet; H, halite; Ht, hydrotalcite; ?, unidentified impurities in Ht and Mx; Mx, meixnerite; *, Kapton foil used to cover samples. The Ht and Mx samples after chloride binding were measured without Kapton foil
In the strätlingite samples after chloride exposure (initial [Cl−] = 1.0 or 1.4 M), Friedel’s salt was detected, but considerable amounts of strätlingite had remained; the XRD patterns for both initial Cl− concentrations were very similar. Again, this observation is in line with the low chloride binding of the strätlingite determined at l/s ≈ 26 mL/g, which was approx. 50% below its theoretical binding capacity at all Cl− concentrations. The monocarbonate, detected as an impurity in the as-synthesized strätlingite, was not present in the chloride-exposed materials, indicating that the monocarbonate had dissolved or transformed to Friedel’s salt. The amount of hydrogarnet in the samples did not change significantly during chloride exposure.
The XRD patterns of the hydrotalcite and the meixnerite after chloride exposure (initial [Cl−] = 1.0 M) were similar to those of their respective starting materials. In the pattern of hydrotalcite after chloride exposure, halite (NaCl; PDF 00-005-0628) was detected. This was likely caused by incomplete removal of the NaOH/NaCl solution before drying the sample and/or formation of NaCl during drying from species adsorbed at the surface of the material (cf. Ref. [29]). No other Cl-bearing phases were identified in the XRD patterns. However, partial exchange of the interlayer CO32− and OH− by Cl− in the hydrotalcite and the meixnerite during chloride exposure cannot be excluded, as the corresponding changes of the XRD patterns of these phases would be only slight. The reported basal spacings of hydrotalcite-like compounds with CO32−, OH−, and Cl− in the interlayer are 7.63–7.65, 7.55–7.68, and 7.65–7.86 Å, respectively [24, 30]; thus, a partial substitution of these interlayer anions is expected to cause only a small shift of the corresponding reflections that would be difficult to identify (cf. Ref. [29]). Thus, the XRD patterns of hydrotalcite and meixnerite after chloride exposure were consistent with their measured chloride binding, which was only 20–30% of their respective theoretical binding capacities.
3.2 Chloride binding by alkali-activated slags
The chloride binding isotherms of the hydrated AAS pastes are shown in Fig. 3. For all AAS, chloride binding increased from 3–4 mg/g at the lowest free [Cl−] to 10–16 mg/g at free [Cl−] ≈ 0.6 M. The bound chloride remained approximately constant at these values for higher chloride concentrations up to 0.9–1.1 M. When free [Cl−] increased further, a steep increase of the bound chloride occurred, reaching values in the range of 21–28 mg/g at free [Cl−] ≈ 1.2 M.

Chloride binding isotherms of the alkali-activated slag (AAS) pastes: A obtained at BAM; B obtained at ETH. The liquid volume/solid mass ratios used for the experiments were approx. 4 mL/g. Empty symbols represent single experiments, and filled symbols represent duplicate experiments. Error bars for Qe represent one standard deviation in each direction for the duplicate experiments. Error bars/standard deviations for free Cl− are not shown, because they were always smaller than the width of the symbols. The complete overlap of some of the data points is caused by rounding errors
The differences in chloride binding behaviour between the different AAS pastes were generally not large. However, it is noted that the data might indicate that AAS Na2SO4 binds slightly more chloride than the other pastes at free [Cl−] ≤ 0.6 M (Fig. 3A), or at least that its chloride binding is comparatively high in this concentration range (Fig. 3B). Pastes AAS Na2SiO3 and AAS Na2O·1.87SiO2 tended to exhibit the highest chloride binding of all AAS pastes at free [Cl−] > 1.0 M. A peculiar feature of the chloride binding isotherms of AAS Na2CO3 was that their plateau-like region extended to free [Cl−] > 1.1 M with an apparent decrease of bound chloride with increasing free Cl− concentration; subsequently, its chloride binding rose steeply.
4 Discussion
The chloride binding isotherms of all studied LDH/AFm phases (monosulfate, strätlingite, hydrotalcite, meixnerite) depended strongly on the l/s ratio employed in the chloride binding experiments (Fig. 1). At l/s ≈ 53 or 108 mL/g, chloride binding approached or slightly exceeded the respective theoretical chloride binding capacities, assuming one Cl− per anion site in the structure, at free [Cl−] ≈ 0.5 M for monosulfate and strätlingite, and at least at free [Cl−] = 1.15 M for meixnerite. For lower l/s ratios of 26 and 13 mL/g, however, the bound chloride was significantly below the respective theoretical chloride binding capacities for free [Cl−] up to approx. 1.4 M. The fact that the data for monosulfate and strätlingite was obtained independently in two different laboratories with two separately produced batches of AFm phases strongly indicates that the lower-than-theoretical chloride binding at low l/s ratios was not an experimental artefact.
In the study of Hirao et al. [28], monosulfate was equilibrated with NaCl solutions at l/s = 10 mL/g for two days. The authors found the maximum bound chloride to be only 55% of the theoretical chloride binding capacity of monosulfate, even up to free [Cl−] ≈ 5 M (Supplementary Fig. S12), and their results indicate that only about half of the monosulfate had transformed to Friedel’s salt during the experiment. In apparent contradiction to this finding, Ke et al. [29] found that the chloride binding capacity of strätlingite and a calcined LDH with composition Mg0.7Al0.3(OH)2(CO3)0.15·0.63H2O (which presumably behaves similar to a Mg–Al–OH LDH in aqueous solution) in NaOH/NaCl solutions considerably exceed their theoretical chloride binding capacities as defined above (Supplementary Fig. S12); however, in these experiments, a l/s ratio of 100 g/g (approx. 96 mL/g) was applied. Taken together, these results are in accord with the present study, where lower-than-theoretical chloride binding was found for low l/s (≤ 26 mL/g), while chloride binding was found to be close to, or slightly higher than, the theoretical binding at high l/s (≥ 53 mL/g). Thus, the present data partly resolves apparent contradictions between previous studies of the chloride binding of LDH/AFm phases. It is noted, however, that Ke et al. [29] assigned some of the chloride binding by strätlingite and most (90–95%) of the chloride binding by the calcined LDH in their study to surface adsorption of Cl−, rather than uptake of Cl− in the structure of the resulting LDH/AFm phases.
Figure 4 shows the predicted thermodynamically stable solid-phase assemblages in the present chloride binding experiments with monosulfate and strätlingite for l/s = 26 and 53 mL/g, calculated with GEMS. The predicted assemblage for strätlingite at l/s = 13 mL/g (Supplementary Fig. S11) was almost identical to that at l/s = 26 mL/g. In all cases, thermodynamic modelling predicts a full transformation of the AFm phase to Friedel’s salt, i.e., attainment of the full theoretical chloride binding, within the range of [Cl−] employed in the experiments to determine the chloride binding isotherms. In the experiments with monosulfate, ettringite was predicted as the by-product of the reactions, i.e., the SO42− released from monosulfate would be taken up by the formation of ettringite. Portlandite was modelled as a minor by-product at low initial [Cl−]. It is noted that, according to the results of Hirao et al. [28], ettringite and portlandite exhibit virtually no chloride binding capacity; thus, these phases can be assumed to not influence the chloride binding in the experiments. The transformation of strätlingite to Friedel’s salt was predicted to involve the formation of natrolite [Na2(Si3Al2)O10·2H2O]. This zeolite would take up the aluminosilicate anions released from the strätlingite. However, this phase does not normally form in cementitious systems [46], and other, possibly amorphous, aluminosilicate phases may have formed in the actual chloride binding experiments. The C–(N–)A–S–H in the systems with strätlingite can be assumed to contribute little to chloride binding, as its predicted amount in the experiments was low, particularly at high [Cl−], and the chloride binding capacities of C–A–S–H and C–S–H have been reported to be considerably lower than, or of the same order of magnitude as, that of LDH/AFm phases [4, 12, 18, 28, 48] (cf. discussion below).

Modelled equilibrium solid-phase assemblages in the chloride binding experiments with: A monosulfate at a liquid volume/solid mass ratio of 26 mL/g; B monosulfate at a liquid volume/solid mass ratio of 53 mL/g; C strätlingite at a liquid volume/solid mass ratio of 26 mL/g; D strätlingite at a liquid volume/solid mass ratio of 53 mL/g
The initial [Cl−] at which complete transformation of the starting AFm phase to Friedel’s salt was predicted were 1.2 M for monosulfate at l/s = 26 mL/g, 1.0 M for monosulfate at l/s = 53 mL/g, 0.75 M for strätlingite at l/s = 26 mL/g, and 0.5 M for strätlingite at l/s = 53 mL/g (Fig. 4). Thus, for both AFm phases thermodynamic modelling predicts that a lower l/s leads to attainment of the full theoretical chloride binding capacity at higher [Cl−]. However, in the actual chloride binding experiments at low l/s (13 and 26 mL/g), the full chloride binding capacity was never reached (Fig. 1) and transformation of the starting LDH/AFm phase remained incomplete (Fig. 2), which is in contradiction to the thermodynamic modelling. Thus, it has to be concluded that the lower-than-theoretical chloride binding in the experiments at low l/s was caused by kinetic restrictions. However, the data obtained in this study does not allow one to examine possible reasons for the kinetic hindrance of the reactions of the LDH/AFm phases.
Studies of the chloride binding of hydrated cement pastes and alkali-activated materials have usually been conducted at considerably lower l/s, in the range of 0.5–10 mL/g, than studies of chloride binding of individual cementitious phases (Supplementary Table S2). Also in the present study, the l/s employed for the experiments with AAS pastes (4 mL/g) was lower than that to study the LDH/AFm phases (13, 26, 53 and 108 mL/g). Considering the results obtained for the latter, it is not clear whether the full chloride binding capacity of these phases was realised in the experiments with AAS pastes within the timeframe of the experiments (3 weeks). Nevertheless, the shape of the chloride binding of isotherms of the AAS pastes (Fig. 3) resembled those of the individual LDH/AFm phases at high l/s (Fig. 1) up to free [Cl−] of approx. 0.9–1.1 M. The influence of C–(N–)A–S–H on the chloride binding isotherms of the AAS pastes in this concentration range is not clear, as different chloride binding isotherms for the related phases C–A–S–H and C–S–H have been reported: Ke [48] measured chloride binding isotherms for C–A–S–H in NaOH/NaCl solutions at l/s ≈ 96 mL/g, which exhibited a small slope and a maximum chloride binding of approx. 60 mg/g in the range [Cl−] = 0–0.45 M, and a subsequent steeper increase to approx. 110 mg/g at free [Cl−] ≈ 0.7 M. Differing from that, Hirao et al. [28] reported chloride binding isotherms for C-S–H in NaCl solutions at l/s = 10 mL/g, that approached its maximum chloride binding already at around [Cl−] = 0.5 M, while the maximum chloride binding capacity was determined to be approx. 18 mg/g, i.e., comparatively low (Supplementary Fig. S13). Wilson et al. [12] found that the chloride binding capacity of C–A–S–H decreased with decreasing Ca/(Al + Si) ratio, as would be the case for binders with high substitution of Portland cement with supplementary cementitious materials (SCMs).
The steep increase of the chloride binding of the AAS pastes at free [Cl−] > 0.9–1.1 M (Fig. 3) appears to differ from what has been observed in previous studies of hydrated cement pastes and alkali-activated materials [5,6,7,8,9,10,11, 15], although in most of these studies the resolution in terms of the number of experiments with different chloride concentrations around 0.9–1.1 M was low. Nevertheless, the behaviour observed in the present study was not expected. Analogously to the LDH/AFm phases, however, the fact that the data was obtained independently in two different laboratories with separately produced AAS pastes strongly indicates that the peculiar features of the chloride binding isotherms were not experimental artefacts.
The strong increase of chloride binding by the AAS pastes at free [Cl−] > 0.9–1.1 M was probably not connected to the formation of calcium hydroxy chloride hydrates, as these are stable only at chloride concentrations above 2–4 M [3]. Thus, it is likely that the increase of chloride binding by one or more of the AAS reaction products was responsible for this behaviour. A possible candidate is the C–(N–)A–S–H gel, as the sodium silicate-activated pastes (AAS Na2SiO3 and AAS Na2O·1.87SiO2) generally showed the highest chloride binding in this concentration range (Fig. 3), and such AAS pastes comprise higher amounts of C–(N–)A–S–H gel than Na2CO3- and Na2SO4-activated slags [18]. In addition, the experiments by Ke [48] indicate that chloride binding by C–(N–)A–S–H is low to moderate at low [Cl−] but starts to increase more strongly at [Cl−] > 0.45 M (Supplementary Fig. S12). However, the experiments by Ke did not extend to chloride concentrations of 0.9–1.1 M, and further studies would be required to confirm that chloride binding increases above these concentrations. No matter what the reason for the strong increase of chloride binding at free [Cl−] > 0.9–1.1 M is, it is noted that the measured chloride binding by the AAS pastes, 10–16 mg/g at free [Cl−] ≈ 0.6 M and 21–28 mg/g at free [Cl−] ≈ 1.2 M, was higher than what has been reported for most conventional cement pastes in NaCl solutions, generally 5–10 mg/g at [Cl−] ≈ 1 M [5,6,7,8,9, 11].
As regards the moderate differences between the chloride binding by the AAS pastes with different activators (Fig. 3), it is likely that these were caused by differences between the kind and amounts of reaction products [18]. The somewhat higher chloride binding by AAS Na2SO4 at free [Cl−] ≤ 0.6 M might be caused by the comparatively high chloride binding capacity of monosulfate (Fig. 1), while differences between the chloride binding at free [Cl−] > 0.9–1.1 M might be related to the amounts of C–(N–)A–S–H in the AAS pastes. Thermodynamic modelling, ideally accompanied by experimental quantification, of the reaction products of the present AAS, and incorporation of the chloride binding isotherms determined in the present experiments and in previous studies would allow this to be investigated in more detail.
5 Conclusions
The chloride binding isotherms and the maximum chloride binding of LDH/AFm phases (monosulfate, strätlingite, hydrotalcite, and meixnerite) in NaOH/NaCl solutions depended strongly on the liquid/solid ratio employed in the chloride binding experiments. At high liquid/solid ratios (l/s ≥ 53 mL/g), the chloride binding capacity of the LDH/AFm phases was similar to their theoretical chloride binding capacity, i.e., assuming that one Cl− ion is taken up in each anion site in the LDH/AFm structure. At low liquid/solid ratios (l/s ≤ 26 mL/g), the maximum chloride binding remained significantly below the theoretical chloride binding capacity up to free [Cl−] ≈ 1.4 M. These findings reconcile previous studies of the chloride binding by LDH/AFm phases, which yielded chloride binding that exceeded the theoretical capacity at high l/s and lower-than-theoretical chloride binding at low l/s.
Thermodynamic modelling showed that in all experiments with the AFm phases (monosulfate and strätlingite), a complete transformation of the AFm phases to Friedel’s salt, i.e., attainment of the full theoretical binding capacity, should be reached within the range of chloride concentrations employed in the experiments. Thus, the observed lower-than-theoretical chloride binding at low l/s has to be assigned to kinetic restrictions. The exact nature of these restrictions remains open at present, and, thus, future studies of this issue are required. Nevertheless, the present results have important implications for the implementation and interpretation of chloride binding experiments on cementitious phases: When implementing such experiments, it must be taken into account that different l/s can lead to different chloride binding isotherms. Considering that chloride binding experiments for cement pastes are usually performed at low l/s, and that chloride binding in real-world concrete elements occurs under even more water-deficient conditions, the present results raise the question how the chloride binding isotherms determined for individual phases should be incorporated into computational models of chloride diffusion in cementitious materials.
The AAS pastes investigated in the present study (activators Na2SiO3, Na2O·1.87SiO2, Na2CO3, and Na2SO4) exhibited chloride binding isotherms whose shape resembled those of conventional cement pastes up to free [Cl−] ≈ 0.9–1.1 M, but with the maximum chloride binding in that concentration range tending to be higher than for conventional cement pastes. At higher chloride concentrations, a steep increase of the chloride binding by all AAS pastes was observed. The cause for this behaviour could not be unequivocally deduced in the present study but may be related to chloride binding by C–(N–)A–S–H. The small to moderate differences between the chloride binding isotherms of the AAS with different activators, with the Na2SO4-activated AAS exhibiting comparatively high chloride binding at free [Cl−] ≤ 0.6 and the sodium silicate-activated AAS pastes demonstrating comparatively high chloride binding at free [Cl−] > 1.0 M, were likely caused by differences between their phase assemblages.
References
Bertolini L, Elsener B, Pedeferri P, Redaelli E, Polder RB (2013) Corrosion of steel in concrete: prevention, diagnosis, repair, 2nd edn. Wiley-VCH, Weinheim
Angst UM (2018) Challenges and opportunities in corrosion of steel in concrete. Mater Struct 51:4. https://doi.org/10.1617/s11527-017-1131-6
Galan I, Glasser FP (2015) Chloride in cement. Adv Cem Res 27:63–97. https://doi.org/10.1680/adcr.13.00067
De Weerdt K, Wilson W, Machner A, Georget F (2023) Chloride profiles: What do they tell us and how should they be used? Cem Concr Res 173:107287. https://doi.org/10.1016/j.cemconres.2023.107287
Tang L, Nilsson L-O (1993) Chloride binding capacity and binding isotherms of OPC pastes and mortars. Cem Concr Res 23:247–254. https://doi.org/10.1016/0008-8846(93)90089-R
Delagrave A, Marchand J, Ollivier J-P, Julien S, Hazrati K (1997) Chloride binding capacity of various hydrated cement paste systems. Adv Cem Based Mater 6:28–35. https://doi.org/10.1016/S1065-7355(97)90003-1
Ipavec A, Vuk T, Gabrovšek R, Kaučič V (2013) Chloride binding into hydrated blended cements: the influence of limestone and alkalinity. Cem Concr Res 48:74–85. https://doi.org/10.1016/j.cemconres.2013.02.010
Machner A, Zajac M, Ben Haha M, Kjellsen KO, Geiker MR, De Weerdt K (2018) Chloride-binding capacity of hydrotalcite in cement pastes containing dolomite and metakaolin. Cem Concr Res 107:163–181. https://doi.org/10.1016/j.cemconres.2018.02.002
De Weerdt K, Colombo A, Coppola L, Justnes H, Geiker MR (2015) Impact of the associated cation on chloride binding of Portland cement paste. Cem Concr Res 68:196–202. https://doi.org/10.1016/j.cemconres.2014.01.027
Ukpata JO, Basheer PAM, Black L (2019) Slag hydration and chloride binding in slag cements exposed to a combined chloride-sulphate solution. Constr Build Mater 195:238–248. https://doi.org/10.1016/j.conbuildmat.2018.11.055
Avet F, Scrivener K (2020) Influence of pH on the chloride binding capacity of Limestone Calcined Clay Cements (LC3). Cem Concr Res 131:106031. https://doi.org/10.1016/j.cemconres.2020.106031
Wilson W, Gonthier JN, Georget F, Scrivener KL (2022) Insights on chemical and physical chloride binding in blended cement pastes. Cem Concr Res 156:106747. https://doi.org/10.1016/j.cemconres.2022.106747
Ismail I, Bernal SA, Provis JL, San Nicolas R, Brice DG, Kilcullen AR, Hamdan S, van Deventer JSJ (2013) Influence of fly ash on the water and chloride permeability of alkali-activated slag mortars and concretes. Constr Build Mater 48:1187–1201. https://doi.org/10.1016/j.conbuildmat.2013.07.106
Ke X, Bernal SA, Hussein OH, Provis JL (2017) Chloride binding and mobility in sodium carbonate-activated slag pastes and mortars. Mater Struct 50:252. https://doi.org/10.1617/s11527-017-1121-8
Zhang J, Shi C, Zhang Z (2019) Chloride binding of alkali-activated slag/fly ash cements. Constr Build Mater 226:21–31. https://doi.org/10.1016/j.conbuildmat.2019.07.281
Florea MVA, Brouwers HJH (2012) Chloride binding related to hydration products. Part I: ordinary Portland cement. Cem Concr Res 42:282–290. https://doi.org/10.1016/j.cemconres.2011.09.016
Baroghel-Bouny V, Wang X, Thiery M, Saillio M, Barberon F (2012) Prediction of chloride binding isotherms of cementitious materials by analytical model or numerical inverse analysis. Cem Concr Res 42:1207–1224. https://doi.org/10.1016/j.cemconres.2012.05.008
Mundra S, Prentice DP, Bernal SA, Provis JL (2020) Modelling chloride transport in alkali-activated slags. Cem Concr Res 130:106011. https://doi.org/10.1016/j.cemconres.2020.106011
Taylor HFW (1997) Cement chemistry, 2nd edn. Thomas Telford, London
Matschei T, Lothenbach B, Glasser FP (2007) The AFm phase in Portland cement. Cem Concr Res 37:118–130. https://doi.org/10.1016/j.cemconres.2006.10.010
Balonis M, Lothenbach B, Le Saout G, Glasser FP (2010) Impact of chloride on the mineralogy of hydrated Portland cement systems. Cem Concr Res 40:1009–1022. https://doi.org/10.1016/j.cemconres.2010.03.002
Bernal SA, Provis JL, Fernández-Jiménez A, Krivenko PV, Kavalerova E, Palacios M, Shi C (2014) Binder chemistry: high-calcium alkali-activted materials. In: Provis JL, van Deventer JSJ (eds) Alkali activated materials: state-of-the-art report, RILEM TC 224-AAM. Springer, Dordrecht, pp 59–92
Myers RJ, Bernal SA, Provis JL (2017) Phase diagrams for alkali-activated slag binders. Cem Concr Res 95:30–38. https://doi.org/10.1016/j.cemconres.2017.02.006
Miyata S (1983) Anion-exchange properties of hydrotalcite-like compounds. Clays Clay Miner 31:305–311. https://doi.org/10.1346/CCMN.1983.0310409
Koritnig S, Süsse P (1975) Meixnerit, Mg6Al2(OH)18·4H2O, ein neues magnesium–aluminium-hydroxid-mineral. Tschermaks Mineral Petrogr Mitt 22:79–87. https://doi.org/10.1007/BF01081303
Myers RJ, Lothenbach B, Bernal SA, Provis JL (2015) Thermodynamic modelling of alkali-activated slag cements. Appl Geochem 61:233–247. https://doi.org/10.1016/j.apgeochem.2015.06.006
Mills SJ, Christy AG, Génin J-MR, Kameda T, Colombo F (2012) Nomenclature of the hydrotalcite supergroup: natural layered double hydroxides. Mineral Mag 76:1289–1336. https://doi.org/10.1180/minmag.2012.076.5.10
Hirao H, Yamada K, Takahashi H, Zibara H (2005) Chloride binding of cement estimated by binding isotherms of hydrates. J Adv Concr Technol 3:77–84. https://doi.org/10.3151/jact.3.77
Ke X, Bernal SA, Provis JL (2017) Uptake of chloride and carbonate by Mg-Al and Ca-Al layered double hydroxides in simulated pore solutions of alkali-activated slag cement. Cem Concr Res 100:1–13. https://doi.org/10.1016/j.cemconres.2017.05.015
Bernard E, Zucha WJ, Lothenbach B, Mäder U (2022) Stability of hydrotalcite (Mg–Al layered double hydroxide) in presence of different anions. Cem Concr Res 152:106674. https://doi.org/10.1016/j.cemconres.2021.106674
Matschei T, Lothenbach B, Glasser FP (2007) Thermodynamic properties of Portland cement hydrates in the system CaO–Al2O3–SiO2–CaSO4–CaCO3–H2O. Cem Concr Res 37:1379–1410. https://doi.org/10.1016/j.cemconres.2007.06.002
Baquerizo LG, Matschei T, Scrivener KL, Saeidpour M, Thorell A, Wadsö L (2014) Methods to determine hydration states of minerals and cement hydrates. Cem Concr Res 65:85–95. https://doi.org/10.1016/j.cemconres.2014.07.009
Baquerizo LG, Matschei T, Scrivener KL, Saeidpour M, Wadsö L (2015) Hydration states of AFm cement phases. Cem Concr Res 73:143–157. https://doi.org/10.1016/j.cemconres.2015.02.011
Henning R, Sturm P, Geddes DA, Keßler S, Walkley B, Gluth GJG (2022) The influence of curing temperature on the strength and phase assemblage of hybrid cements based on GGBFS/FA blends. Front Mater 9:982568. https://doi.org/10.3389/fmats.2022.982568
Miyata S (1980) Physico-chemical properties of synthetic hydrotalcites in relation to composition. Clays Clay Miner 28:50–56. https://doi.org/10.1346/CCMN.1980.0280107
Hibino T, Yamashita Y, Kosuge K, Tsunashima A (1995) Decarbonation behavior of Mg–Al–CO3 hydrotalcite-like compounds during heat treatment. Clays Clay Miner 43:427–432. https://doi.org/10.1346/CCMN.1995.0430405
Mascolo G, Marino O (1980) A new synthesis and characterization of magnesium–aluminium hydroxides. Mineral Mag 43:619–621. https://doi.org/10.1180/minmag.1980.043.329.09
Tongamp W, Zhang Q, Saito F (2007) Preparation of meixnerite (Mg–Al–OH) type layered double hydroxide by a mechanochemical route. J Mater Sci 42:9210–9215. https://doi.org/10.1007/s10853-007-1866-5
Gao W, Li Z (2012) Solubility and KSP of Mg4Al2(OH)14·3H2O at the various ionic strengths. Hydrometallurgy 117–118:36–46. https://doi.org/10.1016/j.hydromet.2012.02.003
Stanimirova T, Piperov N, Petrova N, Kirov G (2004) Thermal evolution of Mg-Al-CO3 hydrotalcites. Clay Miner 39:177–191. https://doi.org/10.1180/0009855043920129
Lothenbach B, Durdziński P, De Weerdt K (2016) Thermogravimetric analysis. In: Scrivener K, Snellings R, Lothenbach B (eds) A practical guide to microstructural analysis of cementitious materials. CRC Press, Boca Raton, pp 177–211
Ke X, Criado M, Provis JL, Bernal SA (2018) Slag-based cement that resists damage induced by carbon dioxide. ACS Sustain Chem Eng 6:5067–5075. https://doi.org/10.1021/acssuschemeng.7b04730
Mundra S, Samson G, Masi G, Achenbach R, Bastidas DM, Bernal SA, Bignozzi MC, Criado M, Cyr M, Gartner N, von Greve-Dierfeld S, Legat A, Nikoonasab A, Provis JL, Raupach M, Gluth GJG (2023) Application of electrochemical methods for studying steel corrosion in alkali-activated materials. Mater Corros 74:988–1008. https://doi.org/10.1002/maco.202313743
Wagner T, Kulik DA, Hingerl FF, Dmytrieva SV (2012) GEM-Selektor geochemical modeling package: TSolMod library and data interface fo multicomponent phase models. Can Mineral 50:1173–1195. https://doi.org/10.3749/canmin.50.5.1173
Kulik DA, Wagner T, Dmytrieva SV, Kosakowski G, Hingerl FF, Chudnenko KV, Berner UR (2013) GEM-Selektor geochemical modeling package: revised algorithm and GEMS3K numerical kernel for coupled simulation codes. Comput Geosci 17:1–24. https://doi.org/10.1007/s10596-012-9310-6
Lothenbach B, Kulik DA, Matschei T, Balonis M, Baquerizo L, Dilnesa B, Miron GD, Myers RJ (2019) Cemdata18: a chemical thermodynamic database for hydrated Portland cements and alkali-activated materials. Cem Concr Res 115:472–506. https://doi.org/10.1016/j.cemconres.2018.04.018
Myers RJ, Bernal SA, Provis JL (2014) A thermodynamic model for C–(N–)A–S–H gel: CNASH_ss. Derivation and validation. Cem Concr Res 66:27–47. https://doi.org/10.1016/j.cemconres.2014.07.005
Ke X (2017) Improved durability and sustainability of alkali-activated slag cements. PhD Thesis, University of Sheffield
Acknowledgements
The authors thank Frank Winnefeld (Empa) very much for making available the tricalcium aluminate used for the synthesis of the monosulfate. Diligent assistance with the laboratory work by Dao Thi Bao Hoa, Solen Garel, Matthias Lindemann, and Claudia Haamkens (BAM) is gratefully acknowledged.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article was prepared within the framework of RILEM TC 283-CAM: Chloride transport in alkali-activated materials. The article has been reviewed and approved by all members of the TC.
TC Membership
Chair: Arnaud Castel
Deputy Chair: Shishir Mundra
Members: Adeyemi Adesina, Muhammed P. A. Basheer, Susan A. Bernal, Maria C. Bignozzi, Yuxin Cai, Frank Collins, Maria Criado, Martin Cyr, Vilma Ducman, Patrycja Duzy, Nina Gartner, Juan Pablo Gevaudan, Ayana Ghosh, Gregor J. G. Gluth, Joost Gulikers, Izabela Hager, Ricky Henning, Petr Hlaváček, R. Doug Hooton, Yuyan Huang, Kazuo Ichimiya, Lesley Suz-Chung Ko, David Law, Wengui Li, Zhenming Li, Qing-Feng Liu, Dirk Lowke, Isabel Martins, Giulia Masi, Sreejith Nanukuttan, Kannan Rajkumar P. R., John L. Provis, Quang Dieu Nguyen, Gabriel Samson, Marijana Serdar, Arezki Tagnit-Hamou, Siska Valcke, Jolien Van Der Putten, Talakokula Visalakshi, Jeanette Visser, Claire White, Frank Winnefeld, Qing Xiang Xiong, Qingliang Yu.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gluth, G.J.G., Mundra, S. & Henning, R. Chloride binding by layered double hydroxides (LDH/AFm phases) and alkali-activated slag pastes: an experimental study by RILEM TC 283-CAM. Mater Struct 57, 72 (2024). https://doi.org/10.1617/s11527-024-02311-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-024-02311-3