
Abstract
Reactive spark plasma sintering has been utilised as a high-throughput processing route for the synthesis of two simulant zirconolite wasteform materials, targeting Ca0.80Ce0.20ZrTi1.60M0.40O7 (M = Fe3+ and Al3+). Materials were processed under 15 MPa uniaxial pressure, with heating/cooling rates of 100 °C/min to 1320 °C, maintained under vacuum. Despite moderate yield (> 80 wt%) of zirconolite-2M, a considerable Ce-rich perovskite phase was formed in both formulations, attributed to complete reduction of the Ce inventory to Ce3+, as determined by Ce L3-edge XANES analysis. The composition charge balanced with Al3+ was favoured on the basis of lower accompanying perovskite fraction.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A strategy combining reuse, immobilisation and disposal has been proposed as a long-term solution for at least some portion of the United Kingdom civil PuO2 inventory, which is forecast to reach ~ 140 teHM (tonnes equivalent heavy metal) once domestic reprocessing operations cease [1]. Prior to placement alongside the existing HLW inventory in a geological disposal facility (GDF) Pu must first be immobilised at the atomic scale in a high-durability matrix, to confer passive safety and improve handling properties. Fundamentally, the primary driver for immobilisation is to prevent Pu migration into the near-field environment over geological timescales such that the overall activity of the waste package at the time of failure is similar to the U ore from which it was derived. On the basis of high chemical durability and radiation stability, crystalline titanate materials (including hollandite, pyrochlore, perovskite, brannerite and zirconolite) are suitable phases for the sequestration of partitioned actinides (Pu, Am, U) and fission products (Cs, Sr) present in many nuclear waste streams. The zirconolite phase (ideally CaZrTi2O7) is considered to be the most suitable matrix for the immobilisation of Pu [2]. The zirconolite-2M parent structure is a derivation of an anion-deficient fluorite superstructure, composed of layered TiO6/TiO5 polyhedra, arranged in a hexagonal-tungsten-bronze (HTB)-type motif interspaced with layers of CaO8/ZrO7 polyhedra, with the overall structure crystallising in the space group C2/c (Z = 8, ρ = 4.47 g/cm3) [3]. The chemical flexibility of the zirconolite phase is one of the primary drivers towards its use as a wasteform as there are five distinct cation receptor sites that are capable of accommodating actinides, neutron poisoning additives (typically Gd3+ and Hf4+ on the basis of compatible oxidation state and ionic radius) and charge compensating species. Typically, larger waste cations such as Pu3+/4+ are most suitably accommodated within the Ca2+ site on the basis of ionic radii, with lower valence charge balancing cations (typically Fe3+ or Al3+) co-substituted in an appropriate molar ratio within the Ti4+ sites. Substitution of tetravalent cations on the Zr4+ site produces a transformation to zirconolite-4M, a superstructure composed of alternating layers of zirconolite-2M and pyrochlore-type modules, resulting in a doubling of the unit cell along the c-axis [4,5,6]. Substitution on the Ca2+ site has been observed, in some instances, to promote the formation of the trigonal variant zirconolite-3T; a number of such solid solutions have been reported in the literature [4, 7,8,9]. In the present study, with Ce utilised as a surrogate for Pu, reactive spark plasma sintering (RSPS) has been deployed as a high-throughput synthesis route to fabricate zirconolite ceramics with the formulation Ca0.80Ce0.20ZrTi1.60M0.40O7 where M = Fe3+ or Al3+. RSPS is a potentially attractive route for the ceramic immobilisation of actinides, given the throughput that can be achieved. A detailed review discussing the application of spark plasma sintering in the context of phosphate wasteform development was recently published [10]. The simultaneous synthesis and consolidation of ceramic materials via RSPS are achieved by rapid DC current pulsing through a compressed powder compact, maintained under uniaxial pressure. This allows the production of monoliths of near theoretical density, even in short processing times, due to heating/cooling rates typically of the order 100 °C/min [11]. The process has previously been demonstrated as suitable for the effective immobilisation of 129I in lead vanadophosphate iodoapatite Pb10(VO4)4.8(PO4)O1.2I2) [12], Cs in hollandite (nominally BaAl2Ti6O16) [13], and more recently Ce in zirconolite [9].
Experimental methodology
Materials synthesis
Oxide powders CaTiO3 (99.9%, Sigma Aldrich), ZrO2 (99.9%, Sigma Aldrich), CeO2 (99.9%, Acros Organics), TiO2 (anatase, 99.9%, Sigma Aldrich), Al2O3 (99.9%, Sigma Aldrich), and Fe2O3 (99.9%, Sigma Aldrich) were calcined at 800 °C for 12 h, prior to addition to a ZrO2-lined milling vessel, in stoichiometric ratios according to the formulations Ca0.80Ce0.20ZrTi1.60Al0.40O7 and Ca0.80Ce0.20ZrTi1.60Fe0.40O7. The mixtures were homogenised using a Fritsch P7 planetary ball mill, with ZrO2 milling media and isopropanol, at 400 rpm for a total of 20 min. Samples were recovered and dried at 80 °C to evaporate excess solvent. Approximately 5 g of each batch was measured and transferred to a 20 mm graphite die, with graphite foil spacers to promote uniform current flow, and compressed under 3 t for approximately 5 min to form loosely bound green bodies. The die was then loaded into a HP-D 1050 SPS system (FCT Systeme GmBH) and ramped at 100 °C/min to 1320 °C, with a constant uniaxial pressure of 15 MPa maintained throughout. A simplified illustration of this configuration is provided in Fig. 1, alongside a photograph of a typical product.

Simplified illustration of RSPS configuration (left) and photograph of a typical ceramic product (right)
Materials characterisation
Post-sintering, pellets were sectioned and prepared for powder X-ray diffraction (XRD) using a Bruker D2 Phaser diffractometer, fitted with Lynxeye position sensitive detector utilising Cu-Kα radiation (λ = 1.5418 Å, Ni filter). Data were collected in the range 10° ≤ 2θ ≤ 80°. Rietveld analysis of powder XRD data was achieved using the Bruker TOPAS package [14]. The true density of each composition was determined by He-gas pycnometry, with material in the powder form. Density measurements were taken using an AccuPyc 1340 II pycnometer, with a total of 20 cycles performed for each specimen under a fill pressure of 12.5 psi. A representative portion of the microstructure was sectioned from the monoliths and prepared for analysis by scanning electron microscopy (SEM) using a Hitachi TM3030 operating at 15 kV accelerating voltage with a working distance of 8 mm. This was fitted with a Bruker Quantax Elemental Dispersive X-ray Spectrometer (EDS) for semi-quantitative compositional analysis. Samples were prepared for SEM–EDS by cold setting in epoxy resin, prior to polishing to a 1 μm optical finish and coating with a conductive amorphous carbon layer to reduce surface-charging effects. Ce L3-edge X-ray absorption near edge spectroscopy (XANES) data were collected at room temperature at Beamline BL-27 at the Photon Factory (PF) accelerator light source (Tsukuba, Japan). The optical arrangement is composed of a double Si(111) monochromator, with slits used to reduce the beam footprint to 3 × 1 mm. Samples were prepared for XANES analysis by suspension of sufficient material to form one absorption length in an inert, low absorbance binder (polyethylene glycol, PEG) in pressed 13 mm discs. Spectra were collected alongside a number of reference compounds of known Ce oxidation state and analysed using the Athena component of the Demeter software package [15].
Results and discussion
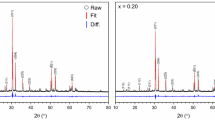
Analysis of powder XRD data (Fig. 2) revealed, for both Al3+ and Fe3+-doped compositions, intense reflections consistent with the zirconolite-2M polytype occupying the dominant fraction of the phase assemblage. When targeting Ca0.80Ce0.20ZrTi1.60Al0.40O7, a number of secondary reflections were identified, the most prominent of which was observed at 2θ = 33.1°, attributed to the (112) reflection of perovskite (nominally CaTiO3). Additional weaker reflections consistent with Al2O3 and ZrO2 were also distinguished. Quantitative phase analysis, derived from Rietveld profile fitting (Table 1) indicated that a yield of 83.5 ± 0.3 wt% zirconolite-2M was obtained. Unit cell parameters for the Al3+ formulation were in good agreement with previous observations, calculated to be: a = 12.4903(5) Å, b = 7.2487(3) Å, c = 11.3762(4) Å, β = 100.656(3)° and V = 1012.22(7) Å3. A greater relative proportion (14.5 ± 0.6 wt%) of perovskite was observed for the Fe3+ formulation demonstrating that, under the given processing conditions, Al3+ performed as a more effective charge compensation cation relative to Fe3+. Rietveld refinement produced unit cell parameters for the Fe3+-doped zirconolite-2M phase that were largely consistent with the Al3+ composition: a = 12.4764(5) Å, b = 7.2634(3) Å, c = 11.4745(4) Å, β = 100.368(2)° and V = 1022.85(7) Å3, yet with a notable increase in unit cell volume. This may be attributed to the ionic radii of Al3+ (53 pm) and Fe3+ (63 pm) relative to Ti4+ (68 pm).

Rietveld analysis of powder diffraction data for Ca0.80Ce0.20ZrTi1.60Al0.40O7 (left) and Ca0.80Ce0.20ZrTi1.60Fe0.40O7 (right) formulations (red line, calculated patterns; black circles, observed patterns; blue line, difference profile). Vertical tick marks indicate theoretical position of zirconolite-2M reflections. Observed zirconolite-2M reflections are labelled with (hkl) values. Perovskite reflections are labelled with closed circles (●)
A representative portion of each microstructure is labelled and displayed in Fig. 3. It was clear that both specimens formed with a fine-grained, high-density microstructure. He-gas pycnometry determined the true density of the formed products to be 4.6071 ± 0.0038 and 4.7976 ± 0.0115 g/cm3 for Al3+ and Fe3+ compositions, respectively. It was clear that when synthesising materials targeting both Al3+ and Fe3+ charge compensation, a moderately heterogeneous phase assemblage was formed, with apparent phase distributions consistent with quantitative phase analyses derived from the Rietveld fitting. Upon inspection of the materials batched as Ca0.80Ce0.20ZrTi1.60Al0.40O7, clear islands of ZrO2 and Al2O3 were present, with regions of fine-grained Ce-substituted CaTiO3 clearly distinguished by complementary EDS phase analysis (not shown). Similar observations were made for the Fe3+-substituted sample; it was also clear that the Ce-perovskite occupied a greater proportion of the microstructure, in agreement with the Rietveld analysis of powder diffraction data discussed above. EDS analysis of perovskite grains present in the material batched as Ca0.80Ce0.20ZrTi1.60Fe0.40O7 demonstrated uptake of a significant portion of Ce (Fig. 4). It was clear in early studies on SYNROC (SYNthetic-ROCk) corrosion that perovskite was the least durable phase in the conventional SYNROC-C wasteform under dissolution conditions [16]. Therefore, partial sequestration of the Pu surrogate fraction within the perovskite phase would be expected to significantly decrease the overall durability of such wasteforms, given the lower chemical durability of this phase relative to the targeted zirconolite matrix. In a recent publication, we demonstrated that, when carefully dissolved under aggressive leaching media, the extent to which a Ce surrogate inventory was extracted into solution could be decreased by a factor of 100 through altering the batch formulation to precluding the formation of an ancillary perovskite phase [17].

Backscattered electron micrograph for representative sections of materials batched targeting the compositions Ca0.80Ce0.20ZrTi1.60Al0.40O7 (left) and Ca0.80Ce0.20ZrTi1.60Fe0.40O7 (right)

EDS analysis of a perovskite cluster present in the material targeting Ca0.80Ce0.20ZrTi1.60Fe0.40O7, presenting a signal corresponding to the Ce Lα emission line
In order to determine the prevalent Ce oxidation state, Ce L3-XANES data were collected alongside the reference compounds Ce4+O2 and Ce3+PO4 (Fig. 5). Both RSPS samples exhibited a single intense asymmetric white line feature consistent with uniform Ce3+ speciation when compared qualitatively with the CePO4 reference compound (in which Ce3+ is ninefold coordinated to oxygen). No features consistent with Ce4+ were presented, confirming that the reducing environment imposed by the graphite SPS die was sufficient to completely reduce the available Ce4+ inventory to Ce3+, and subsequently, promote Ce substitution within the perovskite phase. Moreover, linear combination analysis of Ce L3-XANES data using the spectra of reference compounds was consistent with 100% Ce3+ speciation in the ceramic formulations. These data are consistent with previous studies, wherein sintering of Ce-substituted zirconolite under reducing conditions resulted in the formation of a significant perovskite fraction [18].

Above: Ce L3-edge XANES spectra of materials targeting Ca0.80Ce0.20ZrTi1.60Al0.40O7 (labelled as Al) and Ca0.80Ce0.20ZrTi1.60Fe0.40O7 (labelled as Fe). Below: spectra of the CeO2 and CePO4 reference compounds
Conclusions
Reactive spark plasma sintering has been utilised to synthesise two synthetic zirconolite wasteform materials, targeting Ca0.80Ce0.20ZrTi1.60Al0.40O7 and Ca0.80Ce0.20ZrTi1.60Al0.40O7, with Ce included as a Pu surrogate. Despite the rapid (< 1 h total processing time per sample) formation of high-density ceramic monoliths, a significant portion of the phase assemblage was composed of Ce-rich perovskite, the stability of which was attributed to the complete reduction of the available Ce inventory to Ce3+. These data form a useful contribution towards ongoing efforts to design suitable wasteform compositions for Pu immobilisation and optimisation of processing routes for these materials.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
N.C. Hyatt, Safe management of the UK separated plutonium inventory: a challenge of materials degradation. NPJ Mater. Degrad. 4, 28 (2020)
L.R. Blackburn, N.C. Hyatt, Actinide immobilisation in dedicated wasteforms: an alternative pathway for the long-term management of existing actinide stockpiles, in Encyclopedia of Nuclear Energy, 1st edn., ed. by E. Greenspan (Elsevier, Amsterdam, 2021), pp. 650–662
L.R. Blackburn et al., Review of zirconolite crystal chemistry and aqueous durability. Adv. Appl. Ceram. 120(2), 1–15 (2021)
E.R. Vance et al., Incorporation of uranium in zirconolite (CaZrTi2O7). J. Am. Ceram. Soc. 85(7), 1853–1859 (2002)
B.D. Begg, R.A. Day, A. Brownscombe, Structural effect of Pu substitutions on the Zr-site in zirconolite. Mater. Res. Soc. Symp. Proc. 663, 1–8 (2001)
L.R. Blackburn, S. Sun, L.J. Gardner, E.R. Maddrell, M.C. Stennett, N.C. Hyatt, A systematic investigation of the phase assemblage and microstructure of the zirconolite CaZr1-xCexTi2O7 system. J. Nucl. Mater. 535, 152137 (2020)
M.-X. Zhong et al., Synthesis of Ca1-xCexZrTi2–2xAl2xO7 zirconolite ceramics for plutonium disposition. J. Nucl. Mater. 556, 153198 (2021)
M.R. Gilbert et al., Synthesis and characterisation of Pu-doped zirconolites: (Ca1-xPux)Zr(Ti2–2xFe2x)O7, in IOP Conf. Ser. Mater. Sci. Eng., vol. 9, no. 012007, 2010.
L.R. Blackburn et al., Synthesis and characterisation of Ca1-xCexZrTi2-2xCr2xO7: analogue zirconolite wasteform for the immobilisation of stockpiled UK plutonium. J. Eur. Ceram. Soc. 40(15), 5909–5919 (2020)
A.I. Orlova, Crystalline phosphates for HLW immobilization—composition, structure, properties and production of ceramics. Spark plasma sintering as a promising sintering technology. J. Nucl. Mater. 559, 153407 (2022)
M. Suarez et al., Challenges and opportunities for spark plasma sintering: a key technology for a new generation of materials, in Sintering Applications, 2013.
L. Campayo et al., Relevance of the choice of spark plasma sintering parameters in obtaining a suitable microstructure for iodine-bearing apatite designed for the conditioning of I-129. J. Nucl. Mater. 457, 63–71 (2015)
B.M. Clark, P. Tumurugoti, S.K. Sundaram, J.W. Amoroso, J.C. Marra, K.S. Brinkman, Microstructures of melt-processed and spark plasma sintered ceramic waste forms. Metall. Mater. Trans. E 1E, 341–348 (2014)
A.A. Coelho, J. Evans, I. Evans, A. Kern, S. Parsons, The TOPAS symbolic computation system. Powder Diffr. 26(S1), S22–S25 (2011)
B. Ravel, M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005)
K.L. Smith, G.R. Lumpkin, M.G. Blackford, R.A. Day, K.P. Hart, The durability of synroc. J. Nucl. Mater. 190, 287–294 (1992)
L.R. Blackburn et al., “Influence of accessory phases and surrogate type on accelerated leaching of zirconolite wasteforms. npj Mater. Degrad. 5(24), 1–11 (2021)
B.M. Clark, S.K. Sundaram, S.T. Misture, Polymorphic transitions in cerium-substituted zirconolite (CaZrTi2O7). Sci. Rep. 7(1), 2–10 (2017)
N.C. Hyatt, C.L. Corkhill, M.C. Stennett, R.J. Hand, L.J. Gardner, C.L. Thorpe, The HADES facility for high activity decommissioning engineering & science: part of the UK National Nuclear User Facility, in IOP Conf. Series: Materials Science and Engineering, 2020, vol. 818, pp. 1–8.
Acknowledgments
We acknowledge financial support from the Nuclear Decommissioning Authority (NDA) and EPSRC under Grant Numbers EP/S01019X/1, EP/N017870/1 and EP/R511754/1. This research utilised the HADES/ MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/1 [19]. Collection of the Ce L3-edge XAS data was performed under the approval of the Photon Factory Advisory Committee (Proposal No. 2019G586); the support of Yoshihiro Okamoto (Japanese Atomic Energy Agency) and Noriko Usami (The High Energy Accelerator Research Organisation—Kō Enerugī Kasokuki Kenkyū Kikō) during the experiment is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aldean, I., Sun, SK., Wilkins, M.C.D. et al. Synthesis and characterisation of Ce-doped zirconolite Ca0.80Ce0.20ZrTi1.60M0.40O7 (M = Fe, Al) formed by reactive spark plasma sintering (RSPS). MRS Advances 7, 75–80 (2022). https://doi.org/10.1557/s43580-022-00221-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43580-022-00221-6