
Abstract
A plug flow reactor was constructed to scale the synthesis of metastable, phthalaldehyde-based polymers to achieve production rates of 1–2 kg per day. The flow-induced mixing and in-line polymerization quench and precipitation sequences resulted in improved polymer purity and long-term stability compared to the same materials made from a conventional batch process. Cryogenic rheology was used to probe the complex fluid dynamics encountered during the polymerization of PPA homopolymers. It is envisioned that this continuous flow manufacturing approach could be extended to other low ceiling temperature or aldehyde monomer systems to help implement and support a plastic circular economy.
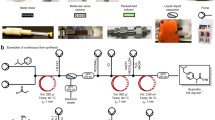
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
The extensive use of synthetic polymers in our lives combined with the lack of robust recycling processes for post-consumer commodity plastics has led to a global plastic waste crisis.[1] Researchers have explored various methods to reuse and recycle polymeric materials to promote a plastic circular economy, including mechanical recycling,[2] valorization,[3] and designing polymers for chemical recycling.[4,5,6]
Polyaldehydes are a promising class of chemically recyclable polymers formed through the chain growth polymerization of aldehyde functional groups. Due to a comparatively low enthalpy of polymerization, the ceiling temperature (Tc) of aldehydes tend to be in the range of − 60 to 60°C, which enables facile depolymerization of the polymers from the solid-state back to their intrinsic monomers in high yields and purity.[7] This behavior makes polyaldehydes suitable candidates to help support a plastic circular economy, although the properties of said polymers, especially toxicity and environmental threats, need to be considered before placing any material in-service. In addition, recent efforts have demonstrated that some aldehydes can be sustainably sourced from renewable, biosynthetic routes.[8,9]
Poly(o-phthalaldehyde) (PPA) has been extensively studied[10] as a resist material for optical and thermal lithography,[11,12,13,14] degradable microcapsules,[15,16,17] and polymer fibers.[18,19] PPA synthesized from cationic polymerizations has a cyclic chain topology and has been shown to possess good shelf-stability even though it is only metastable at ambient conditions.[20,21] Phthalaldehyde has a Tc = − 36°C that necessitates solvents and cryogenic cooling for polymerization to achieve high conversion and molecular weight, which complicates the scale-up of this promising material.[15,16,23]
In this work, we improved the scalability of PPA synthesis from benchtop to industrial manufacturing by developing a continuous flow reactor which can be scaled to high capacity. For the first time, PPA rheology was performed at cryogenic temperatures in dichloromethane (DCM) to gain insight on the fluid dynamics of the polymerization solution at low temperature. These findings provide critical insights into developing polymerization reactors for polyaldehydes and other low Tc monomer systems that are being considered for a plastics circular economy.
Previous research using a microflow reactor has shown that PPA has rapid polymerization kinetics.[24] This concept was extended in this study to show that a plug flow reactor may be suitable for scalable PPA synthesis. The plug flow reactor reported here was constructed inside of a dry ice chest that provided good insulation to help maintain adiabatic conditions at a cryogenic temperature. Stainless-steel pipe (316L, Sulf-inert®) and union-T joints with an outer diameter of 6.35 mm were used as the reaction vessel, Scheme 1. Additional details about the reactor design and construction can be found in the Supporting Information.

(a) Synthesis scheme of phthalaldehyde-based polymers and (b) schematic of the flow reactor used in this investigation.
Monomer, catalyst (0.8 mM boron trifluoride diethyl etherate in DCM), and quenching (0.1 M pyridine in tetrahydrofuran) solutions were prepared in a nitrogen-purged glovebox and stored in 1-L glass bottles with holes for PTFE tubing during operation. Peristaltic pumps were used to pump and mix the reactant solutions (i.e., monomer, catalyst, and quenching agent) into the stainless-steel reactor. The solution reservoirs were kept at room temperature throughout operation, and an inert gas manifold was used to provide a dry nitrogen atmosphere in the headspace. To operate the reactor, first a dry ice and acetone bath at − 78°C filled the dry ice chest containing the reactor. The reactant solutions were pumped at equivalent flow rates into the cold reactor to prevent backflow due to pressure differences. The reactant solution lines were afforded some length to drop in temperature before being mixed at the point where the inlet tubes merged at a union-T joint. The residence time within the reactor was set by the volumetric flowrate of the peristaltic pumps and volume of the reaction zone. The outlet of the reactor was fed into a 3 times volumetric excess of methanol to precipitate and separate the PPA polymer from the catalysts and unreacted monomer. This precipitation solution was stirred for 15 min after the run ended followed by filtration of the solid product. The polymerizations reported here used about 30 g monomer in the feed per sample. Larger volumes of polymer could be synthesized simply by continuously refilling the solution reservoirs that feed into the reactor.
Initially, the flow reactor was operated with only phthalaldehyde in the monomer feed (i.e., homopolymerization) to compare to our previous microflow reactor results. It was found that the initial monomer concentration in the feed required more dilute conditions than used in previous microflow reactor studies ([M]o = 0.746 M) to reduce the solution viscosity within the 20.4-m-long reactor tube. To avoid potentially stalling the pumps, the initial monomer concentration was chosen to be below the critical ‘chain fusion’ concentration ([M]o > 0.5 M) that promotes formation of high molecular weight PPA.[24] These polymerizations were performed with a monomer feed of 0.8 M phthalaldehyde in DCM that is diluted with an equal volume from the catalyst feed at a molar ratio of 1:500 (catalyst to monomer). After this dilution, the resulting initial monomer concentration was 0.4 M and an expected equilibrium conversion of ca. 85%, based on documented equilibrium monomer conversion data.[22,23]
Table I shows the homopolymerization data from 7.5- to 15-min residence times that resulted in polymer molecular weights of 46 and 65 kg/mol, respectively. While these conditions are not identical to those used in the microflow reactor study, the kinetics are still rapid and correspond to a throughput of 66–82 g/hr for this new cryogenic reactor. The polymer production rate was higher for the faster pumping speeds; however, this is at the expense of yield. Optimization studies are underway to find the operating conditions for maximum throughput.
The lower initial monomer concentration used in this reactor is not the sole cause for the reduction in kinetics and yield, shown in Table I. This reactor design mixed the catalyst with the monomer after chilling both to − 78°C that results in lower apparent polymerization rates compared to catalyst mixing at room temperature.[24] Another consideration is that the reactor surface area-to-volume ratio is only 17% of that used in the previous microflow reactor, which reduces the heat transfer rate for the feed solutions. Furthermore, there are likely stochastic events associated with the use of this reactor, specifically start-up losses due to residual nucleophilic impurities (e.g., water) introduced into the reactor between samples that affect run-to-run consistency. This suggests that continual operation of the reactor would improve the overall yield and quality of PPA polymer produced. While these results are slower than the microflow reactor, they still present an improvement compared to conventional batch synthesis that requires long times to chill large volumes to − 78°C and has more opportunities to introduce operator error.
At ambient conditions, PPA is metastable and susceptible to acid- or base-catalyzed hydrolysis of the acetal backbone that results in spontaneous transformation of the polymer to monomer. The long-term stability of PPA is a critical metric for any scalable manufacturing route. To evaluate the long-term stability of the PPA, samples were subjected to accelerated aging in an oven at 45°C to determine their relative stability after synthesis and purification. A PPA batch polymerization sample was prepared in a similar manner as a control. The batch-polymerized PPA control remained stable (at 45°C) for 42 days, which is typical for high-quality, single-step-purified PPA. The PPA samples produced from the new flow reactor were stable at 45°C for more than 82 days, far exceeding the stability of typical batch-prepared samples. It is likely that the intimate mixing of the quencher solution with the product stream in the new flow reactor is a more effective means of residual catalyst removal, which has been identified as the primary cause of premature depolymerization.[21,25]
Cryogenic rheology was performed to evaluate the solution viscosity within the reactor during polymerization. Due to the water sensitivity of the polymerization, it was not possible to determine the increase in viscosity in the rheometer as the polymerization proceeded. Instead, polymer that was previously isolated and purified was redissolved in DCM at the desired polymerization concentration (1 g per 10 mL), which corresponds to the quantitative yield of the reactor. This represents the maximum possible viscosity expected during polymerization of high molecular weight PPA, which is the long-term manufacturing goal. A series of molecular weight PPA samples (32, 158, 240 kg/mol) were chosen to provide insight into the effect of molecular weight on viscosity inside the flow reactor. The samples were subjected to a temperature ramp of 2°C/min from 25 to − 90°C using a strain of 0.1% and a frequency of 1 Hz, followed by a frequency sweep from 0.1 to 10 Hz at 25°C using a strain of 0.1%, Figure 1. Additional information about the rheological experiments can be found in Supporting Information.

(a) Temperature- and (b) frequency-dependent data at 25°C (storage and loss moduli given by solid and dotted lines, respectively) for PPA homopolymers with molecular weights of 32, 158, and 240 kg/mol dissolved at 1 g per 10 mL in DCM.
All the PPA samples behaved like viscoelastic solids with a tan δ less than 1 in the temperature range tested. At room temperature, the samples showed a positive trend between molecular weight and viscosity as expected from increased polymer chain entanglement. As the temperature was lowered, the results showed more complex behavior. Below − 30°C, the 158 kg/mol sample has the highest viscosity, and below − 65°C, the 240 kg/mol sample has a viscosity even lower than the 32 kg/mol sample. Cooling below − 78°C also saw the viscosity of the 158 kg/mol sample become lower than the 32 kg/mol sample. One possible explanation for this behavior is the DCM solvent transitions from a good solvent to a poor solvent for high molecular weight PPA at low temperatures, which causes the high molecular weight PPA to agglomerate or precipitate out of solution resulting in a lower viscosity. This phenomenon has not been observed during batch polymerizations in glass flasks, but the rheology experiments presented here lack the unreacted monomer and polymerization catalyst in solution that could alter the local environment and polymer solubility. The physical origins of these trends are the subject of ongoing investigations. While a plug flow reactor is feasible to produce high molecular weight PPA, other reactor modifications that improve the level of mixing (e.g., in-line mixers) may further benefit the kinetics and energy requirements.
Copolymerizing PPA with aromatic and aliphatic aldehyde monomers has been shown to introduce new physical and chemical functionality.[26,27] The flow reactor was used to synthesize copolymers of phthalaldehyde (oPA) and propanal (PA). These copolymerizations are not amenable to catalyst conditioning at room temperature and require strict isolation until the reactants are fully chilled; otherwise, the Lewis acid polymerization catalyst will also catalyze the formation of a PA trimer that does not participate in the oPA-PA copolymerization.[22,27] The monomer feed to the reactor was a 0.8 M solution of 60 mol% oPA and 40 mol% PA, and was diluted with the catalyst feed at equal volume to the monomer feed and contained enough catalyst to maintain the molar ratio of 1:500 (catalyst to monomer). The residence time was adjusted to determine the yield and incorporation of the PA comonomer. Figure 2 shows the yield and PA incorporation (in mol%) versus residence time. Tabulated results can be found in the Supporting Information (Table S2).

(a) PA content in copolymer, (b) gravimetric yield, and (c) molecular weight and molar mass dispersity for a series of oPA-PA copolymers with different residence times in the reactor.
Once sufficient time was provided for the target PA copolymer to form, the resulting material was remarkably consistent, as shown in Figure 2. Although mol% PA in the product was lower than the 15 mol% identified from small batch experiments, the PA content was consistently 13 ± 1 mol%. It is suspected that the sample with < 10 min residence time did not fully cool to − 78°C before the monomer and catalyst feeds were mixed, which resulted in the low PA content and yield. This problem is unique to the copolymerization system due to the PA trimer byproduct formation at elevated temperatures. Similarly, the target molecular weight and distributions were highly stable at 15 ± 2 kg/mol and Ð = 2.05 ± 0.06. These flow reactor results are equally or more consistent than conventional batch polymerizations, especially given the scales involved in these sensitive, cationic polymerizations.
Interestingly, the yield lagged the incorporation of the PA comonomer after a significant induction period of ~ 30 min. For small batch experiments synthesized in laboratory glassware, the copolymerization is known to equilibrate within 60 min at a [M]o = 0.746 M. A residence time of ~ 70 min was required to reach equilibrium copolymer yield, due to the more dilute monomer concentrations (0.4 M) used in the flow reactor used in this study. A short induction period for PPA homopolymerization has been previously observed.[24] The results reported here are the first to quantify PPA copolymerization kinetics which include a much longer induction period compared to the PPA homopolymer kinetics. The extrapolated throughput from these runs is 27 g/hr, which is lower than the homopolymerization due to the lower equilibrium conversion (40% of the PA is not expected to react) and slower polymerization kinetics.
Previous studies suggest that the incorporation of PA into the copolymer is achieved via propagation at the chain end, and not due to chain transfer.[27] It was previously understood that the PA trimer was an unintended and undesirable byproduct that could only be avoided by polymerizing at a temperature far below the Tc of the coaldehyde-phthalaldehyde mixture. This understanding is consistent with the results shown in the incorporation versus residence time plot. It is likely that polymer chains are fully formed, meaning molecular weight and PA content, before additional chains are initiated. These additional chains then achieve the similar composition and yield given sufficient time. It is unknown whether the activity of a growing polymer chain decreases with PA incorporation or if there are some other thermodynamic or kinetic phenomena driving the statistical nature of a catalyst to continue propagating a chain or dissociating and initiating a new chain. Furthermore, there is no sign that chain transfer reactions are occurring in this copolymer system. Chain transfer reactions have been observed in the homopolymerization, which drastically affect the final molecular weight.[24] Copolymer molecular weight is strongly influenced by the starting monomer concentration and comonomer loading in the feed.[27]
With respect to long-term stability, the oPA/PA copolymers obtained from the flow reactor behaved like the PPA homopolymer samples under accelerated aging conditions. All samples lasted at least 78 days at 45°C before observing depolymerization, which far exceeded the stability of homopolymer or copolymer samples synthesized in a batch reactor. This improved stability is likely due to the improved purification methods in the flow reactor compared to the batch process.
The results presented here have important implications for the scalable manufacturing of chemically recyclable polyaldehydes. A plug flow reactor was constructed to scale the synthesis of metastable, phthalaldehyde-based polymers to achieve production rates of 1–2 kg per day. The flow-induced mixing and combining an in-line polymerization quench and precipitation greatly improved the purification and long-term stability of these polymers when compared to samples made from conventional batch syntheses. Cryogenic rheology was used to probe the complex fluid dynamics encountered during the polymerization of PPA homopolymers. We envision that this continuous flow manufacturing approach could be further extended to other low Tc or aldehyde-based monomer systems to help implement and support a plastic circular economy.
Data availability
The data that support the findings of this study are included in an electronic Supporting Information and available from the corresponding author upon reasonable request.
References
M. Ilyas, W. Ahmad, H. Khan, S. Yousaf, K. Khan, S. Nazir, Plastic waste as a significant threat to environment—a systematic literature review. Rev. Environ. Health 33(4), 383–406 (2018). https://doi.org/10.1515/reveh-2017-0035
Z.O.G. Schyns, M.P. Shaver, Mechanical recycling of packaging plastics: A review. Macromol. Rapid Commun. 2000415, 1–27 (2020). https://doi.org/10.1002/marc.202000415
H. Zhou, Y. Wang, Y. Ren, Z. Li, X. Kong, M. Shao, H. Duan, Plastic waste valorization by leveraging multidisciplinary catalytic technologies. ACS Catal. 12(15), 9307–9324 (2022). https://doi.org/10.1021/acscatal.2c02775
M. Hong, E.Y.X. Chen, Chemically recyclable polymers: A circular economy approach to sustainability. Green Chem. 19(16), 3692–3706 (2017). https://doi.org/10.1039/c7gc01496a
A. Rahimi, J.M. García, Chemical recycling of waste plastics for new materials production. Nat. Rev. Chem. 1(6), 0046 (2017). https://doi.org/10.1038/s41570-017-0046
G.W. Coates, Y.D.Y.L. Getzler, Chemical recycling to monomer for an ideal circular polymer economy. Nat. Rev. Mater. 5(7), 501–516 (2020). https://doi.org/10.1038/s41578-020-0190-4
P. Kubisa, K. Neeld, J. Starr, O. Vogl, Polymerization of higher aldehydes. Polymer (Guildf) 21(12), 1433–1447 (1980). https://doi.org/10.1016/0032-3861(80)90145-7
J. Zhou, Z. Chen, Y. Wang, Bioaldehydes and beyond: Expanding the realm of bioderived chemicals using biogenic aldehydes as platforms. Current Opin. Chem. Biol. (2020). https://doi.org/10.1016/j.cbpa.2020.04.007
I. Ullah, A.L. Khan, L. Ali, A.R. Khan, M. Waqas, J. Hussain, I.J. Lee, J.H. Shin, Benzaldehyde as an insecticidal, antimicrobial, and antioxidant compound produced by Photorhabdus temperata M1021. J. Microbiol. 53(2), 127–133 (2015). https://doi.org/10.1007/s12275-015-4632-4
F. Wang, C.E. Diesendruck, Polyphthalaldehyde: synthesis, derivatives, and applications. Macromol. Rapid Commun. 39(2), 1–21 (2018). https://doi.org/10.1002/marc.201700519
A. Engler, C. Tobin, C.K. Lo, P.A. Kohl, Influence of material and process parameters in the dry-development of positive-tone. Polyaldehyde Photoresist. J Mater Res 35(21), 2917–2924 (2020). https://doi.org/10.1557/jmr.2020.243
P.C. Paul, A.W. Knoll, F. Holzner, M. Despont, U. Duerig, Rapid turnaround scanning probe nanolithography. Nanotechnology 22(27), 275306 (2011). https://doi.org/10.1088/0957-4484/22/27/275306
J. Deng, S. Bailey, S. Jiang, C.K. Ober, High-performance chain scissionable resists for extreme ultraviolet lithography: Discovery of the photoacid generator structure and mechanism. Chem. Mater. 34(13), 6170–6181 (2022). https://doi.org/10.1021/acs.chemmater.2c01444
H. Ito, C.G. Willson, Chemical amplification in the design of dry developing resist materials. Polym. Eng. Sci. 23(18), 1012–1018 (1983). https://doi.org/10.1002/pen.760231807
A.M. Dilauro, A. Abbaspourrad, D.A. Weitz, S.T. Phillips, Stimuli-responsive core-shell microcapsules with tunable rates of release by using a depolymerizable poly (phthalaldehyde) membrane. Macromolecules 46(9), 3309–3313 (2013). https://doi.org/10.1021/ma400456p
V. Eriksson, M. Andersson Trojer, S. Vavra, M. Hulander, L. Nordstierna, Formulation of polyphthalaldehyde microcapsules for immediate UV-light triggered release. J. Colloid Interface Sci. 579, 645–653 (2020). https://doi.org/10.1016/j.jcis.2020.06.024
M.J. Warner, J.P. Lassa, H. Narcross, A. Commisso, K. Ghosh, M. Romero, J.M. Schwartz, A.C. Engler, P.A. Kohl, S.C. Leguizamon, B.H. Jones, Chemical recycling of polybutadiene rubber with tailored depolymerization enabled by microencapsulated metathesis catalysts. ACS Sustain Chem Eng 11(39), 14538–14548 (2023). https://doi.org/10.1021/acssuschemeng.3c03907
S. Li, M.H. Rizvi, B.B. Lynch, J.B. Tracy, E. Ford, Flexible cyclic-poly(phthalaldehyde)/poly(ε-caprolactone) blend fibers with fast daylight-triggered transience. Macromol. Rapid Commun. 2000657, 1–7 (2021). https://doi.org/10.1002/marc.202000657
C. Shi, A. Leonardi, Y. Zhang, P. Ohlendorf, A. Ruyack, A. Lal, C.K. Ober, UV-triggered transient electrospun poly(propylene carbonate)/poly(phthalaldehyde) polymer blend fiber mats. ACS Appl. Mater. Interfaces 10(34), 28928–28935 (2018). https://doi.org/10.1021/acsami.8b06051
J.A. Kaitz, C.E. Diesendruck, J.S. Moore, End group characterization of poly(phthalaldehyde): Surprising discovery of a reversible, cationic macrocyclization mechanism. J. Am. Chem. Soc. 135(34), 12755–12761 (2013). https://doi.org/10.1021/ja405628g
J.M. Schwartz, O. Phillips, A. Engler, A. Sutlief, J. Lee, P.A. Kohl, Stable, high-molecular-weight poly(phthalaldehyde). J. Polym. Sci. A Polym. Chem. 55(7), 1166–1172 (2017). https://doi.org/10.1002/pola.28473
J.M. Schwartz, A. Engler, O. Phillips, J. Lee, P.A. Kohl, Determination of ceiling temperature and thermodynamic properties of low ceiling temperature polyaldehydes. J. Polym. Sci. A Polym. Chem. 56(2), 221–228 (2018). https://doi.org/10.1002/pola.28888
J.P. Lutz, O. Davydovich, M.D. Hannigan, J.S. Moore, P.M. Zimmerman, A.J. McNeil, Functionalized and degradable polyphthalaldehyde derivatives. J. Am. Chem. Soc. 141(37), 14544–14548 (2019). https://doi.org/10.1021/jacs.9b07508
A. Engler, P.A. Kohl, Kinetic investigation on the cationic polymerization of O-phthalaldehyde: Understanding ring-expansion polymerization. Macromolecules 53(5), 1543–1549 (2020). https://doi.org/10.1021/acs.macromol.9b02506
A.M. Feinberg, H.L. Hernandez, C.L. Plantz, E.B. Mejia, N.R. Sottos, S.R. White, J.S. Moore, Cyclic poly(phthalaldehyde): Thermoforming a bulk transient material. ACS Macro Lett. 7(1), 47–52 (2018). https://doi.org/10.1021/acsmacrolett.7b00769
J.A. Kaitz, J.S. Moore, Functional phthalaldehyde polymers by copolymerization with substituted benzaldehydes. Macromolecules 46(3), 608–612 (2013). https://doi.org/10.1021/ma302575s
A. Engler, O. Phillips, R.C. Miller, C. Tobin, P.A. Kohl, Cationic copolymerization of O-phthalaldehyde and functional aliphatic aldehydes. Macromolecules 52(11), 4020–4029 (2019). https://doi.org/10.1021/acs.macromol.9b00740
Funding
This work was funded by America’s Seed Fund through the National Science Foundation SBIR program under award number 2231988. We acknowledge collaboration with Sandia National Laboratories, a multimission laboratory managed and operated by the National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA-0003525, via a Laboratory Directed Research and Development program.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Reactor construction and data collection were performed by J.M.S. All authors contributed to analyzing data and writing the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
J.M.S, P.A.K., and A.E. own stock in a start-up company commercializing polyaldehyde copolymers.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schwartz, J.M., Kohl, P.A. & Engler, A. Scaling the polymerization of polyaldehydes through continuous flow synthesis. MRS Communications (2024). https://doi.org/10.1557/s43579-024-00608-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1557/s43579-024-00608-6