
Abstract
CsPbBr3 represents a highly attractive material for perovskite light-emitting diodes (PeLEDs) in the green spectral range. However, the lack of deposition tools for reproducible and scalable growth of perovskite films is one of the major obstacles hindering PeLED commercialization. Here, we employ the highly scalable showerhead-assisted chemical vapor deposition (CVD) method to produce uniform pinhole-free CsPbBr3 films for PeLED application. The precursors CsBr and PbBr2 are evaporated under low vacuum in N2 carrier gas. By adjusting the PbBr2 sublimation temperature, process conditions for CsBr-rich, stoichiometric, and PbBr2-rich CsPbBr3 layer growth have been developed. A substrate temperature of 160 °C enables direct growth of these CsPbBr3 films on a polymeric hole transport layer (HTL), finally yielding PeLEDs with a maximum luminance of 125 cd/m2. Although the device efficiency still lags behind solution-processed counterparts, our approach presents the first demonstration of PeLEDs containing CsPbBr3 films processed in a perovskite showerhead-assisted CVD reactor.
Graphic abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Beyond photovoltaics, which has been the main targeted market of metal halide perovskites in the last decade [1,2,3,4], photonic applications have emerged as another appealing field [5,6,7]. Perovskite films demonstrate readily tunable emission colors and sharp photoluminescence spectra [6, 8,9,10]. In PeLED application, high external quantum efficiencies exceeding 20% are achieved [6], rendering these compounds auspicious candidates for photonics as well as for next-generation displays. Most popular perovskite emitters are methylammonium, formamidinium, or cesium lead trihalides (MAPbX3, FAPbX3 or CsPbX3, X = I, Br, Cl). CsPbX3 as a fully inorganic compound naturally features superior thermal and chemical stability [11, 12]. However, the lack of deposition tools with precise process control on an industrial scale [13] remains a critical obstacle for the commercialization of PeLEDs. High-quality perovskite emissive films are typically fabricated via spin-coating, suffering from low reproducibility and a limitation of substrate size. More advanced methods based on solution processes, including doctor-blading and printing, yield perovskite films with a poor morphology [14, 15]. Furthermore, solution processing is not compatible with established state-of-the-art LED display patterning technologies, is inefficient in terms of precursor consumption, and makes use of toxic solvents strictly regulated in industrial fabrication. Thus, vacuum thermal evaporation (VTE) and CVD as solvent-free technologies featuring accurate reproducibility and intrinsic material purity are promising for commercial perovskite deposition [16, 17]. Using VTE, Li et al. demonstrated the formation of dense and smooth CsPbBr3 layers deposited by co-evaporation of the precursors CsBr and PbBr2 [18]. Obtained PeLEDs exhibited a maximum EQE of 3.3% and a luminance of 9442 cd/m2. As an alternative VTE approach, Ajjouri et al. applied single-source evaporation of previously prepared CsPbX3 powder to obtain emissive perovskite thin films [19]. Compared to VTE, CVD promises higher material usage efficiency and improved film uniformity, especially on large-area substrates [20]. However, CVD has been much less investigated than VTE, owing to the lack of applicable CVD technology. So far, CVD tools for perovskite deposition are usually based on tube furnace systems [21], with limited process control and scalability. Nevertheless, PeLEDs fabricated in such simple reactors demonstrated efficiencies up to 1.6% and a luminance approaching 5,000 cd/m2 [22].
In this work, we introduce a custom-developed perovskite hot-wall showerhead-assisted CVD tool along with an optimized low-vacuum (2 hPa) CVD process for the growth of dense CsPbBr3 emissive films. Fully vapor-deposited CsPbBr3 layers are obtained by simultaneous sublimation of both halide precursors (CsBr and PbBr2). The influence of sublimed precursor ratio on film crystallinity, morphology, and optical properties is studied. The first PeLED containing fully inorganic CsPbBr3 films deposited via showerhead-assisted CVD is demonstrated, paving the way for large-area PeLED production.
CVD technology
CsBr (beads, Sigma-Aldrich, 99.999%) and PbBr2 (beads, Alfa Aesar, 99.999%) are used as precursor materials for CsPbBr3 layers. Perovskite films are deposited on an established PeLED anode stack consisting of glass/ITO (100 nm)/PTAA (poly(bis(4-phenyl)(2,4,6-trimethylphenyl)amine), 10 nm). The entire PeLED stack as well as an energy diagram are depicted in Fig. 1. CVD of CsPbBr3 is performed in a custom-designed hot-wall reactor at 2 hPa system pressure by simultaneous sublimation of both precursors in two Creaphys thermal evaporators at temperatures of 600 °C for CsBr and 340–360 °C for PbBr2.


(a) Simplified flow diagram of the CVD tool including MFC, two sources for simultaneous sublimation of CsBr and PbBr2, a mixing unit, run-vent valves, and the reaction chamber. (b) Cross-section of the CVD reaction chamber.

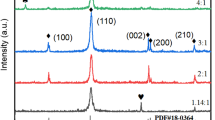
XRD measurement results of CVD films on glass/ITO/PTAA substrates with different PbBr2 crucible temperatures ranging from 340 to 360 °C, and a reference pattern of spin-coated CsPbBr3.

SEM top-view images of layers deposited by CVD with different PbBr2 temperatures of 340 °C, 350 °C, and 360 °C compared to a CsPbBr3 reference sample processed via spin-coating. Corresponding cross-section images are shown in the insets. ITO is located between the red dashed lines.

Normalized room temperature steady-state PL spectra of films grown by CVD with different PbBr2 temperatures of 340 °C, 350 °C, and 360 °C compared to a solution-processed CsPbBr3 reference.

(a) Absorption coefficients and (b) time-resolved PL intensities of the CVD sample (CsBr-rich, 340 °C) and the spin-coated CsPbBr3 reference.

(a) Operating pixel (0.11 cm2) of the CVD-PeLED (CsBr-rich) at 120 cd/m2. (b) EL spectra of the CsBr-rich CVD-PeLED and the spin-coated reference measured at 100 cd/m2 and 6000 cd/m2, respectively. (c) L-V curves of both devices. (d) J-V characteristics of PeLED with a CsPbBr3 layer processed by CVD (CsBr-rich, stoichiometric, PbBr2-rich) and via spin-coating.
The simplified flow diagram of the system is depicted in Fig. 2a. The total N2 carrier gas flow is set to 700 standard cubic centimeters per minute (sccm) (500 sccm for CsBr, 100 sccm each for PbBr2 and purge flow) by mass flow controllers (MFC). Both gas streams loaded with sublimed precursors impinge perpendicularly and are merged in a static forced-mixing unit. A high-temperature run-vent system is employed to prevent undesired deposition during heat-up and cool-down phases. The heated showerhead (Fig. 2b) spreads the carrier gas flow, enabling homogeneous deposition on temperature-controlled substrates (160 °C) up to 108 cm2 in area (12 cm × 9 cm). On this substrate size, a maximum center-to-edge perovskite layer thickness deviation of 9% has been determined (measured for a methylammonium bismuth iodide film with 100 nm thickness in the substrate center). In situ monitoring of the deposition rate is performed using a quartz crystal microbalance (QCM) mounted over the showerhead. The employed parameters for the QCM measurement are: CsBr density 4.44 g/cm3 [23], PbBr2 density 6.66 g/cm3 [24], Z-factor 1.0. The deposition time for each layer is in the range of 100–110 min.
Results and discussion
The CVD system has originally been developed for the deposition of hybrid organic–inorganic perovskites, requiring relatively low temperatures of 500 °C for piping and reactor interior heating to prevent parasitic condensation. However, to deposit all-inorganic CsPbBr3 layers, temperatures exceeding the reactor limit of 500 °C are necessary. To sublime CsBr precursor molecules, a crucible temperature of 600 °C (vapor pressure curve shown in Fig. S1) has to be applied, which would require wall temperatures > 600 °C. Consequently, parasitic condensation and re-evaporation of CsBr on the inner system surfaces will occur. For that reason, a limited and less controllable CsBr deposition rate is expected. As initial experiments, single-precursor deposition of CsBr (600 °C crucible temperature) is performed to determine its growth rate. A high N2 carrier gas flow of 500 sccm through the CsBr source and a low base pressure of the system of 2 hPa are applied to achieve a high gas velocity and thus, to reduce parasitic CsBr condensation. Consequently, a reasonable CsBr deposition rate of 0.02 nm/s is achieved. An XRD spectrum of a deposited film (Fig. S2) confirms that CsBr is successfully deposited. In the case of PbBr2, its lower sublimation temperature prevents parasitic condensation (vapor pressure curve shown in Fig. S1). In contrast to CsBr, this allows an accurate control of the PbBr2 deposition rate by adjusting the crucible temperature. In the following, for the growth of perovskite films, the CsBr temperature is fixed at 600 °C, whereas the PbBr2 temperature is varied in the range of 340–360 °C. This allows us to control the CsBr to PbBr2 ratio during the process. The qualitative evaluation of film composition and perovskite phases for three samples processed at PbBr2 crucible temperatures of 340 °C, 350 °C and 360 °C is performed by XRD analyses (Fig. 3).
The diffractograms of the CVD films are compared to that of a solution-processed CsPbBr3 reference (also deposited on glass/ITO/PTAA). All three CVD samples exhibit characteristic features indicating an orthorhombic CsPbBr3 phase [29], as also observed in the solution-processed reference sample. The signal at 35.4° can be assigned to the (400) planes of ITO [30]. It can be seen that the CVD samples processed with PbBr2 temperatures of 340 °C and 360 °C in addition show multiple reflections which are not observed for the reference film. In general, the co-existence of CsPb2Br5 and Cs4PbBr6 phases along with stoichiometric CsPbBr3 can be expected due to their high thermodynamic stability and similar formation enthalpies [31, 32]. The XRD peaks of the CVD sample processed with the highest PbBr2 temperature of 360 °C can be assigned either to the orthorhombic CsPbBr3 (green dots) or to CsPb2Br5 (blue squares) phases. Based on the CsBr-PbBr2 phase diagram, the CsPb2Br5 phase is obtained under PbBr2-rich conditions (33% CsBr, 67% PbBr2) [33] and crystallizes in a 2D layered structure [34]. Therefore, by reducing the PbBr2 temperature, stoichiometric CsPbBr3 or CsBr-rich phases are likely to be formed. Indeed, the XRD pattern of the CVD sample processed at 350 °C matches that of the reference film and shows all characteristic peaks arising from crystal planes of the pure orthorhombic CsPbBr3 phase [29]. It should be noted that PbBr2 or CsBr residues cannot be fully excluded due to the limited sensitivity of grazing-incidence XRD analysis and the possible formation of amorphous phases. As expected by further lowering the PbBr2 temperature to 340 °C, in addition to CsPbBr3, the CsBr-rich 0D perovskite phase Cs4PbBr6 becomes prominent [35]. Interestingly, the reference layer from liquid phase does not exhibit any peaks related to Cs4PbBr6, although it is processed with an excess of CsBr in the perovskite precursor solution (PbBr2:CsBr molar ratio of 1.0:1.7).
As shown by the SEM images in Fig. 4, all CVD layers are fully closed, which is required to prevent short circuits in PeLED stacks. Unlike solution processing, for which the PTAA film requires N2 plasma treatment in order to ensure wettability of the precursor solution to achieve a closed layer, CVD process does not require any pretreatment [36]. This confirms that poor precursor wetting on hydrophobic surfaces is not an issue in CVD processes, opening more flexibility regarding the choice of novel electrode substrates and charge carrier selective films for PeLED optimization. The SEM cross-section analysis (insets in Fig. 4) further demonstrates layer thicknesses in the range of 100—180 nm and smooth perovskite films, allowing a homogeneous deposition of electron transport layer (ETL) and LiF/Al cathode. For all CVD samples, the morphology is characterized by rounded grains, similar to those of the solution-processed reference sample and films in earlier reports [37]. The CsBr-rich sample (340 °C, dual CsPbBr3-Cs4PbBr6 phase) exhibits less defined domains, which might correspond to CsPbBr3 crystallites embedded in a matrix of Cs4PbBr6. Ling et al. have also observed both phases and assigned the small grains to CsPbBr3 and the larger domains to Cs4PbBr6 [38]. The average domain size determined by the linear intercept method [39] is calculated to be about 100 nm. For the CVD samples with higher PbBr2 content, an increase in crystallite size is observed. The stoichiometric sample (350 °C) exhibits an average grain size of 110 nm, while the PbBr2-rich sample shows an increased domain size of 140 nm. Thus, in CVD processes, the average domain size is affected by the ratio of the sublimed precursors CsBr and PbBr2. It should be noted that these stoichiometry-dependent characteristics can also be identified for other CVD layers (not shown here) which are of thicknesses different from those shown in Fig. 4.
In Fig. 5, the normalized room temperature steady-state PL spectra of the CVD films and the solution-processed reference are depicted. The PL emission maximum of the reference layer is localized at 520 nm, matching the optical bandgap of CsPbBr3 between 2.3 and 2.4 eV [40]. The luminescence of the CsBr-rich CVD sample and the reference processed at CsBr-rich conditions are centered at the same wavelength of 520 nm. A full width at half maximum (FWHM) of 75 meV and 92 meV is determined for the reference and the CsBr-rich CVD sample, respectively. In literature, a comparable FWHM of 82 meV has been reported [41]. A stepwise increase of the PbBr2 content in perovskite films leads to a redshift of the PL spectrum to 521 nm (stoichiometric sample) and 525 nm (PbBr2-rich sample). A similar behavior has also been observed in solution-processed CsPbBr3 films [42]. Most likely, this red shift is caused by a higher density of shallow defects present in perovskite films with a higher Pb content. The absence of additional emission peaks arising from Cs4PbBr6 or CsPb2Br5 can be explained by their optical bandgaps. CsPb2Br5 possesses an indirect bandgap [31], inhibiting efficient radiative recombination processes. In the case of Cs4PbBr6, its direct wide bandgap (3.9 eV) [31] hinders optical excitation of charge carriers due to insufficient energy of the excitation laser (3.1 eV, 405 nm). Interestingly, despite different compositions, all CVD samples show similar absolute PL intensities without a clear dependence on phase composition. Compared to the reference film, however, the CVD samples exhibit 3 orders of magnitude lower absolute PL intensities. Considering that CVD- and solution-processed films share comparable morphologies, the absolute PL intensity can be correlated to the radiative recombination efficiency. Hence, it can be deduced that CVD samples also contain a significant density of deep trap states, leading to dominant non-radiative recombination processes. According to literature, the presence of dual CsPbBr3-CsPb2Br5 or CsPbBr3-Cs4PbBr6 phases can enhance the PL intensity via defect passivation (halide vacancies, under-coordinated Pb2+) in perovskite films [34, 38]. However, no PL enhancement effect is observed for the CVD samples. It is worth noting that the interaction between different perovskite phases and their effect on PL intensity remains enigmatic in such multiphase perovskite films and further optimization and studies are required.
To further investigate the intrinsic material differences between the CVD sample grown at CsBr-rich conditions and the CsPbBr3 reference film, absorbance measurements are performed. The recorded spectra are depicted in Fig. 6. Both samples feature comparable peak extinction coefficients ranging from 2.2 × 104 to 2.4 × 104 cm−1 (Fig. 6a), which are consistent with literature data [43]. Thus, differences in the absolute PL intensities cannot be correlated to a distinctive absorption behavior. As already stated above, a lower internal quantum efficiency (IQE) of luminescence in the case of the CVD samples might explain the limited PL intensities due to strong contribution of non-radiative recombination. The characteristic excitonic absorption peaks for both samples are located at 2.4 eV. This agrees with the observations made in literature for polycrystalline CsPbBr3 films and illustrates the generation of excitons being relevant for their absorption behavior [44]. In halide perovskites, the role of excitons during emission is controversially discussed. Most likely, for a holistic understanding of the recombination behavior, free charge carrier as well as excitons have to be considered [45,46,47]. The energy of the absorption maximum is only 30 meV higher than the emission maximum. This Stokes shift could be attributed to the existence of shallow defects or inhomogeneous broadening.
Time-resolved PL decay curves of the CsBr-rich CVD and the reference samples are depicted in Fig. 6b. It can be seen that the CVD sample exhibits an initial rapid decay of its PL intensity with a time constant < 1 ns, which is in the range of the response time of the analyzer. Thus, the actual recombination process may be even faster and is likely related to dominant non-radiative recombination kinetics induced by Shockley–Read–Hall recombination of free carriers or exciton quenching [48, 49]. After the fast initial drop by two orders of magnitude, the PL intensity declines slower, following an exponential behavior with a time constant of 10 ns. This is an indication of a different dominant recombination mechanism at lower non-equilibrium carrier densities. In contrast, the reference film features a slower decay of PL intensity (time constant 10 ns), which fits to the efficient radiative recombination evidenced by its high PL intensity. The rapid non-radiative recombination mechanism in the CVD-CsPbBr3 film is most likely caused by a high density of defects. Such defects, located in the bulk, grain boundaries or at interfaces to adjacent layers, could be caused by the CVD process itself. However, defects might just as well be induced as a consequence of the limited heating capabilities of the CVD tool. This circumstance could have adverse effects on film purity as prematurely deposited CsBr might react with humidity or oxidize when opening the system to ambient air (e.g., during source reloading). As parasitically deposited material re-evaporates in subsequent deposition experiments, oxidized CsBr species can potentially be incorporated into the growing films. Their concentration is expected to be maximal in early stages of film deposition, i.e., close to the HTL/perovskite interface, which will be shown below to be of special relevance for PeLED operation.
As a next step, we realized a PeLED based on the Cs-rich CVD-processed CsPbBr3 film as emissive layer. The PeLED architecture consists of ITO/PTAA/perovskite/TPBi/LiF/Al, as illustrated in Fig. 1a. A picture of the operating device is shown in Fig. 7a. A pronounced green electroluminescence (EL) with a brightness of 120 cd/m2 at a driving voltage of 4.3 V is obtained. To our knowledge, this demonstrates the first operational PeLED based on CsPbBr3 deposited by showerhead-assisted CVD. Regardless of similar PL characteristics for all CVD samples, only the CsBr-rich CVD-PeLED and the reference device emit detectable EL. The EL spectra of the CVD-PeLED are compared to that of a solution-processed reference device in Fig. 7b. Both spectra show similar center emission wavelengths of 523 nm and 520 nm with narrow FWHM of 23 nm and 17 nm for the PeLED with CVD- and solution-processed CsPbBr3 films, respectively. The absence of detectable EL in stoichiometric and PbBr2-rich CVD-PeLEDs might be caused by a lack of the Cs4PbBr6 phase in the perovskite film. As has been reported in literature, Cs4PbBr6 could lead to defect passivation [38]. In our case, the presence of Cs4PbBr6 does not appear to show impact on the absolute PL intensity, whereas its presence in the film is most likely the reason for light emission of the CsBr-rich CVD-PeLED. This could be related to different carrier densities and spatial distributions during optical and electrical excitation. In contrast to optical excitation from the sample top during PL measurements, electrical excitation probably leads to a recombination zone localized closed to the HTL/perovskite interface due to electron accumulation in CsPbBr3 [50]. Thus, EL is more prone to non-radiative loss channels located at the HTL/perovskite interface (reasons for a high level of impurity incorporation in this region have been discussed above). The CVD sample reaches a maximum luminance of 125 cd/m2 (Fig. 7c), which is around two orders of magnitude lower compared to that of the spin-coating reference (11,500 cd/m2). In literature, PeLEDs achieve a maximum luminance between 0.5 and 9442 cd/m2 in the case of thermally evaporated CsPbBr3 [18, 51] and 5,025 cd/m2 for CVD-based CsPbBr3 [22]. The external quantum efficiency (EQE) is calculated to be 0.003%, which is several orders of magnitude lower compared to that of the reference PeLED (1.1%). The inferior PeLED performance can be traced back to the still high defect densities in the perovskite films especially at the HTL/perovskite interface.
When the PeLEDs are operated at high driving current densities (> 100 mA/cm2), defect migration and device overheating can be induced, degrading device performance [52]. Compared to the PeLED reference with a current density of 200 mA/cm2 at 4.5 V, the CsBr-rich CVD-PeLED exhibit significantly larger current densities > 1 A/cm2 (Fig. 7d). For higher voltages, the luminance of the CVD-PeLED starts dropping as can be seen in Fig. 7c. Comparing the threshold voltages for current onset (Vth, defined at a current density of 0.1 mA/cm2) of all J-V curves, Vth is in the range of 1.3 V to 1.5 V for the CVD samples compared to 2.4 V for the spin-coating reference device. Thus, devices consisting of CVD-CsPbBr3 most likely suffer from charge imbalance and carrier leakage. Based on the energy alignment of the PeLED stack (Fig. 1b), electron injection from TPBi into CsPbBr3 is quasi-barrier-free, whereas holes have to overcome a certain energy step to be injected into the emissive layer [50]. A possible cause for the onset of current at too low Vth values might be the high defect density in the perovskite films, especially at the HTL/perovskite interface. Efficient electron injection is expected to cause electron accumulation at the HTL/EML interface, facilitating direct non-radiative recombination processes via local defects (w/o injection into the perovskite valence band). Samples with thermally more robust inorganic NiOx as HTL (Fig. S3) show comparable properties and suggest that such low values of Vth are related to the perovskite film rather than to a potential HTL degradation.
Conclusion
In this work, we employed a custom-built showerhead-assisted CVD tool for the deposition of CsPbBr3 films. We demonstrated the control of film stoichiometry by varying the PbBr2 source temperature in the range of 340–360 °C with constant CsBr source temperature of 600 °C. The formation of compact and uniform perovskite films with grain sizes between 100 and 140 nm enables their application in thin-film electronic devices. We demonstrate the first CsPbBr3-based PeLEDs deposited by showerhead-assisted CVD with a maximum luminance of 125 cd/m2. Studies on the optical properties of perovskite films employing steady-state and time-resolved PL spectroscopy indicate lower PL intensity and a reduced effective lifetime of excited species for the CVD sample compared to the spin-coating reference. This was traced back to a high density of deep trap states as a major cause for the dominant non-radiative recombination processes in CVD-CsPbBr3. Ongoing work on intrinsic material differences between the CVD- and solution-processed CsPbBr3 films is expected to clarify the reasons for the high defect densities and indicate potential paths for improvement. Besides, further upgrades and optimizations of the custom-built CVD tool will enable investigations into a broader range of growth conditions and eliminate parasitic CsBr deposition. Subsequently, CVD-PeLEDs with higher luminance are aspired, matching the efficiencies of the spin-coating reference. The presented study highlights the feasibility of depositing dense CsPbBr3 films by CVD directly into device stacks combined with process scalability and precise control, rendering this technology a favorable candidate for commercial large-area production.
PeLED fabrication
For PeLED fabrication, pre-structured ITO-on-glass substrates (2.5 cm × 2.5 cm, Automatic Research) are cleaned with acetone and isopropyl alcohol in an ultrasonic bath. Afterward, the substrates are thoroughly cleaned with O2 plasma for 15 min at 100 W. All following steps are conducted either in purified N2 atmosphere or under vacuum conditions. PTAA (Sigma-Aldrich, 99.99%) is prepared by dissolving PTAA powder in anhydrous toluene with a concentration of 2 mg/ml. 50 µl of PTAA solution is then spin-coated at 6000 rpm for 30 s and annealed for 10 min at 100 °C to form the HTL. For the solution-processed CsPbBr3 emission layer (EML) serving as a reference, CsBr and PbBr2 are dissolved with a molar ratio of 1:1.7 in dimethylsulfoxide (DMSO) to obtain an 11 wt% solution [36]. Before spin-coating, we expose the PTAA film to N2 plasma for 20 s to ensure full coverage with CsPbBr3. 100 µl of the precursor solution is spin-coated at 2000 rpm for 40 s, followed by thermal annealing at 80 °C for 10 min. CVD of CsPbBr3 is performed in a custom-designed hot-wall reactor as described above without any plasma treatment of PTAA. After transferring the substrates into an AIXTRON OVPD Gen1 cluster tool without exposure to air, the ETL TPBi (2,2',2''-(1,3,5-benzinetriyl)-tris(1-phenyl-1-H-benzimidazole), Sigma-Aldrich, 99.99%, 30 nm) is deposited at a base pressure of 0.9 hPa and a substrate temperature of 20 °C. Lastly, LiF (1 nm) and Al (100 nm) are deposited via VTE through shadow masks. The entire PeLED stack is depicted in Fig. 1. The substrate contains 8 PeLED pixels with an area of 0.11 cm2 each.
Characterization methods
Structural characterization of the deposited CsPbBr3 films is carried out by grazing-incidence X-ray diffraction (XRD) 2Θ scans with a fixed angle of incidence using a Philips XPERT PRO tool. Scanning electron microscopy (SEM, Zeiss Sigma) is performed to evaluate layer morphology and to measure film thickness. For steady-state photoluminescence (PL) measurements, a Horiba iHR320 spectrometer combined with a thermoelectrically cooled Synapse CCD detector and a 405 nm diode laser (Z-Laser GmbH) are used. Further optical analysis is conducted by UV–VIS absorption measurements and time-resolved PL spectroscopy. Absorption spectra are recorded using a Shimadzu UV2550 double-beam spectrometer in combination with an integration sphere in transmission geometry to reduce scattering. Time-resolved PL analysis is performed utilizing a Becker & Hickl HPM-100-40C hybrid detector and a pulsed 405 nm laser diode (PicoQuant with a pulse width < 50 ps and a repetition rate of 40 MHz).
For PeLED characterization, EL and J–V–L (current density–voltage–luminance) characteristics are measured using a Red-Tide spectrometer and an HP Agilent 66332A sourcemeter.
Data availability
The data of this study are available from the corresponding author upon reasonable request.
Change history
11 October 2021
This article was updated to add Open Access funding note.
References
Q. Jiang, Y. Zhao, X. Zhang, X. Yang, Y. Chen, Z. Chu, Q. Ye, X. Li, Z. Yin, J. You, Surface passivation of perovskite film for efficient solar cells. Nat. Photon. 13, 460 (2019)
M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M.K. Nazeeruddin, S.M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt, M. Grätzel, Cesium-containing triple cation perovskite solar cells: improved stability, reproducibility and high efficiency. Energy Environ. Sci. 9(6), 1989 (2016)
A. Kojima, K. Teshima, Y. Shirai, T. Miyasaka, Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131(17), 6050 (2009)
NREL: Efficiency chart, https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies.20200406.pdf. Accessed 30 Dezember 2020.
Z. Xiao, R.A. Kerner, L. Zhao, N.L. Tran, K.M. Lee, T.-W. Koh, G.D. Scholes, B.P. Rand, Efficient perovskite light-emitting diodes featuring nanometre-sized crystallites. Nat. Photon. 11(2), 108 (2017)
K. Lin, J. Xing, L.N. Quan, F.P.G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, C. Di Zhang, W. Yan, X. Li, Y. Liu, J. Lu, E.H. Kirman, Q.X. Sargent, Z. Wei, Perovskite light-emitting diodes with external quantum efficiency exceeding 20 %. Nature 562(7726), 245 (2018)
N. Wang, L. Cheng, R. Ge, S. Zhang, Y. Miao, W. Zou, C. Yi, Y. Sun, Y. Cao, R. Yang, Y. Wei, Q. Guo, Y. Ke, M. Yu, Y. Jin, Y. Liu, Q. Ding, D. Di, G. Le Yang, H. Xing, C. Tian, F. Jin, R.H. Gao, J.W. Friend, W. Huang, Perovskite light-emitting diodes based on solution-processed self-organized multiple quantum wells. Nat. Photon. 10(11), 699 (2016)
S. Adjokatse, H.-H. Fang, M.A. Loi, Broadly tunable metal halide perovskites for solid-state light-emission applications. Mater. Today 20(8), 413 (2017)
Y.-H. Kim, H. Cho, J.H. Heo, T.-S. Kim, N. Myoung, C.-L. Lee, S.H. Im, T.-W. Lee, Multicolored organic/inorganic hybrid perovskite light-emitting diodes. Adv. Mater. 27(7), 1248 (2015)
Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang, W. Huang, Perovskite light-emitting diodes based on spontaneously formed submicrometre-scale structures. Nature 562(7726), 249 (2018)
H. Cho, J.S. Kim, Y.-H. Kim, T.-W. Lee, Influence of A-site cation on the thermal stability of metal halide perovskite polycrystalline films. J. Inf. Disp. 19(1), 53 (2018)
Y. Zhou, Y. Zhao, Chemical stability and instability of inorganic halide perovskites. Energy Environ. Sci. 12(5), 1495 (2019)
R. Swartwout, M.T. Hoerantner, V. Bulović, Scalable deposition methods for large-area production of perovskite thin films. Energy Environ. 2(2), 119 (2019)
Z. Wei, H. Chen, K. Yan, S. Yang, Inkjet printing and instant chemical transformation of a CH3NH3PbI3/nanocarbon electrode and interface for planar perovskite solar cells. Angew. Chem. 53(48), 13239 (2014)
Y. Deng, E. Peng, Y. Shao, Z. Xiao, Q. Dong, J. Huang, Scalable fabrication of efficient organolead trihalide perovskite solar cells with doctor-bladed active layers. Energy Environ. Sci. 8(5), 1544 (2015)
P.-S. Shen, J.-S. Chen, Y.-H. Chiang, M.-H. Li, T.-F. Guo, P. Chen, Low-pressure hybrid chemical vapor growth for efficient perovskite solar cells and large-area module. Adv. Mater. Interfaces 3(8), 1500849 (2016)
M.R. Leyden, L.K. Ono, S.R. Raga, Y. Kato, S. Wang, Y. Qi, High performance perovskite solar cells by hybrid chemical vapor deposition. J. Mater. Chem. A 2(44), 18742 (2014)
J. Li, P. Du, S. Li, J. Liu, M. Zhu, Z. Tan, M. Hu, J. Luo, D. Guo, L. Ma, Z. Nie, Y. Ma, L. Gao, G. Niu, J. Tang, High-throughput combinatorial optimizations of perovskite light-emitting diodes based on all-vacuum deposition. Adv. Funct. Mater. 29(51), 1903607 (2019)
Y. El Ajjouri, F. Palazon, M. Sessolo, H.J. Bolink, Single-source vacuum deposition of mechanosynthesized inorganic halide perovskites. Chem. Mater. 30(21), 7423 (2018)
M. Heuken, N. Meyer, Organic Vapor Phase Deposition, in Organic Electronics (Wiley, New York, 2006), pp. 203–232
M.M. Tavakoli, L. Gu, Y. Gao, C. Reckmeier, J. He, A.L. Rogach, Y. Yao, Z. Fan, Fabrication of efficient planar perovskite solar cells using a one-step chemical vapor deposition method. Sci. Rep. 5, 14083 (2015)
Z. Shi, L. Lei, Y. Li, F. Zhang, Z. Ma, X. Li, Wu. Di, T. Xu, Y. Tian, B. Zhang, Z. Yao, G. Du, Hole-injection layer-free perovskite light-emitting diodes. ACS Appl. Mater. Interfaces 10(38), 32289 (2018)
N. Kurata, N. Kubota, Y. Takei, H. Nanto, Eu-doped CsBr phosphor as a new optically-stimulable phosphor material for medical X-ray imaging sensor. Radiat. Prot. Dosimetry 119(1–4), 398 (2006)
S.V. Myagkota, A.V. Gloskovskii, A.S. Voloshinovskii, Photo-and X-ray luminescence spectra of CsPbX3 microcrystals dispersed in a PbX2 (X=Cl, Br) matrix. Opt. Spectrosc. 88(4), 538 (2000)
C. Xu, Z. Liu, E.-C. Lee, High-performance metal oxide-free inverted perovskite solar cells using poly(bis(4-phenyl)(2,4,6-trimethylphenyl)amine) as the hole transport layer. J. Mater. Chem. C 6(26), 6975 (2018)
S. Ahn, K. Yabumoto, Y. Jeong, K. Akagi, Low bandgap poly(thienylenemethine) derivatives bearing terarylene moieties in the side chains. Polym. Chem. 5(24), 6977 (2014)
S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem, K. Leo, White organic light-emitting diodes with fluorescent tube efficiency. Nature 459(7244), 234 (2009)
Z. Wei, A. Perumal, R. Su, S. Sushant, J. Xing, Q. Zhang, S.T. Tan, H.V. Demir, Q. Xiong, Solution-processed highly bright and durable cesium lead halide perovskite light-emitting diodes. Nanoscale 8(42), 18021 (2016)
C.C. Stoumpos, C.D. Malliakas, J.A. Peters, Z. Liu, M. Sebastian, J. Im, T.C. Chasapis, A.C. Wibowo, D.Y. Chung, A.J. Freeman, B.W. Wessels, M.G. Kanatzidis, Crystal growth of the perovskite semiconductor CsPbBr3: a new material for high-energy radiation detection. Cryst. Growth Des. 13(7), 2722 (2013)
F.H. Alsultany, N.M. Ahmed, M.Z. Matjafri, Effects of CW CO2 laser annealing on indium tin oxide thin films characteristics. SNL 04(04), 83 (2014)
J. Yin, H. Yang, K. Song, A.M. El-Zohry, Y. Han, O.M. Bakr, J.-L. Brédas, O.F. Mohammed, Point defects and green emission in zero-dimensional perovskites. J. Phys. Chem. Lett. 9(18), 5490 (2018)
S. Caicedo-Dávila, H. Funk, R. Lovrinčić, C. Müller, M. Sendner, O. Cojocaru-Mirédin, F. Lehmann, R. Gunder, A. Franz, S. Levcenco, A.V. Cohen, L. Kronik, B. Haas, C.T. Koch, D. Abou-Ras, Spatial phase distributions in solution-based and evaporated Cs–Pb–Br thin films. J. Phys. Chem. C 123(29), 17666 (2019)
S. Caicedo-Dávila, R. Gunder, J.A. Márquez, S. Levcenko, K. Schwarzburg, T. Unold, D. Abou-Ras, Effects of postdeposition annealing on the luminescence of mixed-phase CsPb2Br5/CsPbBr3 thin films. J. Phys. Chem. C 124(36), 19514 (2020)
J. Shamsi, A.S. Urban, M. Imran, L. de Trizio, L. Manna, Metal halide perovskite nanocrystals: synthesis, post-synthesis modifications, and their optical properties. Chem. Rev. 119(5), 3296 (2019)
L. Wang, H. Liu, Y. Zhang, O.F. Mohammed, Photoluminescence origin of zero-dimensional Cs4PbBr6 perovskite. ACS Energy Lett. 5(1), 87 (2020)
G. Simkus, S. Sanders, D. Stümmler, A. Vescan, H. Kalisch, M. Heuken, High-intensity perovskite LED using poly(bis(4-phenyl)(2,4,6-trimethylphenyl)amine) as hole transport and electron-blocking layer. MRS Adv. 5(8), 411 (2020)
G. Tong, L.K. Ono, Y. Qi, Recent progress of all-bromide inorganic perovskite solar cells. Energy Technol. 8(4), 1900961 (2020)
Y. Ling, L. Tan, X. Wang, Y. Zhou, Y. Xin, B. Ma, K. Hanson, H. Gao, Composite perovskites of cesium lead bromide for optimized photoluminescence. J. Phys. Chem. Lett. 8(14), 3266 (2017)
G.F. Vander Voort, A.M. Gokhale, Comments on “grain size measurements using the point-sampled intercept technique.” Scr. Metall. Mater 26(10), 1655 (1992)
J. Almutlaq, J. Yin, O.F. Mohammed, O.M. Bakr, The benefit and challenges of zero-dimensional perovskites. J. Phys. Chem. Lett. 9(14), 4131 (2018)
J. Si, Y. Liu, Z. He, H. Du, K. Du, D. Chen, J. Li, M. Xu, H. Tian, H. He, D. Di, C. Lin, Y. Cheng, J. Wang, Y. Jin, Efficient and high-color-purity light-emitting diodes based on in situ grown films of CsPbX3 (X = Br, I) nanoplates with controlled thicknesses. ACS Nano 11(11), 11100 (2017)
J. Xu, W. Huang, P. Li, D.R. Onken, C. Dun, Y. Guo, K.B. Ucer, C. Lu, H. Wang, S.M. Geyer, R.T. Williams, D.L. Carroll, Imbedded nanocrystals of CsPbBr 3 in Cs4PbBr6: kinetics, enhanced oscillator strength, and application in light-emitting diodes. Adv. Mater. 29, 43 (2017)
A. Haque, T.D. Chonamada, A.B. Dey, P.K. Santra, Insights into the interparticle mixing of CsPbBr3 and CsPbI3 nanocubes: halide ion migration and kinetics. Nanoscale 12(40), 20840 (2020)
S. Kondo, M. Kakuchi, A. Masaki, T. Saito, Strongly enhanced free-exciton luminescence in microcrystalline CsPbBr3 films. J. Phys. Soc. Jpn. 72(7), 1789 (2003)
C. Wolf, T.-W. Lee, Exciton and lattice dynamics in low-temperature processable CsPbBr3 thin-films. Mater. Today Energy 7, 199 (2018)
M. Dendebera, Y. Chornodolskyy, R. Gamernyk, O. Antonyak, I. Pashuk, S. Myagkota, I. Gnilitskyi, V. Pankratov, V. Vistovskyy, V. Mykhaylyk, M. Grinberg, A. Voloshinovskii, Time resolved luminescence spectroscopy of CsPbBr 3 single crystal. J. Lumin. 225, 117346 (2020)
Y. Yuan, M. Chen, S. Yang, X. Shen, Y. Liu, D. Cao, Exciton recombination mechanisms in solution grown single crystalline CsPbBr 3 perovskite. J. Lumin. 226, 117471 (2020)
M. Gerhard, B. Louis, R. Camacho, A. Merdasa, J. Li, A. Kiligaridis, A. Dobrovolsky, J. Hofkens, I.G. Scheblykin, Microscopic insight into non-radiative decay in perovskite semiconductors from temperature-dependent luminescence blinking. Nat. Commun. 10(1), 1698 (2019)
J. Chen, N.-G. Park, Causes and solutions of recombination in perovskite solar cells. Adv. Mater. 31(47), 1803019 (2019)
D. Luo, Q. Chen, Y. Qiu, M. Zhang, B. Liu, Device engineering for all-inorganic perovskite light-emitting diodes. Nanomaterials 9, 7 (2019)
S. Xie, A. Osherov, V. Bulović, All-vacuum-deposited inorganic cesium lead halide perovskite light-emitting diodes. APL Mater. 8(5), 51113 (2020)
Q. Dong, L. Lei, J. Mendes, F. So, Operational stability of perovskite light emitting diodes. J. Phys. Mater. 3(1), 12002 (2020)
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sanders, S., Simkus, G., Riedel, J. et al. Showerhead-assisted chemical vapor deposition of CsPbBr3 films for LED applications. Journal of Materials Research 36, 1813–1823 (2021). https://doi.org/10.1557/s43578-021-00239-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-021-00239-w