
Abstract
Spontaneous noble metal dopant segregation in an oxide lattice can lead to the formation of metallic clusters and extended acicular inclusions. In a thin-film process, the shape and orientation of such noble metal inclusions are governed by the crystal growth direction, giving rise to a composite material with lattice-matched metal nanopillars embedded vertically in an insulating or semiconducting oxide matrix. An interesting application of such composites is in photoelectrochemical cell electrodes, where the metallic nanopillars take on three distinct roles: forming a Schottky junction with the host matrix, providing a low-loss current path from bulk to surface, and creating an efficient electrocatalytic active site on the electrode surface. In particular, we discuss the application of vertically aligned metal–oxide nanopillar composites in photoelectrochemical water-splitting cells used for direct solar-powered hydrogen generation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ubiquitous availability of water and sunlight on Earth makes hydrogen gas an excellent fuel candidate for a clean and sustainable energy cycle, provided that the hydrogen fuel is obtained in a sustainable solar-powered process. Among the various options for harvesting and storing solar energy in the form of hydrogen, photoelectrochemical (PEC) water splitting, illustrated in Figure 1, is the most direct method, where the splitting of water and the evolution of either hydrogen or oxygen gas occur on the surface a semiconductor photoelectrode, which is essentially a simplified solar cell placed in water. In terms of band structure, an oxygen evolution photoelectrode consisting of an n-type semiconductor, such as SrTiO3, exhibits an upward band bending in the space–charge layer that forms at the water interface. This internal field drives the photogenerated holes to the electrode surface where oxygen gas is produced in an electrocatalytic water oxidation reaction. In a similar manner, a p-type semiconductor would be used for a hydrogen evolution electrode, where photogenerated electrons arriving at the water interface reduce water to hydrogen gas. A practical PEC cell requires two reacting surfaces to achieve water splitting into hydrogen and oxygen. The hydrogen gas can be collected at the PEC electrode surface and stored or transported in liquefied bulk form, to be consumed at a time and location where it is needed for a particular application, such as powering a fuel cell vehicle or feeding ammonia synthesis in a fertilizer plant (Figure 1). Ultimately, the consumption of the hydrogen leads to the generation of water, and the energy harvesting and consumption cycle can repeat.

A photoelectrochemical water-splitting cell contains a photoactive semiconductor electrode in water. Carriers generated by light are transported to the surface by the internal field close to the water interface where electrons and holes can lead to the evolution of hydrogen and oxygen, respectively. In a hydrogen fuel cycle, the hydrogen can be transported and used by fuel cell vehicles and the chemical industry. Water released when hydrogen is consumed can be returned to the environment.
Solar-energy collection efficiency
Despite the conceptual simplicity of the design of a PEC electrode, numerous unsolved materials challenges remain and even for the best photoelectrodes reported to date, the efficiency of energy conversion from sunlight to hydrogen is either too low or the working life too short for PEC water splitting to replace methane reforming or water electrolysis as a major source of hydrogen.1,2 Considering solar-powered water electrolysis, assuming a solar-panel efficiency of 20% and water electrolysis efficiency of 50%, a PEC water-splitting cell would have to reach an overall energy-conversion efficiency of about 10% to be competitive. The materials challenges that limit the efficiency of practical PEC electrode materials can be understood by looking at the band-edge energy versus depth diagram in Figure 1, which illustrates the operation of n-type SrTiO3 as a PEC oxygen evolution electrode. Doped SrTiO3 is one of the best model semiconductors for this application and sufficiently stable in water for long-term operation.3
It is instructive to trace the various losses that are inevitable in the PEC energy-conversion process.4 Starting with sunlight shining on a SrTiO3 electrode in water, a reflective light loss of 2% occurs at the air–water interface (nwater = 1.33) and another 8% is lost at the water–SrTiO3 interface (nSTO = 2.4). Once a photon has entered the SrTiO3 crystal, useful absorption that generates carriers in the conduction and valence bands can only occur if the photon energy exceeds the band gap, which is 3.2 eV for SrTiO3 but can be reduced to a more suitable 2 eV by doping. The smallest usable band gap is 1.23 eV, which is the energy barrier for splitting water, but a slightly larger energy of about 2 eV is targeted in practical doped semiconductors used in PEC electrodes. For the standard sunlight photon flux spectrum, the fraction of photons with an energy exceeding 3.2 eV is only 1.6 percent. This fraction increases to 21% if the band gap is reduced to 2 eV and thus, at best, 19% of available photons generate a photocarrier. The ratio is slightly more favorable in terms of energy. Although any extra photon energy above the band gap is lost to heat though rapid thermalization to the conduction band bottom after the absorption event, the available solar photon flux is strongly skewed toward the infrared part of the spectrum, which means that about 26% of the incident light energy can be converted to photocarriers in a doped semiconductor photoelectrode. Band-gap engineering is therefore of great importance in optimizing the energy-harvesting efficiency of oxide photoelectrodes.
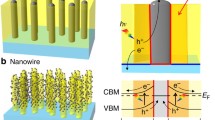
Once a photon is absorbed and an electron–hole pair is formed, only a small fraction of the excited charge carriers can actually participate in the water-splitting reaction at the surface. We can identify three regions in the photoelectrode heterostructure, as illustrated in Figure 2a. A space–charge region generally forms at the semiconductor–water interface due to the alignment of the chemical potential in the semiconductor with the redox potentials of water. The illustration shows an n-type semiconductor that exhibits carrier depletion at the water interface and thus promotes oxygen evolution in a water oxidation reaction driven by the photoexcited holes. The direction of band bending would be reversed for a p-type semiconductor but the essential aspects of the carrier transport are the same. Efficient carrier separation and transport of electrons or holes to the surface can only occur in the space–charge region. Due to the low intensity of sunlight and hence the low carrier density, the space–charge region depth is primarily determined by the permittivity of the semiconductor and can be estimated to be a few tens of nanometers for titanate semiconductors. The electric field in the deeper layers of the semiconductor is negligible, and excited charge movement is primarily diffusive, similar to a thick single-crystal silicon solar cell, with the exception that the nonequilibrium carrier lifetime is much shorter in oxides than in Si-reported values are in the nano- or even picosecond range.5,6 The primary reasons for the short carrier lifetime are trapping at shallow trap sites that may be related to oxygen vacancies or other lattice defects and the presence of unoccupied mid-gap states in nominally p-type systems, such as Rh:SrTiO3. Trap-mediated carrier recombination is the dominant excited carrier loss mechanism even in nondoped SrTiO3 crystals, as shown by a sharp increase of photoconductivity at low temperatures,7 similar to defect-rich CdSe and CdS semiconductors.

(a) Carrier transport is driven by the internal field in the space–charge region close to the surface of a semiconductor in water. No internal field is present in the diffusive transport region where recombination probability is high. Transport is Ohmic in the conducting substrate region. Light absorption length is typically much larger than the electrode thickness. (b–e) Several possible nanostructure designs that have been studied to resolve the conflict between short charge-carrier escape length and long light absorption length.
Besides the space–charge layer thickness, the total thickness of the electrode layer is defined by the light absorption length. The absorption coefficient of intrinsic SrTiO3 above the band-gap energy is on the order of 105 cm−1,8 which means that to absorb most of the incident light, the semiconductor layer thickness should be on the order of 100 nm. However, since intrinsic SrTiO3 has a band gap of 3.2 eV, this absorption level only applies to interband transitions induced by ultraviolet light. Doping is required to reduce the band gap and obtain visible light absorption down to 2 eV, but doping levels must be limited to a few percent to avoid carrier trapping and recombination at doping-induced defects. The visible light absorption is, thus, at least an order of magnitude lower than for band-gap excitations and would require an even larger light absorbing layer thickness, on the order of a micrometer. This length scale mismatch between the carrier extraction and light absorption lengths is illustrated in the bottom part of Figure 2a.
Finally, the heterostructure requires a mechanical and electrical support, which forms a third layer in the structure and may be provided by a metallic Nb-doped SrTiO3 substrate as shown in Figures 1 and 2a or a metallic electrode layer that is a good epitaxial lattice match for the photoactive part of the heterostructure. For SrTiO3-based compositions, a SrRuO3 or a Sr2RuO4 electrode layer may be used.9 The PEC electrode can then be attached to an electric circuit that connects the photoelectrode to a passive metallic counter electrode or in a tandem configuration to another PEC electrode of opposite polarity.2 Regardless of the particular cell design, the electrode support has to be a good metal to minimize Ohmic losses in the circuit that connects the two electrodes. For thin-film PEC devices, sufficiently low defect densities in the surface space–charge region can be achieved only by growing the electrodes epitaxially on single-crystal substrates, such as Nb-doped SrTiO3. However, even moderately highly doped SrTiO3 is not optically inert—although the Nb doping raises the Fermi level to the bottom of the conduction band, a clear photoresponse can still be seen in an oxygen evolution reaction on the surface of Nb:SrTiO3 crystals. If a SrTiO3 PEC electrode is grown on a heavily doped Nb:SrTiO3 substrate while gradually reducing the Nb doping level from 1% to 0, the local Ohmic conductivity of the electrode can be matched to the depth-dependent carrier mobility. In this way, maximum charge extraction efficiency can be achieved in the space–charge region where carrier drift is driven by the strong internal electric field, while Ohmic losses are minimized in the drift region of the electrode by Nb doping.10 Other methods of improving carrier mobility have also been studied, such as making the use of exciton mobility11 or co-doping to reduce the density of unoccupied traps.5,6
The design of an efficient oxide energy-harvesting material, thus, presents us with a seemingly irreconcilable mix of mutually exclusive properties. The most serious conflict is the requirement of doping to achieve the optimal band gap and visible light absorption while any doping scheme also carries the risk of creating structural defects that lead to trapping or reduce the lifetime and mobility of photoexcited carriers. The search for a high-performance water-splitting photocatalyst materials and related PEC electrode development has a long history, starting with the original report in 1972 by Fujishima and Honda on hydrogen generation from water on the surface of TiO2.12 So far, no perfect material has been found that has a sufficiently narrow band gap, a suitable alignment of the conduction- and valence-band edges with the water redox potentials, and sustains high photocarrier mobility near room temperature. It is still an open question whether this is a fundamentally impossible proposition, as halide perovskites do show exceptionally long carrier lifetimes,13 but unfortunately, degrade rapidly in the presence of water. Developing a water-compatible oxide material with similarly efficient defect screening has so far failed, which means that the only feasible approach is to reduce the length scale of excited carrier transport by suitable nanostructuring.
Nanostructured photoelectrochemical electrodes
A number of nanostructured light-harvesting materials designs, illustrated in Figure 2b–e, have been proposed and tested. The most obvious approach from a synthesis and cost point of view is the use of a layer of nanoparticles on a conducting support (Figure 2b).14 The classic approach is to synthesize a semiconductor material in bulk powder form in a ceramic process and spread the powder with a squeegee on a carrier, such as a glass sheet coated with an indium tin oxide (ITO) electrode. This surprisingly simple technique also creates a heterostructure between the water-exposed surface of a particle and the contact point with the ITO electrode. This is probably the most widely studied method of nanostructuring due to the technical simplicity and low cost of synthesis. Numerous improvements can be made while maintaining the basic powder design, such as using core–shell nanoparticles with additional internal space–charge regions for more efficient carrier separation, utilizing the work function differences between different facets of single-crystalline nanoparticles, and the coating of nanoparticle surfaces with uniform or even site-selective cocatalysts.3 Despite a large effort that has been put into the development of nanoparticle-based photoelectrodes, the light absorption length mismatch has been only partly solved. Although light transmitted through a single nanoscale particle can be absorbed in another particle, the surface reflection and scattering losses tend to negate any benefits of increased absorption.
To minimize scattering and reflection losses, it is possible to use nanopillars, nanotubes, or other elongated nanostructures (Figure 2c) that are electrically connected to a conducting substrate. As with the nanoparticles on a glass sheet, the nanopillars or nanotubes minimize the transport of excited carriers in the radial direction. For a diameter of less than 100 nm, essentially the whole volume of a nanopillar is taken up by the surface space–charge region, and electron–hole separation is driven by the large internal field. When such a nanopillar array is illuminated along the surface normal of the substrate, the light absorption length is defined by the length of the nanopillars, which can be designed to match the visible light absorption length. The charge collection inside the nanopillars can be further enhanced by using a multilayer design, forming a coaxial heterostructure.15,16 A major drawback of the pillar designs is the complexity of the synthesis process, especially for multilayer pillars16 and the short working life of a pillar array. From a cost perspective, a hydrogen-generation panel should operate over similar time scales as a photovoltaic solar panel and it is unlikely that surface fouling can be prevented to sustain the water-splitting reaction over a period of several years. An additional issue with closely spaced nanostructures is the release of gas. Since the photoelectrode is a gas generation device, gas bubbles need to be quickly removed from the surface. Any hydrogen or oxygen bubbles attaching to the electrode surface reduce the effective reaction area.
The third approach to solve the light absorption length mismatch problem is to use an artificial composite material with many internal interfaces, as illustrated in Figure 2d.17 Light-induced electron transfer from donor to acceptor regions naturally separates the charges, and the bicontinuous network of heterojunctions provides a current extraction path.
It is possible to combine the favorable aspects of all the structures described here by embedding metallic nanopillars inside the photoactive semiconductor matrix, as shown in Figure 2e.18 The light absorption length is defined in this structure by the thickness of the nanopillar composite, a cylindrical space–charge region can form coaxially around each pillar if the work function of the pillar material is chosen suitably, and the charge extraction length is defined by half of the average distance between the metallic nanopillars. A nanopillar composite, thus, effectively separates the charge generation region in the bulk of the composite film from the electrocatalytically active regions at the tips of the nanopillars that protrude into water.
The key to synthesizing such nanopillar composites without resorting to an expensive lithographic microfabrication process is self-organization, whereby two materials spontaneously phase separate. If the phase separation occurs continuously at the growth front of a thin film, extended vertical pillars of the segregated minority phase may form in the dominant host matrix. Indeed, there are a number of examples of such structures forming spontaneously in various materials systems. For our discussion, the interest is primarily in oxide hosts and either oxide19,20 or metal21,22 nanopillars forming in an oxide host lattice.
Choice of materials for water splitting
Testing of many photocatalyst and PEC electrode materials has shown that doped SrTiO3 is an excellent starting point, with the best candidate materials being Rh:SrTiO3, Ir:SrTiO3, Al:SrTiO3, and various co-doped systems, such as (Rh, La)SrTiO3.3,14,23 Among these candidate materials, Al:SrTiO3 is primarily interesting as a model system, since Al doping does not reduce the band gap of the SrTiO3 host, and it can thus only absorb in the weak ultraviolet end of the solar spectrum.
Besides aluminum, the choice of noble metals as dopants in an oxide host system may appear counterintuitive at first, since, as the name implies, these elements tend not to oxidize and are expensive or rare, something that should be avoided in a large-scale energy-harvesting application. However, there are several good reasons why noble metals such as Pt, Ir, Pd, Au, and Rh, might be used in PEC electrodes. Most importantly, doping SrTiO3 with Rh or Ir reduces the band gap from 3.2 to 2.7 eV and 2 eV, respectively, which means that light in the visible part of the solar spectrum can be absorbed.24,25 These dopant materials are quite special in the context of applications that rely on the high energy of electrons photoexcited to the conduction band because for both Rh and Ir, the dopant-related 4d or 5d states are located just above the valence band top of the SrTiO3 host. This means that the band gap is reduced by raising the energy of the valence-band edge, rather than changing the Ti 3d-character conduction band of SrTiO3. This scheme is particularly favorable for hydrogen evolution electrode materials where the relatively high energy of the conduction band edge relative to the water redox potentials is essential for transferring electrons from the semiconductor to water. An additional motivation that combines an oxide semiconductor host material with noble metals is the excellent electrocatalytic performance of metallic Ir and Pt in the water-splitting reaction.26 This means that noble metal doping can be a viable approach even for electrodes that collect holes for oxygen evolution and transfer electrons to a metallic counter electrode for hydrogen generation.
Synthesis challenges
From the point of view of oxide thin-film growth, combining noble metals with transition-metal oxides such as titanates presents several interesting challenges related to oxidation kinetics and oxide volatility. Thin films of suitable oxide host materials, particularly titanates such as SrTiO3, are normally grown at temperatures of around 600°C to 800°C and ambient oxygen pressures of anywhere from 10−7 Torr to 100 mTorr. However, thin films grown under these conditions are generally not suitable for solar-energy-harvesting applications due to the high density of intrinsic defects that lead to rapid carrier trapping and recombination. Improved crystal quality can be achieved by growing films by molecular beam epitaxy27 or by pulsed laser deposition at very high temperature of 1300°C and strongly reducing oxygen pressure of just 10−7 Torr.28,29 These conditions are problematic for many potential candidate dopant metals due to the high vapor pressure of the corresponding oxides, such as PdO, PtO2, RhOx, or IrOx.30 Growing a noble-metal-doped SrTiO3 is thus difficult in the sense that too low growth temperatures will lead to a defect-rich material in which the recombination rate is too high for practical energy harvesting, whereas high-temperature growth will lead to evaporative loss of noble metal dopants. Likewise, for oxygen pressure, low oxygen pressure is preferred for titanate growth to avoid cation segregation,31,32 but it may lead to the formation of segregated metal, while too high oxygen pressures promote rapid oxidation and evaporative loss of the noble metal oxides. This problem is particularly severe for high-temperature growth of Ir-doped titanates due to the formation of IrO3 at just slightly higher oxygen pressures than the formation of stable IrO2. Since IrO3 has no stable solid phase, even slightly over-oxidizing conditions will lead to the evaporative loss of Ir from the film.33 For many other potential metal dopants, suppression of evaporative loss can be achieved by increasing the oxygen pressure to the mTorr range, albeit at the risk of generating Sr vacancies.31
Segregation mechanism
Segregation of a secondary phase in thin-film or bulk-ceramic synthesis is usually considered a sign of failure. Nevertheless, nanoscale metal segregation offers an interesting route to the synthesis of metal oxide composite materials in a simple single-step process that does not require microfabrication or patterning. In the context of catalysis, for example, spontaneous metal segregation is the basis of the so-called self-regenerating catalyst design, which responds to the ambient atmosphere by reversibly oxidizing or segregating Pd metal nanoparticles in an oxide host.34
The segregation of a secondary metal phase during oxide crystal growth is controlled mainly by the growth temperature and the ambient oxygen pressure. Selecting a suitably low oxygen pressure that prevents full oxidation of the dopant metal is therefore necessary. For thin-film growth, there is the important additional parameter of growth rate, which for pulsed laser deposition, PLD is on the order of 10 s for a single perovskite unit cell layer of 4 Å thickness. Since the oxidation of the film and the surface migration of adatoms occur on similar time scales,35,36 the growth rate can be used to control the self-organized formation of metal clusters during film deposition. This effect can be seen in Figure 3 for the growth of Ir-doped SrTiO3 at 700°C and an oxygen pressure of 1 mTorr when the deposition rate is varied from 8 to 312 s/nm. The segregated metal clusters are visible in the atomic force microscope (AFM) images as white dots on the surfaces of the films. While the three-dimensional structure of the segregated metal cannot be determined from the AFM images, it is clear that the metal clusters are not present on the surface for the slowest growth rate, presumably lost due to gradual evaporation of slowly forming IrO3.

(a–d) Atomic force microscope images of 20-nm-thick Ir:SrTiO3 films grown at various deposition rates at 700°C and 1 mTorr of oxygen. The growth rates are shown in the images. The image area is 1 µm × 1 µm. Fast growth produces a nanodot array as shown in (e). Bright regions in the film are shown to be Ir rich by in situ energy-dispersive x-ray spectroscopy. Slower growth rates (b, c) produce an array of nanopillars in the film (f, h). A plan view of (h) is shown in (g). The horizontal white lines in the cross-sectional transmission electron microscope images mark the substrate–film interface. No metal segregation is observed in (d).18,37,39
A transmission electron microscope (TEM) image of a film, where metal segregation does occur (Figure 3e), shows that the clusters form directly at the substrate interface and take the shape of a nanodots.37 The most interesting structure is obtained in the intermediate growth rate range if the conditions are such that a stable surface concentration of the metal adatoms can be maintained. In this case, vertically aligned nanopillars will form, as shown in Figure 3f, h.18 The nanopillar diameter is uniform throughout the composite crystal, as is the lateral pillar spacing. The image also shows that the pillar growth may spontaneously terminate, presumably because the surface density of the Ir atoms does not remain constant (Figure 3f).37
At this point, we can consider the mechanisms that lead to the formation of a vertical aligned nanopillar (VAN) composite structure. For oxide–oxide nanocomposites, the driving force for spontaneous phase separation may be related to miscibility limits or the preference for different crystal structures by the two component materials, such as a perovskite and a spinel or rutile and corundum.20,38 For noble metals and perovskites, the lattice matching is nearly perfect, since most noble metals form a face-centered cubic (fcc) lattice that has a mismatch of less than 5% with SrTiO3. Platinum, in particular, is an interesting case because while at room temperature, the Pt lattice parameter is larger than that of SrTiO3 (3.92 Å versus 3.905 Å), its thermal expansion coefficient is smaller, which means that at about 700°C, Pt and SrTiO3 are perfectly lattice matched and the system is completely strain free. Despite the nonexistent strain, platinum still forms faceted islands when deposited on SrTiO3 at around 700°C with island sizes in the hundreds of nanometers range with similar island spacing length scales.39,40 This shows that the migration rate of the metal adatoms on the surface is much larger than for oxide species. SrTiO3 itself would exhibit layer-by-layer growth on a stepped substrate surface at around 700°C,36 which means that the migration length of oxide species is on the order of tens of nanometers (i.e., an order of magnitude lower than for metals such as Pt or Ir under identical temperature and oxygen pressure (Figure 4)].

In PLD film growth, periods of near-instantaneous deposition with high supersaturation of adatoms are followed by long periods of surface relaxation during which the deposited adatoms can migrate on the surface. Metal pillar formation can occur when the noble metal (gray) migration length is much larger than the oxide host material (green) migration length. The migration process ends when the next deposition pulse arrives on the surface.
It is, thus, reasonable to interpret the phase separation and the formation of metal nanopillars in an oxide host as being enabled by the large difference in the surface migration rates of the component adatom species. While long-range migration of unoxidized noble metal atoms leads to the formation of large metal clusters, the oxide host lattice grows in the layer-by-layer mode due to the much smaller length scale of clustering associated with the lower migration rate of the oxide species. The segregation of a noble metal codeposited with an oxide can be viewed as a type of kinetic phase separation,41 which is known to occur for crystallization of multicomponent materials from a melt. In this case, however, no liquid state is present, but the nanopillar formation shows that the nonequilibrium surface layer of a composite film that exists for the brief periods of relaxation between the laser ablation pulses behaves in a sense more like a liquid than a solid or a gas. It is also important to note that outside of the growth window for pillar formation (temperature, oxygen pressure, growth rate), a homogeneously doped thin film is obtained instead.18,39 Bulk-ceramic synthesis of the Ir (5 at.%):SrTiO3 target pellet also leads to a homogeneous material with no indication in x-ray diffraction scans of macroscopic metal segregation, which suggests that the 5% doping does not exceed the solubility limit of Ir in SrTiO3. Indeed, for Ir, bulk metal segregation is unlikely because SrIrO3 exists as a stable solid phase under similar process conditions.42,43 The metal segregation that leads to nanopillar growth only happens in a strongly nonequilibrium surface migration process during thin-film growth in a metal-specific process parameter window, and the mechanism is, thus, different from well-known bulk phase-separation mechanisms, such as the spinodal separation of TiO2 and VO2.44 Composition analysis by in situ energy-dispersive x-ray spectroscopy (EDX) shows that the average Ir content in the film is 4 at.%, slightly below the original 5 at.% doping level of the PLD target. EDX analysis is usually not possible for a doped SrTiO3 film grown on a SrTiO3 substrate. However, when polished into a wedge shape, it is possible to obtain a TEM plan-view sample where the substrate part has been completely removed close to the sample edge. This type of measurement is illustrated in Figure 5 for a nanopillar composite film. The EDX analysis indicates that the kinetic phase separation leads to approximately half of the Ir being conglomerated in the nanopillars, reducing the average film doping level to 2 at.% Ir. This remaining doping level has important implications for the PEC performance of the nanocomposite film.

Energy-dispersive x-ray spectroscopy (EDX) analysis of nanopillar composite film composition on the edge of a wedge-polished thin-film sample. Wide-area average Ir content is 4 at.%. Narrow-beam film composition far from nanopillars shows 2 at.% Ir content. Pillar composition on the edge of the film is 47 at.% Ir. It should be noted that while the electron beam is narrower than a nanopillar, secondary electron scattering means that the EDX probes the average composition over a slightly wider area than the electron beam width.18
Besides the surface migration kinetics, the second aspect that appears to be important for the formation of vertically aligned pillars as opposed to some form of laterally expanded structure or random nanodot formation is the anisotropic surface energy of noble metal clusters on an oxide surface.45,46,47 Surface energy anisotropy defines the shapes of various self-organized metal or oxide nanostructures and is not limited to the noble metals discussed here.48,49
Near-perfect lattice matching between the fcc metal and perovskite occurs in a cube-on-cube orientation that would expose the metal (001) plane at the surface. This lattice configuration does indeed occur inside the composite crystal but is unfavorable at the surface due to the significantly lower surface energy of the fcc (111) facets.50 As shown in Figure 6, the facet formation is visible at the exposed tips of Ir, Pt, Pd, and Rh nanopillars in SrTiO3. The preference for the (111) facets is particularly clear in the case of Pt, which is perfectly lattice matched with SrTiO3 at the growth temperature. When Pt metal is deposited on an atomically flat SrTiO3 (001) surface at high temperature, the metal forms epitaxial faceted islands instead of a uniform two-dimensional layer.39 This behavior explains why pillar growth is favored at moderately low growth rates, where metal adatoms have time to find the (111) facets of nanopillars on the film surface and, thus, promote the growth of existing pillars, rather than form new isolated metal clusters. If the growth rate is increased, the available migration time for surface adatoms is reduced before the next batch of deposited adatoms arrives and random nanodot formation dominates over pillar formation, as was shown in Figure 3.

Faceting at the tips of Ir, Pt, Pd, and Rh nanopillars. The face-centered-cubic lattices of these metals prefer to expose the (111) facets due to the lower surface energy. The Pd and Rh images show the exposed tips of pillars at the film surface. Ir and Pt images show terminated pillar growth inside the film. All images are 30 × 20 perovskite unit cells (12 nm × 8 nm).18,39
Despite the preference for exposing the (111) facets at the pillar growth front, the metal lattice of the pillars is growing approximately in the [001] direction, as can be seen in an x-ray diffraction Ir (111) pole figure in Figure 7b. Four slightly elongated diffraction peaks appear at 45° positions relative to the [100] and [010] in-plane directions of the SrTiO3 host lattice. This shows that the Ir metal does indeed grow in the \(\left[ {100} \right]_{{{\text{Ir}}}} || \, \left[ {100} \right]_{{{\text{SrTiO}}_{3} }}\) cube-on-cube configuration illustrated in Figure 7a and shown in the cross-sectional and plan views of Ir nanopillars in Figure 7c, d.

The metal nanopillars grow in a cube-on-cube configuration (a), as shown by the presence of four Ir (111) diffraction peaks, offset by 45° from the SrTiO3 [100] direction, in an X-ray diffraction pole figure (b). The cross-sectional (c) and plan-view (d) scanning transmission electron microscope images of a small (c) and a large (d) pillar show the presence of the Ir metal planes. Due to strain relaxation, the metal lattice is tilted from the host lattice by up to about 1°, which is why individual metal atom columns cannot be distinguished, but spread out into line shapes. Lattice plane tilting is visible in (c). Both pillars contain multiple grains, with each grain tilted around the a-, b-, or c-axis direction to relax the lattice mismatch strain between the metal and the SrTiO3 host crystal.18
Although noble metals are fairly well lattice matched with SrTiO3, strains of up to 5% are still present at room temperature. The strain relaxation inside a thin nanopillar occurs by lattice tilting,51 which can be observed in both cross-section and plan-view STEM images as a spread of the metal lattice into streaks corresponding to atomic planes, rather than showing individual atomic columns similar to the undistorted SrTiO3 lattice. The Ir pillars in particular show that multiple metal grains are present with different tilt directions. Presumably, the multigrain structure allows strain to be relaxed in all three lattice directions. The plan-view image of a large-diameter pillar in Figure 7d also shows that the pillars are not round but show distinct in-plane faceting along the (100) and (110) lattice planes. The shape of the nanopillars is mainly determined by the competition between the strain energy and interfacial energy. The strain energy dominates over interfacial energy in large pillars, and the pillars exhibit square or faceted shapes. The interfacial energy dominates in small pillars, which, therefore, exhibit a circular shape (Figure 7).21,22
Benefits of nanopillars for water splitting
The functional benefits of the nanopillar composite material for a PEC electrode can be seen in a cyclic voltammetry experiment in a PEC cell that is shown in Figure 1. A bias is applied on a test electrode in an electrolyte, and the current is recorded as shown in Figure 8a. The current is proportional to the charge generated by light and injected into the electrolyte (or water), and it is proportional to the number of hydrogen or oxygen molecules that are generated. The magnitude of the current is, thus, a measure of how well an electrode performs under identical light illumination conditions. In this case, several Ir nanopillar composite films are compared with homogeneously doped Ir:SrTiO3 films, and it is clear that the observed current is larger by nearly an order of magnitude. No current flows for either type of electrode in dark conditions, when no photocarriers are generated.

(a) Comparison of the photoelectrochemical performance of homogeneously doped Ir:SrTiO3 water-splitting electrodes and Ir pillar—Ir:SrTiO3 nanocomposite electrodes. The observed current density of the composite electrodes is an order of magnitude higher than for the homogeneous films. The measurement is done with visible light illumination from a 1 kW xenon lamp filtered with a 420-nm-long pass filter in a 0.1 M KOH electrolyte.18 (b) Only the surface layer is active for a homogeneous film. (c) Charge from the whole film volume can be collected in the nanocomposite device.
In a homogeneously doped film, shown in Figure 8b, only a thin surface layer sustains a space–charge layer and is, thus, electronically active, while most of the incident light is absorbed deeper in the thin-film electrode where the generated photocarriers are lost to recombination. In contrast, the high work function of Ir metal ensures that a Schottky junction forms at every nanopillar interface with the n-type Ir (2%):SrTiO3 host lattice, forming coaxial space–charge regions around each nanopillar as shown in Figure 8c. In this composite material, essentially the whole volume of the film becomes electronically active while the electron collection length in the semiconductor is minimized and the Ir metal nanopillars provide an efficient charge transport path from deep layers of the film to the surface. Luckily, Ir is also an excellent electrocatalyst,26 promoting the water-splitting reaction at the tips of the nanopillars. The nanocomposite material, thus, separates the functional regions of light absorption and charge generation from the site where water splitting occurs but does so without the need for patterning or heterostructure growth. The material is formed by self-organized nanopillar segregation in a film growth process that is identical to conventional oxide thin-film growth by PLD.
The excellent PEC performance of the nanopillar composite films also shows that the rule of thumb that catalysts require large surface areas is not necessarily true for composite PEC electrodes where the water splitting occurs mainly at the tips of the Ir nanopillars. The nanocomposite structure, thus, focuses charge collected from a large area to the nanoscale reaction points, and even though the total area of the nanopillar tips is only about 1% of the total film surface area, the total current injected into water is an order of magnitude higher than for a planar film.
Summary
The PEC response of the Ir nanopillar–Ir:SrTiO3 composite electrodes is in a sense a lucky case, because Ir doping of SrTiO3 forms a semiconductor with a sufficiently high Fermi level to form a Schottky junction with Ir metal. This behavior is not universal for other noble metals that we have looked at. Based on powder photocatalyst studies, Rh is likely to be the best dopant for constructing a p-type hydrogen evolution electrode. While a Rh nanopillar composite can be grown (Figure 6), the work function of Rh metal matches the deep Fermi level position of Rh:SrTiO3, which means that no Schottky-type space–charge regions form. In the case Pt, which has the best lattice match with SrTiO3, we have observed excellent nanopillar growth, but since Pt doping does not lead to gap narrowing of SrTiO3, the composite material is not useful as a visible light PEC electrode. Several metals are known to form nanopillars in SrTiO3, and the list of elements is likely to be broader than the four examples shown here (Pt, Pd, Ir, Rh). A clear difference between the various pillars is the growth habit. Pt clearly follows the crystallographic [001] direction in SrTiO3, while other metals, especially Pd, appear to prefer growth along the [011] direction, creating meandering pillars.18 While nanopillars other than Ir appear to have no direct applications for water-splitting applications, these nanowires create nanoscale Schottky point contacts with an n-type substrate such as Nb:SrTiO3 at the bottom of each pillar. It is also possible to electrochemically etch the metal nanopillars, forming well-defined pores with a diameter of a few nanometers but a length of tens or hundreds of nanometers in the oxide host material.
References
Z. Wang, C. Li, K. Domen, Chem. Soc. Rev. 48, 2109 (2019)
J. Luo, J.-H. Im, M.T. Mayer, M. Schreier, M.K. Nazeeruddin, N.-G. Park, S.D. Tilley, H.J. Fan, M. Grätzel, Science 345, 1593 (2014)
T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi, K. Domen, Nature 581, 411 (2020)
K.T. Fountaine, H.J. Lewerenz, H.A. Atwater, Nat. Commun. 7, 13706 (2016)
K. Furuhashi, Q. Jia, A. Kudo, H. Onishi, J. Phys. Chem. C 117, 19101 (2013)
D.H.K. Murthy, H. Matsuzaki, Q. Wang, Y. Suzuki, K. Seki, T. Hisatomi, T. Yamada, A. Kudo, K. Domen, A. Furube, Sustain. Energy Fuels 3, 208 (2019)
N. Osawa, R. Takahashi, M. Lippmaa, Appl. Phys. Lett. 110, 263902 (2017)
M.I. Cohen, R.F. Blunt, Phys. Rev. 168, 929 (1968)
R. Takahashi, M. Lippmaa, ACS Appl. Mater. Interfaces 9, 21314 (2017)
S. Kawasaki, R. Takahashi, M. Lippmaa, J. Phys. Chem. C 123, 15551 (2019)
W. Zhang, D. Yan, K. Appavoo, J. Cen, Q. Wu, A. Orlov, M.Y. Sfeir, M. Liu, Chem. Mater. 29, 4036 (2017)
A. Fujishima, K. Honda, Nature 238, 37 (1972)
L. Zhang, M. Zhou, Z. Zhang, J. Yuan, B. Li, W. Wen, J. Tian, J. Mater. Chem. A 7, 22229 (2019)
A. Kudo, S. Yoshino, T. Tsuchiya, Y. Udagawa, Y. Takahashi, M. Yamaguchi, I. Ogasawara, H. Matsumoto, A. Iwase, Faraday Discuss. 215, 313 (2019)
Y. Yan, D. Wang, P. Schaaf, Dalton Trans. 43, 8480 (2014)
Y. Pihosh, V. Nandal, T. Minegishi, M. Katayama, T. Yamada, K. Seki, M. Sugiyama, K. Domen, ACS Energy Lett. 5, 2492 (2020)
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger, Science 270, 1789 (1995)
S. Kawasaki, R. Takahashi, T. Yamamoto, M. Kobayashi, H. Kumigashira, J. Yoshinobu, F. Komori, A. Kudo, M. Lippmaa, Nat. Commun. 10, 11818 (2016)
H. Song, C. Li, C.N. Van Nguyen, H.-J. Liu, R. Qi, R. Huang, Y.-H. Chu, C.-G. Duan, J. Mater. Res. 32, 2790 (2017)
A. Imai, X. Cheng, H.L. Xin, E.A. Eliseev, A.N. Morozovska, S.V. Kalinin, R. Takahashi, M. Lippmaa, Y. Matsumoto, V. Nagarajan, ACS Nano 7, 11079 (2013)
L. Mohaddes-Ardabili, H. Zheng, S.B. Ogale, B. Hannoyer, W. Tian, J. Wang, S.E. Lofland, S.R. Shinde, T. Zhao, Y. Jia, L. Salamanca-Riba, D.G. Schlom, M. Wuttig, R. Ramesh, Nat. Mater. 3, 533 (2004)
A. Chen, Q. Su, H. Han, E. Enriquez, Q. Jia, Adv. Mater. 31, 1803241 (2019)
A. Kudo, Y. Miseki, Chem. Soc. Rev. 38, 253 (2009)
S. Kawasaki, R. Takahashi, K. Akagi, J. Yoshinobu, F. Komori, K. Horiba, H. Kumigashira, K. Iwashina, A. Kudo, M. Lippmaa, J. Phys. Chem. C 118, 20222 (2014)
S. Kawasaki, K. Akagi, K. Nakatsuji, S. Yamamoto, I. Matsuda, Y. Harada, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, C. Sakai, H. Niwa, M. Oshima, K. Iwashina, A. Kudo, J. Phys. Chem. C 116, 24445 (2012)
T. Reier, M. Oezaslan, P. Strasser, ACS Catal. 2, 1765 (2012)
J. Son, P. Moetakef, B. Jalan, O. Bierwagen, N.J. Wright, R. Engel-Herbert, S. Stemmer, Nat. Mater. 9, 482 (2010)
Y. Kozuka, Y. Hikita, C. Bell, H.Y. Hwang, Appl. Phys. Lett. 97, 012107 (2010)
M. Lippmaa, N. Nakagawa, M. Kawasaki, S. Ohashi, Y. Inaguma, M. Itoh, H. Koinuma, Appl. Phys. Lett. 74, 3543 (1999)
S. Kawasaki, K. Nakatsuji, J. Yoshinobu, F. Komori, R. Takahashi, M. Lippmaa, K. Mase, A. Kudo, Appl. Phys. Lett. 101, 033910 (2012)
R. Meyer, R. Waser, J. Helmbold, G. Borchardt, J. Electroceram. 9, 101 (2002)
K. Szot, W. Speier, U. Breuer, R. Meyer, J. Szade, R. Waser, Surf. Sci. 460, 112 (2000)
X. Hou, R. Takahashi, T. Yamamoto, M. Lippmaa, J. Cryst. Growth 462, 24 (2017)
Y. Nishihata, J. Mizuki, T. Akao, H. Tanaka, M. Uenishi, M. Kimura, T. Okamoto, N. Hamada, Nature 418, 164 (2002)
X.D. Zhu, W. Si, X.X. Xi, Q. Jiang, Appl. Phys. Lett. 78, 460 (2001)
M. Lippmaa, N. Nakagawa, M. Kawasaki, S. Ohashi, H. Koinuma, Appl. Phys. Lett. 76, 2439 (2000)
M. Lee, R. Arras, R. Takahashi, B. Warot-Fonrose, H. Daimon, M.-J. Casanove, M. Lippmaa, ACS Omega 3, 2169 (2018)
C. Viti, M. Ferrari, Eur. J. Mineral. 18, 823 (2006)
M. Lippmaa, S. Kawasaki, R. Takahashi, T. Yamamoto, Jpn. J. Appl. Phys. 59, 010501 (2020)
A. Altberg, G. Atiya, V. Mikhelashvili, G. Eisenstein, W.D. Kaplan, J. Mater. Sci. 49, 3917 (2014)
J.H. Los, W.J.P. van Enckevort, E. Vlieg, E. Flöter, J. Phys. Chem. B 106, 7321 (2002)
T.J. Anderson, S. Ryu, H. Zhou, L. Xie, J.P. Podkaminer, Y. Ma, J. Irwin, X.Q. Pan, M.S. Rzchowski, C.B. Eom, Appl. Phys. Lett. 108, 151604 (2016)
K.T. Jacob, T.H. Okabe, T. Uda, Y. Waseda, J. Alloys Compd. 288, 188 (1999)
Z. Hiroi, H. Hayamizu, T. Yoshida, Y. Muraoka, Y. Okamoto, J. Yamaura, Y. Ueda, Chem. Mater. 25, 2202 (2013)
A. Chen, Z. Bi, Q. Jia, J.L. MacManus-Driscoll, H. Wang, Acta Mater. 61, 2783 (2013)
W. Zhang, A. Chen, Z. Bi, Q. Jia, J.L. MacManus-Driscoll, H. Wang, Curr. Opin. Solid State Mater. Sci. 18, 6 (2014)
J.L. MacManus-Driscoll, Adv. Funct. Mater. 20, 2035 (2010)
G. Zhong, F. An, Y. Bitla, J. Wang, X. Zhong, M. Ye, Y. Zhang, W. Gao, X. Pan, S. Xie, J. Li, J. Mater. Chem. C 6, 4854 (2018)
R. Takahashi, H. Misumi, T. Yamamoto, M. Lippmaa, Cryst. Growth Des. 14, 1264 (2014)
R. Tran, Z. Xu, B. Radhakrishnan, D. Winston, W. Sun, K.A. Persson, S.P. Ong, Sci. Data 3, 160080 (2016)
X. Weng, M. Hennes, A. Coati, A. Vlad, Y. Garreau, M. Sauvage-Simkin, E. Fonda, G. Patriarche, D. Demaille, F. Vidal, Y. Zheng, Phys. Rev. Mater. 2, 106003 (2018)
Acknowledgments
The work was supported in part by JSPS Kakenhi Grant Nos. 17H04895, 20H02622, and JST-Mirai Program Grant No. JPMJMI18G5. A part of this work was supported by Nagoya University Microstructural Characterization Platform as a Program of “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lippmaa, M., Kawasaki, S., Takahashi, R. et al. Nanopillar composite electrodes for solar-driven water splitting. MRS Bulletin 46, 142–151 (2021). https://doi.org/10.1557/s43577-021-00030-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43577-021-00030-6