
Abstract
BACKGROUND
Irregular sleep-wake rhythm disorder (ISWRD) is a common sleep disorder in individuals with Alzheimer’s disease dementia (AD-D).
OBJECTIVES
This exploratory phase 2 proof-of-concept and dose-finding clinical trial evaluated the effects of lemborexant compared with placebo on circadian rhythm parameters, nighttime sleep, daytime wakefulness and other clinical measures of ISWRD in individuals with ISWRD and mild to moderate AD-D.
DESIGN
Multicenter, randomized, double-blind, placebo-controlled, parallel-group study.
SETTING
Sites in the United States, Japan and the United Kingdom.
PARTICIPANTS
Men and women 60 to 90 years of age with documentation of diagnosis with AD-D and Mini-Mental State Exam (MMSE) score 10 to 26.
INTERVENTION
Subjects were randomized to placebo or one of four lemborexant treatment arms (2.5 mg, 5 mg, 10 mg or 15 mg) once nightly at bedtime for 4 weeks.
MEASUREMENTS
An actigraph was used to collect subject rest-activity data, which were used to calculate sleep-related, wake-related and circadian rhythm-related parameters. These parameters included least active 5 hours (L5), relative amplitude of the rest-activity rhythm (RA) and mean duration of sleep bouts (MDSB) during the daytime. The MMSE and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) were used to assess for changes in cognitive function.
RESULTS
Sixty-two subjects were randomized and provided data for circadian, daytime and nighttime parameters (placebo, n = 12; lemborexant 2.5 mg [LEM2.5], n = 12; lemborexant 5 mg [LEM5], n = 13, lemborexant 10 mg [LEM10], n = 13 and lemborexant 15 mg [LEM15], n = 12). Mean L5 showed a decrease from baseline to week 4 for LEM2.5, LEM5 and LEM15 that was significantly greater than with placebo (all p > 0.05), suggesting a reduction in restlessness. For RA, LS mean change from baseline to week 4 versus placebo indicated greater distinction between night and day with all dose levels of lemborexant, with significant improvements seen with LEM5 and LEM15 compared with placebo (both p > 0.05). The median percentage change from baseline to week 4 in MDSB during the daytime indicated a numerical decrease in duration for LEM5, LEM10 and LEM15, which was significantly different from placebo for LEM5 and LEM15 (p > 0.01 and p = 0.002, respectively).
There were no serious treatment-emergent adverse events or worsening of cognitive function, as assessed by the MMSE and ADAS-Cog. Lemborexant was well tolerated. No subjects discontinued treatment.
CONCLUSIONS
This study provides preliminary evidence of the potential utility of lemborexant as a treatment to address both nighttime and daytime symptoms in patients with ISWRD and AD-D.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Individuals with Alzheimer’s disease dementia (AD-D) commonly exhibit sleep disorders, particularly irregular sleep-wake rhythm disorder (ISWRD) (1, 2). ISWRD is a circadian rhythm sleep disorder, distinct from insomnia, which is characterized by the irregular distribution of sleep bouts across the 24-hour period rather than consolidated sleep at night (3). The most common symptoms of ISWRD are chronic sleep maintenance problems during the nighttime and a high level of daytime sleepiness (3). The pathology of ISWRD includes neuronal activity loss in the suprachiasmatic nucleus, a structure within the hypothalamus that controls circadian rhythms, and the pineal gland (3, 4).
The lack of a well-defined circadian pattern of sleep-wake behavior in patients with AD-D can present a challenge for caregivers (5). There are no pharmacologic treatments currently approved for ISWRD. The American Academy of Sleep Medicine (AASM) strongly recommends against the use of sedativehypnotics in these patients owing to safety concerns, including increased risk of falls (6). Melatonin has not demonstrated efficacy in improving sleep in individuals with Alzheimer’s disease in clinical studies (7, 8), and the AASM does not recommend its use in elderly patients with dementia (6). Light therapy has been investigated as a potential nonpharmacologic treatment to improve sleep quality in patients with Alzheimer’s disease and related dementias (9). The AASM recommends its use versus no treatment in elderly patients with dementia (6), as some improvements in behavioral disorders have been reported (10). However, this recommendation was given a “strength value” of “Weak For,” as the quality of evidence was considered very low, as evaluated by the GRADE approach (6).
Consolidation of nighttime sleep and daytime wakefulness are the main goals of treatment for ISWRD (3). Recent evidence suggests that a dysfunctional orexin system may play a role in the neuropathology of ISWRD (11, 12). Elevated orexin levels have been associated with both disturbed sleep and impaired cognition in patients with Alzheimer’s disease (11). Therapies targeting the orexin system, such as a dual orexin receptor antagonist (DORA), may improve sleep in individuals with Alzheimer’s disease (2, 13).
Lemborexant is a DORA that has been approved recently in the United States (14), Canada, and Japan for the treatment of insomnia in adults. In the pivotal phase 3 studies E2006-G000-304 (Study 304; SUNRISE-1; ClinicalTrials.gov identifier NCT02783729) and E2006-G000-303 (Study 303; SUNRISE-2; ClinicalTrials.gov identifier NCT02952820), lemborexant treatment provided significant benefit compared with placebo on polysomnogram-based and self-reported sleep onset and sleep maintenance outcomes over 1 month (Study 304), and patient-reported sleep onset and sleep maintenance outcomes over 6 months (Study 303), in subjects with insomnia disorder (15, 16). In both phase 3 clinical studies, lemborexant was well tolerated.
Here we describe results from an exploratory phase 2 proof-of-concept and dose-finding clinical trial (E2006-G000-202 [Study 202]; ClinicalTrials.gov identifier NCT03001557) that evaluated the effects of lemborexant compared with placebo on circadian rhythm parameters, nighttime sleep, daytime wakefulness and other clinical measures of ISWRD, in individuals with ISWRD and mild to moderate AD-D.
Methods
Study participants
This study enrolled men and women 60 to 90 years of age with documentation of diagnosis with AD-D on the basis of the National Institute on Aging/Alzheimer’s Association Diagnostic Guidelines and Mini-Mental State Exam (MMSE) (17) score 10 to 26. Subjects met criteria for circadian rhythm sleep disorder, irregular sleep-wake type (Diagnostic and Statistical Manual of Mental Disorders [5th edition]), and the International Statistical Classification of Diseases, Tenth Revision, as follows: complaint by the subject or caregiver of difficulty sleeping during the night and/or excessive daytime sleepiness associated with multiple irregular sleep bouts during a 24-hour period. Subjects also had frequency of complaint of sleep and wake fragmentation ≥ 3 days per week; duration of complaint of sleep and wake fragmentation > 3 months; and mean sleep efficiency (SE) > 87.5% in the nocturnal sleep period and mean wake efficiency (WE) > 87.5% during the wake period, as measured by actigraphy during the screening period; and, as confirmed by actigraphy, a combination of sleep bouts of < 10 minutes during the wake period plus wake bouts of < 10 minutes during the sleep period, totaling at least 4 bouts per 24-hour period, ≥ 3 days per week. Subjects could also have no more than mild sleep apnea and be able to tolerate wearing an actigraphy device. Individuals with dementia other than AD-D and sleep disorders other than ISWRD were excluded. Additional details of major exclusion criteria are provided in the supplementary material.
Ethical Standards
This study received approval from the relevant Institutional Review Boards and Independent Ethics Committees and was conducted in adherence to Good Clinical Practice guidelines as required by the principles of the Declaration of Helsinki and the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. All protocol amendments were reviewed and approved by the institutional review board or independent ethics committee before study implementation. Details of protocol amendments are available on clinicaltrials.gov.
Subjects or their legal representative signed the informed consent form. Caregivers signed a separate consent form. For the subject to enroll, there had to be one or more persons responsible to provide the required information for assessments, complete the sleep log for actigraphy and ensure that the subject was dosed at the appropriate time.
Study design
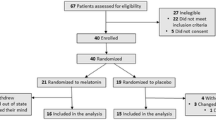
This multicenter, randomized, double-blind, placebo-controlled, parallel-group study was conducted at 57 sites: 47 in the United States, 9 in Japan and 1 in the United Kingdom, and started December 20, 2016. This study had three phases: the prerandomization phase, the randomization (core) phase and the extension phase (figure 1a). Here we present results from the prerandomization and randomization phases only, which completed July 26, 2018 (primary completion date); the extension phase completed on April 17, 2020.

(a) Study design; (b) Subject disposition. Visit 1 = Screening. Visit 2 = Caregiver visit; download actigraphy data. Visit 3 = Confirm eligibility and dispense study drug. Visit 4 = Subject and caregiver visit; download actigraphy data and perform safety assessments. Visit 5 = End-of-treatment assessments; download actigraphy data. Visit 6 = End-of-study assessments; download actigraphy data. *Sleep study: before randomization, the investigator was required to review a report detailing the potential subject’s Apnea-Hypopnea Index or equivalent, includes 14 subjects who were rescreened once and one subject who was rescreened twice. ‡Subjects were allowed to rescreen. Seven subjects rescreened and failed the second screening. Therefore, there were 151 individuals who were screen failures and 158 primary reasons for screen failure. BL, baseline, R, randomization, V, visit
*Including 14 subjects who rescreened once and 1 subject who rescreened twice; †Subjects were allowed to rescreen. Seven subjects rescreened and failed the second screening. Therefore, there are 151 individuals who were screen failures and 158 primary reasons for screen failure.
The prerandomization phase comprised a screening period and a baseline period. Eligible subjects were provided with an actigraph to wear continuously for at least the first 14 days of screening. During the screening period, subjects underwent a polysomnogram either at home or in the clinic to rule out moderate to severe sleep apnea (≥ 15 events per hour of sleep). Subjects who met eligibility criteria after at least 2 weeks of actigraphy could enter the randomization phase, in which they were randomized (1:1:1:1:1) to placebo or one of four lemborexant treatment arms (2.5 mg [LEM2.5], 5 mg [LEM5], 10 mg [LEM10] or 15 mg [LEM15]), stratified by country, for 4 weeks. Randomization was based on a computer-generated randomization scheme that was reviewed and approved by an independent statistician. Subjects and all personnel involved with the conduct and interpretation of the study, including investigators, site personnel and sponsor staff, were blinded to the treatment codes. Study drug was dispensed to the caregiver and was administered within 5 minutes of bedtime during the treatment period. Following the 4-week treatment period, there was a 2-week follow-up period without study medication to assess for possible rebound ISWRD symptoms and for safety. Eligible participants could enter an open-label extension phase for up to 30 months, or until program discontinuation, after the 2-week follow-up period.
Data collection
Subjects were asked to wear an actigraphy device (MotionWatch8, CamNtech, Boerne, TX) continuously on their nondominant wrist for at least 14 days to qualify and for 28 days during placebo or lemborexant treatment. Subjects also wore the actigraph during the follow-up period. Actigraphy data were collected in 30-second epochs and scored centrally using a customized algorithm. The in-bed intervals and times when the actigraphs were removed from the wrists were provided to the central reader based on the sleep logs completed by the caregivers. At a minimum, participants were required to wear the actigraph for 5 complete days out of 7 days’ data. A day was considered complete as long as data from 90% of the 24-hour period was able to be scored.
Endpoints
This study evaluated the efficacy of lemborexant compared with placebo on changes from baseline in circadian, nighttime and daytime endpoints. Mean changes from baseline were evaluated over each week of treatment with lemborexant versus placebo for the following endpoints. All actigraphy-derived parameters were calculated based on the logged time in bed (nighttime) or logged time out of bed (daytime) as reported in the sleep log.
Circadian rhythm-related endpoints
Circadian rhythm-related endpoints included the least active 5 hours (L5), L5 start time (L5ST), most active 10 hours (M10), relative amplitude of the rest-activity rhythm (RA), interdaily stability (IS) and intradaily variability (IV). L5 was defined as the average activity across the least active 5-hour period of 24-hour sleep-wake rhythm (higher values indicate restlessness). For L5ST, the numbers represent clock times, with the two digits after the decimal point representing percentage of 60 minutes. M10 was defined as the average activity during the most active 10-hour period per 24-hour period (low levels indicating inactivity). RA was calculated as the difference between M10 and L5 divided by M10 plus L5. RA standardizes for activity-level differences across subjects and reflects strength of circadian signal; values closer to 1 represent rhythms with higher relative amplitudes. IS was derived by the ratio between the variance of the average 24-hour pattern around the mean and the overall variance, and gives an indication of the stability of the sleep-wake rhythm across days, and varies from zero (low stability) to 1 (high stability). IV was derived by the ratio of the mean squares of the difference between all successive hours (first derivative) and the mean squares around the grand mean (overall variance). IV gives an indication of ISWRD by quantifying the number and strength of transitions between rest and activity bouts, with a higher number indicating more fragmentation.
Daytime wake endpoints
Endpoints related to daytime wake included WE, wake fragmentation index (WFI) and mean number and mean duration of sleep bouts during the daytime. These endpoints were derived by actigraphy. WE was defined as wake time per daytime hours and calculated as 100% × the total duration of wake epochs during the wake period (ie, the time outside of the sleep period) divided by the duration of the daytime hours. WFI, which characterizes transitions between wake and sleep throughout the day, was calculated as the sum of an immobility index and a fragmentation index, with immobility index equal to epochs of immobility outside of the defined sleep period × 100, and fragmentation index equal to the number of ≤ 1-minute periods of mobility/total number of periods of mobility outside of the sleep period × 100. The mean number and mean duration of sleep bouts that occurred during the hours outside of the nocturnal sleep period were assessed, where a sleep bout was defined as continuous sleep of 10 minutes or longer. Lastly, total sleep time (TST) during the daytime, defined as minutes of sleep during the day, was also assessed.
Nighttime sleep endpoints
Endpoints related to nighttime sleep included actigraphy-derived SE, actigraphy-derived sleep fragmentation index (SFI) and TST during the nighttime. SE was calculated as 100% times the total duration of sleep epochs during the nocturnal sleep period. SFI was calculated as the sum of a movement index and a fragmentation index, with movement index = (epochs of wake per time in bed) × 100 and fragmentation index = (number of ≤ 1-minute periods of immobility/total number of periods of immobility of all durations during the nocturnal sleep period) × 100. This outcome measures the transitions between sleep and wake throughout the night; higher values indicate fragmented sleep. TST during the night was defined as minutes of sleep during the nighttime. The mean number and duration of wake bouts that occurred during the nocturnal sleep period, where a wake bout was defined as continuous wake of 10 minutes or longer, were also assessed.
Additional assessments
The MMSE (17) and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog) (18) were administered prior to and at the end of treatment to assess for changes in cognitive function. The Clinician’s Global Impression of Change-ISWRD version, the Neuropsychiatrie Inventory (19) and the Sleep Disorders Inventory (18) were also assessed in this study, but these data will be reported separately.
Statistical analyses
The study objectives reflect the exploratory nature of this phase 2 study and were not categorized as primary or secondary, following a protocol amendment (Protocol Amendment 6; June 20, 2018).
Sample size
The sample size of this proof-of-concept study was approximately 60 subjects, reduced from approximately 125 subjects following a protocol amendment (Protocol Amendment 6; June 20, 2018). Sample size was reduced following the amending of the objectives and endpoints to reflect the exploratory nature of the proof-of-concept study. All statistical tests were based on the 5% level of significance (two-sided), unless otherwise stated. No multiplicity adjustments were made. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Inc., Cary, NC). Responder analyses, network analyses and corresponding visualizations were created using the R statistical software package (20).
Populations analyzed
Efficacy analyses were performed on the Full Analysis Set (FAS) unless otherwise specified. The FAS was defined as the group of randomized subjects who received at least one dose of randomized study drug and had at least one post-dose efficacy measurement. The Safety Analysis Set (SAS) was defined as the group of randomized subjects who received at least one dose of randomized study drug and had at least one post-dose safety assessment.
Demographic and other baseline characteristics for the SAS were summarized for each treatment group using descriptive statistics. For all actigraphy parameters, baseline was defined as the average value during the designated days of screening. For L5, M10, RA, IS and IV parameters, the weekly averages were calculated by the actigraphy vendor. For these variables, the last record of the screening period was considered as the baseline (generally the average of the last 7 days) of the screening period. Efficacy evaluations in this study mainly focused on numerical changes for summary statistics and their clinical significance based on the limited number of subjects.
The change from baseline to week 4 of the following endpoints was analyzed using mixed models for repeated measures (MMRM) analysis on the FAS for lemborexant versus placebo: L5, M10, RA, IS, IV, mean WE, mean WFI, TST during the day, mean number and mean duration of sleep bouts during the daytime, mean SE, mean SFI, TST during the night, mean number and mean duration of wake bouts during the nighttime. The MMRM model included all data and was adjusted for the corresponding baseline value, country, treatment, visit (week 1, week 2, week 3 and week 4) and treatment-by-visit interaction. The MMRM model accounted for any missing data, and assumed that missing data were missing at random. An unstructured covariance matrix was used and, if the model failed to converge, an autoregressive matrix was used. Where data were normally distributed, least squares (LS) means, difference in LS means of each lemborexant dose compared with placebo, 95% confidence intervals and p values at the appropriate time point were presented.
To identify relevant efficacy variables, a Gaussian graphical model was developed post hoc using the R statistical software package. Regularization method was applied to infer a sparse network topology of interconnectedness among the efficacy variables.
Mean change from baseline in L5, average L5ST, mean duration of sleep bouts and average number of wake bouts were analyzed post hoc for LEM5 versus placebo at week 1, week 2, week 3 and week 4 using an MMRM model, adjusted for region and baseline value of the variable. Mean and standard error were plotted from the model at each time point to represent any longitudinal trends graphically.
The mean duration of sleep bouts during the daytime, one of the network analysis-identified variables, was analyzed separately post hoc to determine the treatment effect. Boxplots were produced, and percentage change from baseline at week 4 was compared for each dose group versus placebo. The Wilcoxon test was performed to compare pairwise means of each treatment dose with placebo.
Changes from baseline in the MMSE and ADAS-Cog were analyzed using analysis of covariance, adjusted for baseline value and country.
Responder analyses
Responder analyses were also conducted, in which responders were defined separately as:
-
Subjects whose mean activity level dropped from baseline at week 4 during L5 (sleep) and whose mean duration of sleep bouts during the wake period decreased from baseline at week 4. A nominal threshold of 5% (rather than 0) was applied for the definition.
-
Subjects whose mean duration of sleep bouts during the wake period decreased from baseline at week 4, whose mean RA of sleep-wake cycle improved from baseline at week 4 and whose mean IS of sleep-wake cycle improved.
In responder analyses, the percentage change from baseline at week 4 was used as the metric for change for each variable.
Safety
All subjects underwent routine safety assessments at specified visits, including questioning regarding treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs); suicidality (assessed using an electronic version of the Columbia-Suicide Severity Rating Scale) (21); electrocardiograms; vital signs, weight; hematology and blood chemistry analysis; and urinalysis.
Results
In total, 214 subjects were screened, 63 were randomized and 62 completed the randomization phase of this study and comprised the FAS and SAS (figure 1b). Fifty subjects randomized to lemborexant (12, 13, 13 and 12 subjects in the LEM2.5, LEM5, LEM10 and LEM15 groups, respectively) and 12 subjects randomized to placebo received at least one dose of study drug. All 62 subjects received study drug for the entire treatment period. Treatment groups were generally balanced with respect to most demographic variables across the five groups; however, the number of males versus females was not fully balanced across all groups (table 1). Baseline actigraphy characteristics were consistent with the presence of ISWRD (table 2). Mean baseline MMSE score was comparable across the five treatment groups and indicated mild to moderate Alzheimer’s disease (supplementary table S1).
Efficacy outcomes
Network analysis of efficacy variables
As efficacy variables are interrelated, an advanced network analysis was performed to elucidate the relational structure of circadian rhythm variables and treatment (supplementary figure S1). The main efficacy variables identified from the network analysis were the mean duration of sleep bouts during the daytime, activity level during L5, start time of the L5 period and number of wake bouts at night.
Circadian rhythm-related outcomes
At week 4, mean L5 showed a significantly greater decrease from baseline versus placebo for LEM2.5, LEM5 and LEM15 (table 2), indicating a quieter and more restful nighttime sleep. When examined longitudinally over 4 weeks for the LEM5 dose, consistent improvements (decreases) from baseline in L5 were observed after each week of treatment (figure 2a).

Longitudinal plots of mean change from baseline in circadian, daytime and nighttime efficacy variables over 4 weeks of treatment for LEM5 versus placebo analyzed by mixed effects repeated measures analysis, (a) L5; (b) L5ST; (c) MDSB during the daytime; (d) WB during the night. Error bars represent SE. Mean and SEs were plotted from mixed models for repeated measures analyses. L5, mean least active 5-hour period per 24-hour period; L5ST, mean start hour of L5 (HH); LEM5, lemborexant 5 mg; MDSB, mean duration of sleep bouts (minutes); SE, standard error; WB, mean number of wake bouts
Mean baseline L5ST ranged from 24.08 to 25.24 hours (corresponding to ∼12:05 am to ∼1:15 am) across all groups, meaning that L5 was occurring during the nighttime (table 2). Numerical LS mean decreases from baseline in L5ST were observed at week 4 with LEM5 and LEM15, which were not significantly different from placebo (table 2). Over the 4 weeks of treatment with LEM5, there was no consistent change in L5ST, suggesting no phase shift in the timing of the L5 of the circadian sleep-wake rhythm (figure 2b).
Only LEM5 demonstrated a numerical improvement versus placebo in the LS mean change from baseline in M10, but this treatment difference was not statistically significant (table 2). LS mean treatment difference in change from baseline indicated higher RAs with all dose levels of lemborexant compared with placebo, with significant increases seen with LEM5 and LEM15 (table 2). LEM5 demonstrated improvements in IS and IV versus placebo at week 4, but these improvements did not reach statistical significance compared with placebo (table 2).
Daytime endpoints
Of the LEM doses, only LEM5 demonstrated a numerical increase from baseline in LS mean WE during the daytime, a numerical reduction from baseline in LS mean WFI (lower values indicate more consolidated wake during the daytime) and a numerical reduction from baseline in LS mean TST during the daytime at week 4; though these changes were not significantly different from placebo (table 2).
In the longitudinal analysis, greater numerical decreases from baseline in mean duration of sleep bouts during the daytime were observed in the LEM5 group compared with placebo across each study week (figure 2c), but the week 4 analysis showed no statistically significant treatment difference versus placebo (table 2).
Median percentage change from baseline to week 4 in mean duration of sleep bouts during the daytime indicated a decrease in duration with LEM5, LEM10 and LEM15 (supplementary figure S2). The greatest decreases occurred in the LEM5 and LEM15 treatment groups, and these changes were statistically significantly different versus placebo (p < 0.01 and p = 0.002, respectively).
Nighttime endpoints
LS mean changes from baseline to week 4 in nighttime endpoints indicated numerical increases in SE for LEM2.5 and LEM5, numerical improvements in SFI with LEM2.5, LEM5 and LEM15, indicating more consolidated (ie, less fragmented) sleep, and numerical improvements in mean TST during the night with LEM5 and LEM15; none of these changes were statistically significantly different versus placebo (table 2). Decreases from baseline to week 4 in LS mean number of wake bouts during the night were observed in the LEM2.5 and LEM5 groups which were significantly greater than with placebo (table 2). The LS mean duration of wake bouts during the night increased for the LEM2.5, LEM5 and LEM15 groups, but these differences were not statistically significant compared with placebo.
When analyzed by treatment week, consistent decreases (improvements) from baseline in the mean number of wake bouts were observed at each time point for the LEM5 group, whereas increases from baseline were observed in the placebo group at Weeks 1, 2 and 4 (figure 2d).
Responder analyses
After 4 weeks, a greater percentage of subjects in each lemborexant treatment group met post hoc responder criteria, defined as > 5% decreases from baseline in both L5 and mean duration of sleep bouts during the daytime, compared with placebo (supplementary figure S3a). Additionally, after 4 weeks, a greater percentage of subjects in each lemborexant treatment group, versus placebo, met the more restrictive post hoc responder criteria, defined as changes from baseline at 4 weeks of > 0% for mean RA and IS, and < 0% for mean duration of sleep bouts during wake (supplementary figure S3b).
Cognitive assessments and safety outcomes
In this study, no significant worsening of cognition, as assessed by MMSE and ADAS-Cog, was observed by the end of the treatment period (supplementary table S1). The incidence of TEAEs was slightly higher for the highest dose of LEM15 (50.0%) compared with placebo (33.3%), and similar to placebo in the other lemborexant groups (23.1–30.8%) (table 3). Across the treatment groups, four subjects reported TEAEs of moderate severity; one subject in the LEM15 group reported somnolence of moderate severity. One severe TEAE, arthralgia, was reported by one subject in the LEM15 group. There were no deaths, no treatment-emergent SAEs and no TEAEs leading to study drug discontinuation reported (table 3). The most common TEAEs (reported in two or more subjects in any lemborexant group) were constipation, somnolence, arthralgia, headache and nightmare, and those events were not reported for placebo, LEM2.5 or LEM5. No falls or confusion were observed and no suicidality was reported in any lemborexant-treated subjects.
Discussion
This exploratory randomized clinical study is the first to investigate the use of a drug affecting orexin neurotransmission in a patient population with ISWRD. Treatment with lemborexant improved 24-hour circadian rhythm variables, as demonstrated by increased RA, and helped to consolidate nighttime sleep by decreasing L5. Subjects were able to have longer, more restful and less fragmented sleep, a key goal in the treatment of ISWRD (3). Lemborexant exhibited treatment benefit, as detected by the interconnected efficacy variables in ISWRD patients on their circadian rhythm. Results of this study provide preliminary evidence that treatment with lemborexant may improve both 24-hour circadian rhythm variables and nocturnal sleep variables and impact the duration of daytime unplanned naps in subjects with ISWRD and AD-D. Additionally, these results suggest that proof-of-concept was established, as objective endpoints were identified that both characterized ISWRD in this patient population and were clinically relevant.
LEM5 appeared to be the most consistently effective dose in improving circadian rhythm-related, wake-related and sleep-rated actigraphy variables in this study. LEM5 demonstrated significant treatment differences versus placebo at week 4 in improving L5, RA and mean number of wake bouts during the night. LEM5 also resulted in less daytime sleep, as demonstrated by the greater numerical decreases from baseline in mean duration of sleep bouts during the day compared with placebo during each study week. Importantly, numerically higher RAs in circadian sleep-wake rhythms (ie, more distinction between night and day) were seen with all lemborexant dose levels.
Lemborexant was generally well tolerated in this population of individuals with Alzheimer’s disease and ISWRD. The rate of TEAEs was low, no treatment-emergent SAEs were reported and no new safety concerns were identified in this study. The safety profile in this study population was consistent with that observed in adult subjects with insomnia (15, 16). Additionally, treatment with lemborexant did not worsen the cognitive functions of this population of subjects with ISWRD and AD-D.
Dysregulation of the sleep-wake cycle is a common problem in patients with Alzheimer’s disease (22). One potential consequence for patients with Alzheimer’s disease who suffer from sleep disorders is an increased likelihood of institutionalization (23). However, at this time, the lack of approved pharmacologic treatments for patients with ISWRD and AD-D represents an unmet medical need. Some evidence is available to support the use of nonpharmacologic interventions, such as light therapy, behavioral techniques and increased social and physical activity during the daytime, to improve sleep in patients with Alzheimer’s disease (9, 10, 24). Both the American Geriatric Society and the AASM discourage the use of benzodiazepines in older adults (6, 25), as this drug class has been shown to be significantly associated with falls in the elderly population (26).
DORAs, which block the orexin system, may have the potential to improve sleep in patients with AD-D. Data regarding the treatment of insomnia (not ISWRD) in patients with mild to moderate Alzheimer’s disease have recently been added to the prescribing information for the DORA suvorexant (27).
Strengths of this study include the use of actigraphy, which can capture the full 24-hour sleep-wake pattern in treatment trials and has been a common method for assessing sleep in individuals with Alzheimer’s disease (28). Study limitations include the small sample size, which was, in part, due to slow recruitment. Additionally, the study duration was only 1 month.
These results provide important new information regarding the potential utility of lemborexant to address both nighttime and daytime symptoms that affect sleep-related quality of life of patients with ISWRD and AD-D, as well as reduce the burden of patients’ sleep disturbances on their caregivers and families. Further evaluation in future clinical trials is warranted to confirm the value of lemborexant in this patient population.
References
Brzecka A, Leszek J, Ashraf GM, et al. Sleep disorders associated with Alzheimer’s disease: a perspective. Front Neurosd 2018;12:330.
Musiek ES, Xiong DD, Holtzman DM. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med 2015;47:e148.
Zee PC, Vitiello MV. Circadian rhythm sleep disorder: irregular sleep wake rhythm type. Sleep Med Clin 2009;4:213–218.
Zhu L, Zee PC. Circadian rhythm sleep disorders. Neurol Clin 2012;30:1167–1191.
Gehrman P, Gooneratne NS, Brewster GS, Richards KC, Karlawish J. Impact of Alzheimer disease patients’ sleep disturbances on their caregivers. Geriatr Nurs 2018;39:60–65.
Auger RR, Burgess HJ, Emens JS, Deriy LV, Thomas SM, Sharkey KM. Clinical practice guideline for the treatment of intrinsic circadian rhythm sleep-wake disorders: advanced sleep-wake phase disorder (ASWPD), delayed sleep-wake phase disorder (DSWPD), non-24-hour sleep-wake rhythm disorder (N24SWD), and irregular sleep-wake rhythm disorder (ISWRD). An update for 2015: an American Academy of Sleep Medicine clinical practice guideline. J Clin Sleep Med 2015;11:1199–1236.
Serfaty M, Kennell-Webb S, Warner J, Blizard R, Raven P. Double blind randomised placebo controlled trial of low dose melatonin for sleep disorders in dementia. Int J Geriatr Psychiatry 2002;17:1120–1127.
Singer C, Tractenberg RE, Kaye J, et al. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer’s disease. Sleep 2003;26:893–901.
Figueiro MG, Plitnick BA, Lok A, et al. Tailored lighting intervention improves measures of sleep, depression, and agitation in persons with Alzheimer’s disease and related dementia living in long-term care facilities. Clin Interv Aging 2014;9:1527–1537.
Mishima K, Okawa M, Hishikawa Y, Hozumi S, Hori H, Takahashi K. Morning bright light therapy for sleep and behavior disorders in elderly patients with dementia. Acta Psychiatr Scand 1994;89:1–7.
Liguori C, Nuccetelli M, Izzi F, et al. Rapid eye movement sleep disruption and sleep fragmentation are associated with increased orexin-A cerebrospinal-fluid levels in mild cognitive impairment due to Alzheimer’s disease. Neurobiol Aging 2016;40:120–126.
Liguori C, Romigi A, Nuccetelli M, et al. Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol 2014;71:1498–1505.
Duncan MJ, Farlow H, Tirumalaraju C, et al. Effects of the dual orexin receptor antagonist DORA-22 on sleep in 5XFAD mice. Alzheimers Dement 2019;5:70–80.
Dayvigo (lemborexant) tablets [package insert]. Woodcliff Lake, NJ: Eisai Inc.; 2019.
Rosenberg R, Murphy P, Zammit G, et al. Comparison of lemborexant with placebo and zolpidem tartrate extended release for the treatment of older adults with insomnia disorder: a phase 3 randomized clinical trial. JAMA Netw Open 2019;2:e1918254.
Karppa M, Yardley J, Pinner K, et al. Long-term efficacy and tolerability of lemborexant compared with placebo in adults with insomnia disorder: results from the phase 3 randomized clinical trial SUNRISE 2. Sleep 2020.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198.
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984;141:1356–1364.
Trzepacz PT, Saykin A, Yu P, et al. Subscale validation of the neuropsychiatric inventory questionnaire: comparison of Alzheimer’s disease neuroimaging initiative and national Alzheimer’s coordinating center cohorts. Am J Geriatr Psychiatry 2013;21:607–622.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2013; http://www.R-project.org/.
Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 2011;168:1266–1277.
Liguori C, Spanetta M, Izzi F, et al. Sleep-wake cycle in Alzheimer’s disease is associated with tau pathology and orexin dysregulation. J Alzheimers Dis 2020;74:501–508.
Pollak CP, Perlick D. Sleep problems and institutionalization of the elderly. J Geriatr Psychiatry Neurol 1991;4:204–210.
McCurry SM, Gibbons LE, Logsdon RG, Vitiello MV, Teri L. Nighttime insomnia treatment and education for Alzheimer’s disease: a randomized, controlled trial. J Am Geriatr Soc 2005;53:793–802.
American Geriatrics Society 2019 Beers Criteria Update Expert Panel. American Geriatrics Society 2019 Updated AGS Beers Criteria(R) for Potentially Inappropriate Medication Use in Older Adults. J Am Geriatr Soc 2019;67:674–694.
Woolcott JC, Richardson KJ, Wiens MO, et al. Meta-analysis of the impact of 9 medication classes on falls in elderly persons. Arch Intern Med 2009;169:1952–1960.
Belsomra (suvorexant) tablets [package insert]. Whitehouse Station, NJ: Merck & Co., Inc.; 2020.
Camargos EF, Louzada FM, Nóbrega OT. Wrist actigraphy for measuring sleep in intervention studies with Alzheimer’s disease patients: application, usefulness, and challenges. Sleep Med Rev 2013;17:475–488.
Acknowledgement
Medical writing assistance was provided by Rebecca Jarvis, PhD, of ProScribe — part of the Envision Pharma Group and was funded by Eisai Inc. Envision Pharma Group’s services complied with international guidelines for Good Publication Practice (GPP3).
Funding
This study was sponsored by Eisai Inc. The sponsor participated in the design and conduct of the study; the collection, analysis and interpretation of data; and the preparation, review and approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Declaration of conflicting interests
Drs Moline, Rabbee, Filippov and Dhadda are employees of Eisai Inc. Dr Bsharat is formerly an employee of Eisai Inc. Mr Kubota is an employee of Eisai Co. Ltd. Dr Thein is the director and founder of Pacific Research Network, which received funding from the study sponsor, Eisai Inc., for the conduct of this study. Mr Kemethofer is an employee of The Siesta Group, the central actigraphy scoring vendor.
Electronic supplementary material
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
About this article

Cite this article
Moline, M., Thein, S., Bsharat, M. et al. Safety and Efficacy of Lemborexant in Patients With Irregular Sleep-Wake Rhythm Disorder and Alzheimer’s Disease Dementia: Results From a Phase 2 Randomized Clinical Trial. J Prev Alzheimers Dis 8, 7–18 (2021). https://doi.org/10.14283/jpad.2020.69
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.14283/jpad.2020.69