
Abstract
In the dynamic landscape of pharmaceutical advancements, the strategic application of active pharmaceutical ingredients to the skin through topical and transdermal routes has emerged as a compelling avenue for therapeutic interventions. This non-invasive approach has garnered considerable attention in recent decades, with numerous attempts yielding approaches and demonstrating substantial clinical potential. However, the formidable barrier function of the skin, mainly the confinement of drugs on the upper layers of the stratum corneum, poses a substantial hurdle, impeding successful drug delivery via this route. Ultradeformable vesicles/carriers (UDVs), positioned within the expansive realm of nanomedicine, have emerged as a promising tool for developing advanced dermal and transdermal therapies. The current review focuses on improving the passive dermal and transdermal targeting capacity by integrating functionalization groups by strategic surface modification of drug-loaded UDV nanocarriers. The present review discusses the details of case studies of different surface-modified UDVs with their bonding strategies and covers the recent patents and clinical trials. The design of surface modifications holds promise for overcoming existing challenges in drug delivery by marking a significant leap forward in the field of pharmaceutical sciences.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Drug delivery outlines the formulations, innovations, methodologies, and procedures used to transport a pharmaceutical compound within the body to produce the intended therapeutic outcome [1, 2]. Drug delivery methods are designed tools often utilized to release their medicinal content in a controlled way [3]. Recent advancements in pharmaceutical delivery systems have concentrated mainly on innovative drug delivery that focuses on administering the drug at the proper time, location, and dosage for maximum effectiveness and safety [4]. Developing novel drug delivery systems has received considerable attention in recent years. The term “novel drug delivery systems” refers to a new strategy for drug delivery that attempts to address the drawbacks associated with conventional drug delivery methods [5]. It is primarily involved in developing newer pharmaceutical systems with optimized properties like higher permeability, reduced particle size, and improved site-selective targeting [6]. Over one-third of pharmaceuticals currently under clinical testing are related to skin delivery. It has several advantages over conventional systems, such as evasion of first-pass metabolism, gastrointestinal side effects, lesser fluctuations in the plasma levels of the drug, and increased patient compliance.
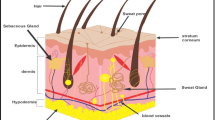
Skin delivery represents the optimal drug administration route for attaining dermal (local) or transdermal delivery (systemic effects) [7]. Despite its potential benefits, conventional transdermal and topical delivery systems encounter difficulties in penetration via deeper skin layers [8]. During the last decades, in the 1980s, traditional vesicular carriers have drawn much attention for the delivery of drugs via the skin [9]. The key barrier for transdermal drug delivery is generally the stratum corneum (SC), which is 30 µm thick & its penetration is regarded as the rate-limiting step, which causes the vesicles to confine on the SC’s upper layer [10, 11]. Thus, at the beginning of the 1990s, novel lipid vesicles called ultra-deformable vesicles, also referred to as flexible (elastic) or deformable liposomes, were designed to circumvent these limitations [12, 13]. Ultradeformable vesicles or carriers (UDVs), a specialized type of liposomal formulation, exhibit unique elasticity and flexibility due to their composition of phospholipids and highly hydrophilic mobile detergents [14]. It has demonstrated remarkable efficacy in traversing the undamaged skin barrier and facilitating the targeted delivery of encapsulated therapeutic agents to both the epidermal & dermal layers and the systemic circulation, as they are more deformable than conventional liposomes [15]. The two primary prerequisites in skin pharmacotherapy are transdermal delivery with enhanced skin permeation and targeted delivery of drugs. Ethosomes [16], transfersomes [17], bilosomes [18], cerosomes [19, 20], flexosomes [21], menthosomes [22], invasomes [23], and transethosomes [24] are the new ultra-deformable vesicles with high flexibility or elasticity that have been developed in and gained considerable interest in past few years. Figure 1 provides the publication trends and statistics data on different UDVs.

a Statistical representation depicting the distribution of Ultradeformable vesicle publications across various categories such as reviews, research articles, and book chapters. b A graphical depiction showcasing the evolution of publications on ultradeformable vesicles over time. * Ongoing Publications
The plain nano-carriers, such as ethosomes and liposomes, used for topical drug delivery lack site-specific targeting toward skin disease. Their mechanism primarily relies on drug accumulation and diffusion during topical absorption. Moreover, the stability and lower encapsulation efficiency of liposomal vesicles pose significant obstacles to the commercial production feasibility of UDVs [7, 25]. Thus, it necessitates additional treatment strategies to achieve targeted drug delivery to its specific action site. Surface modification of nanocarriers has emerged as a promising strategy for enhancing the effectiveness of UDVs. This constitutes a fundamental advancement in the treatment of several diseases. Among various approaches, surface modification holds significant potential in addressing the challenge of deformable vesicles, and it represents a notable breakthrough in the field of transdermal targeting. The modification in the surface of ultra-deformable vesicles alters its properties with better drug-targeting capability and localization as they establish an effective therapeutic concentration at the intended site for an extended period [26]. Different surface modification strategies, including polymers, peptides, small molecules, and surfactants, have been used to improve the UDV’s delivery and targeting capacity. Many reviews have been published focusing on targeted delivery and surface modifications of liposomes over a few decades [2, 27]. Recently published papers have summarized surface modifications utilizing different nanocarriers for enhancing drug delivery through different routes [26, 28]. However, none of these papers have focused on wide transdermal applications using surface modifications for different UDVs. A meticulous literature survey revealed a lack of articles on this topic, its patents, and clinical trials. Hence, the present review provides an overview of the case studies on different surface-modified UDVs, emphasizing the different surface coating agents and focusing on the recent therapeutic patents and clinical trials of UDVs in this early research field. The current review also addresses these research gaps and delineates the specific areas that require further attention to study the potential of surface-modified UDVs.
Surface Modification Strategies for Transdermal Targeting
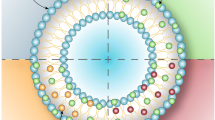
Surface modification of UDVs encompasses diverse approaches. These strategies enhance UDVs’ functional properties and targeting capabilities, thus facilitating precise and efficient drug delivery [26]. The subsequent section comprehensively overviews the studies utilizing the various surface-modifying agents with their applications in achieving targeted skin delivery (Fig. 2). Multiple strategies exist to integrate polymers, peptides, and small molecules into the structural framework of UDVs, which could be achieved through non-covalent and covalent interactions.

Representation of currently utilized ultradeformable vesicles with varied surface modifying agents
Non-Covalent Bonding
Non-covalent surface modification of UDVs occurs via non-covalent forces, including hydrogen bonding, electrostatic interactions, van der Waal’s strengths, and hydrophobic interactions [29]. Utilizing these interactions allows for the attachment of targeting ligands or functional molecules onto the surface of nanocarriers. Adsorption materials, such as polyelectrolytes and saccharides, modify UDVs through electrostatic interaction or hydrogen bonding or by combining both without complex covalent conjugation chemistry (Fig. 3). The attraction between the charges on the carrier and the polyelectrolyte leads to an electrostatic film surrounding the vesicular systems [28]. Hence, non-covalent bonding strategies offer several advantages by enabling the surface modifiers or targeting ligands to dissociate from the nanocarriers upon reaching the target site, ensuring efficient drug release. Secondly, the methods employed in non-covalent modifications are more flexible than covalent bonding approaches. These methods help maintain the integrity, stability, and functionality of the UDVs and the attached surface modifier [25, 30, 31]. It also offers the advantage of simplicity and minimal impact on the molecular structure and interactions with biological targets. However, non-covalent modifications are susceptible to various external factors, such as ionic strength and pH changes, with a lack of instability and reproducibility [28, 32].

General method of preparation of surface-modified ultradeformable vesicles with covalent and non-covalent bonding strategies
Covalent Bonding
The covalent bonding approach enables the direct conjugation of small molecules and peptides to UDVs in a single-step process, often involving organic solvents. It is a commonly employed technique for modifying the nanovesicle’s surface [33, 34]. Different covalent bond chemical modification strategies are widely used for the surface alteration of UDVs, including carbodimide chemistry and esterification reactions [25, 30, 31]. EDC (1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide) is a water-soluble compound derived from carbodiimide and is commonly employed with N-hydroxysuccinimide (NHS) to facilitate carbodiimide coupling reactions. This reaction involves intermediate active ester formation through condensation between the NHS and carboxylic groups. The active ester reacts with an amine group, forming an amide bond (Fig. 3). The interaction between UDVs and targeting ligands relies on forming amide bonds among the OH drug and NH2 ligands [35]. Polymer conjugation with ligands can also be formed through an esterification reaction. Esterification is a frequently observed chemical reaction where an ester is produced as the final product through the reaction between two reactants, namely alcohol and acid. The reaction involves 1,1′-carbonyldiimidazole as a coupling agent and a catalyst as 4-dimethylaminopyridine (DMAP) [36,37,38]. Covalent bonds are utilized for ligand binding to facilitate targeting or mitigate the toxicity profile of vesicles [32]. Compared to non-covalent bonding approaches, covalent bonding methodologies augment vesicular solubility in both aqueous and non-aqueous solvents, elevating stability, biocompatibility, and biodistribution [32, 39]. Despite these merits, it faces a few drawbacks, such as necessitating the incorporation of a linker to add a coating moiety and the possible potential side reactions. In the presence of other agents, chemical bonding may lose its stability during the reaction [28].
Laboratory-Scale Manufacturing of Surface-Modified Ultradeformable Vesicles
The thin film hydration technique is the most generally used preparation technique for surface modifications of UDVs, and the other method is ethanol injection.
Synthesis of Conjugated Surface Modifier
The surface modifying agent with 1-hydroxypyrrolidine-2,5-dione (NHS) and (EDC)
1-(3-Dimethylamino-propyl)-3-ethylcarbodiimide hydrochloride was dissolved in distilled water, and the reaction was carried out for 3 h at 37°C by adjusting the solution pH to 7.5 with sodium hydroxide solution. A solution containing dioleoyl phosphoethanolamine (DOPE) was introduced into the reaction mixture, and the reaction proceeded within a temperature-controlled water bath at 37°C for 24 h. The resultant solution was subjected to dialysis against double-distilled water and subsequently lyophilized, forming the conjugated surface modifier [25].
Thin Film Hydration Technique
The lipid component, cholesterol, with other excipients mixture, was dissolved in the organic mixture in a round bottom flask. In the covalent bonding strategy, a surface modifier was added in the initial stage with the excipient mixture. The organic solution was subjected to evaporation under reduced pressure using a rotary evaporator at a suitable temperature and revolutions per minute (rpm) for a particular duration, forming a thin, dry film. The film was hydrated using a buffer or distilled water at a suitable rpm. The resulting suspension was either bath or probe sonicated to reduce the vesicles’ size and convert large multilamellar vesicles to small unilamellar vesicles to form surface-modified UDVs (Fig. 3) [40].
In the non-covalent bonding strategy, the surface modifier was added dropwise to the initially prepared UDVs under magnetic stirring at room temperature, forming surface-modified UDVs through the adsorption phenomenon [40].
Ethanol Injection Method
The drug, lipid, surface modifier, and cholesterol were dissolved in ethanol and heated using a water bath until complete dissolution was achieved. This solution was then gradually introduced into double-deionized water at 60°C under magnetic stirring at 500 rpm. After 15 min of stirring, the mixture was ultrasonicated in an ice bath using a probe sonicator to yield surface-modified UDVs [41].
Large-Scale Production in Industrial Manufacturing
The industrial manufacturing processes for UDVs remain unexplored, but they share procedural similarities with liposome production methods. While numerous techniques exist for laboratory-scale liposome synthesis, only some are suitable for commercial manufacturing, primarily due to the need for precise control over critical quality attributes. In large-scale production of parenteral liposomes, the ethanol injection method followed by extrusion stands out as the preferred method due to its reproducibility in polydispersity index and particle size [42]. However, large-scale liposome manufacturing involves numerous tests and unit operations and is labor intensive, which includes buffer preparation, filtration, lipid hydration, extrusion, diafiltration, and sterile filtration. Each step necessitates stringent in-process controls such as particle size analysis, filter integrity testing, pH monitoring, and bioburden assessment. Large-scale manufacturing of liposomes involves using the ethanol injection method followed by the extrusion technique. Approximately nine-unit operations are involved in both hydrophilic and lipophilic drug models. Additionally, conducting quality controls for each step adds complexity, making the process labor-intensive and lengthy [43].
Manufacturing liposomes involves numerous energy- and time-intensive steps, particularly membrane extrusion, lipid hydration, and diafiltration, which demand rigorous in-process controls and specialized expertise. Addressing these challenges could streamline the process and enhance its robustness. One promising approach is the antisolvent/nanoprecipitation technique, characterized by LeciPlex®, wherein a stabilizer and a phospholipid dispersed in a biocompatible solvent immediately form sub-micron vesicles upon exposure to water. These self-assembled technologies offer precise control over polydispersity index and particle size, potentially eliminating the need for diafiltration and extrusion and avoiding using organic solvents. Alternatively, controlled precipitation techniques, such as microfluidics, enable liposome formation with desired particle sizes without the need for lipid hydration or extrusion, thus significantly simplifying the process [43]. The same technology can be implemented in the large-scale production of UDVs.
Surface Modifications of Ultradeformable Vesicular Carriers
Polymer-Coated Ultradeformable Vesicles
Polymeric surface modification of UDVs presents a novel and promising approach for enhancing transdermal drug delivery. This technique entails the application of a polymer coating onto the vesicle surface or its inclusion during the preparation stage, aiming to improve their interaction with the skin and facilitate efficient drug permeation through the dermal barrier. This segment extensively explores surface modification techniques employed to enhance the delivery of different drugs to the skin via UDVs by highlighting the utilization of a range of polymers. It includes Hyaluronic acid (HA), dextran sulphate, PEG (Polyethylene glycol), a combination of PEG with monoterpenes, HA-conjugated Octadecylamine (ODA), HA-conjugated-DOPE, Galactose conjugated chitosan and Poly ethylenimine in conjunction with Sodium cholate.
Hyaluronic acid (HA) is a naturally existing linear glycosaminoglycan, constituting a hydrophilic, negatively charged polysaccharide consisting of recurring units of N-acetyl-D-glucosamine disaccharides and glucuronic acid. Research has shown that low molecular weight (5–50 kDa) HA exhibits a more pronounced enhancement of permeation compared to its medium (100–300 kDa) & high molecular weight counterparts (600–1200 kDa) [44, 45]. Due to its excellent biocompatibility, biodegradability, and hydrophilicity, HA is widely utilized in biomedicine. It is a prominent and significant constituent of the extracellular matrix and is abundantly found in the skin [46]. HA employs multiple mechanisms to enhance skin penetration, including HA receptor-facilitated transport, skin hydration, & hydrophobic interaction due to viscoelastic properties and stratum corneum interactions. Its exceptional hygroscopicity allows for hydration of both the SC & the dermis, resulting in the swelling of corneocytes, alterations in intercellular lipid microstructure, and ultimately facilitating permeation through the well-hydrated skin [47]. Furthermore, HA’s hydrophobic domain can engage with the SC, reducing the skin’s barrier function. HA’s viscoelastic characteristics also contribute to the prolonged retention of drugs within the epidermis [44, 48].
Yuan et al. engineered HA-modified indomethacin (IND)-loaded transfersomes (IND-HTs) to improve the percutaneous permeation (Table I). A covalent bond between HA & octadecylamine (ODA) was established to facilitate the effective modification of HA on the transfersome surface, which involved synthesizing HA-ODA through a coupling reaction, where the amino groups of ODA are chemically linked with the HA’s carboxyl moieties. Subsequently, the HA-ODA was incorporated into a carbopol 940 hydrogel to form IND-HTs/Gel. In vitro drug release study of IND-HTs/Gel showed a 9.7% drug release in the initial 2 h and 68.8% at 48 h, indicating the slow drug diffusion into the skin layers from the transfersomes than the premature release of the drug before permeation of the vesicles into the skin. The in vitro skin penetration study across porcine skin exhibited a 1.73-fold enhancement, with a cumulative drug permeation of 171.73 ± 30.29 µg/cm2, & a 2.54-fold higher drug deposition compared with IND-Ts/Gel [44]. A study by Xie et al. showed that HA-containing ethosomes exhibited 172.03 ± 0.90 μg/cm2 h of in vitro skin flux. Concurrently, a fluorescence intensity of 17.3 demonstrated penetration distribution within rat skin with 90% of the survival rate of cells across various concentrations. It indicates effective penetration of the drug through the SC to the deeper dermal skin layers [46]. In 2021, Albash et al. studied the effect of HA-enriched spironolactone-loaded cerosomes (OHAEC) on ex vivo and in vivo distribution for the topical delivery of the drug in the skin. The optimized formulation of OHAEC demonstrated a substantial increase in drug deposition over 24 h, reaching (55.45 ± 0.35 µg/cm2), resulting in a remarkable local accumulation efficiency index that was 18.5 times higher than the suspension. The AUC0–10, obtained from the in vivo dermatokinetic study of 402.9 µg h/cm2, showed a 1.6-fold higher spironolactone deposition than (248.3 µg h/cm2) suspension [20]. Zhang et al. conjugated HA with DOPE to possess inter and intramolecular interactions through an EDC-NHS reaction to modify curcumin-loaded ethosomes. The study reported that after 8 h of topical administration, the HA-ES (Ethosome) and ES groups showed significantly higher cumulative transdermal curcumin levels (3.32- and 2.30-fold, respectively) and increased skin retention compared to the (Propylene glycol-based curcumin solution) PGS group. In vitro skin permeation analysis indicated a 3 to sixfold increase in curcumin concentration in the epidermis and dermis with HA-synthesized polymer micelles compared to non-polymeric micelle solutions. (High-Performance Liquid Chromatography) HPLC analysis post-in vivo application confirmed notably increased skin drug retention of curcumin in the HA-ES group compared to the PGS group. The HA-ES group exhibited the lowest levels, highlighting its efficacy in enhancing topical drug absorption. The HA-ES group exhibited a reduction in inflammation symptoms, including decreased levels of IL-17A, IL-1β mRNA, IL-22, TNF-α, & reduced expression of CCR6 protein in comparison to PGS & ES groups [25].
(TCI) Transcutaneous immunization offers a compelling substitute for vaccination, characterized by reduced discomfort and enhanced efficiency [58, 59]. It capitalizes on the skin’s rich population of antigen-presenting cells, notably dendritic cells, to initiate an immune response. This unique feature allows for administering lower drug doses than oral delivery and injections [58,59,60]. (SF) Silk fibroin is a highly favorable biomaterial due to its superior biocompatibility, adjustable structural properties, and notable oxygen permeability [61,62,63]. SF nanofibrous mats, encapsulated with ethosomes (ESNFM) and modified with galactosylated chitosan (GC) and hyaluronic acid, show promise for transdermal drug delivery, as they target dendritic cells and leverage HA’s skin-friendly properties, making them an attractive option for TCI. The study hypothesized that nanofibrous mats comprising SF incorporated with GC-modified Eths & HA presents a potentially optimal solution for TCI, using ovalbumin (OVA) as a model drug to assess Eth-HA-GC’s targeting and (dendritic cells) DC activation capabilities. In vitro experiments confirmed that Eth-HA-GC was superior in bone-marrow-derived DCs uptake compared to Eth-HA or bare Eth, emphasizing GC’s role in DC targeting. A novel ESNFM, OVA@Eth-HA-GC/SF, was created via green electrospinning, displaying excellent skin compatibility and transdermal performance. The positively charged surface of ethosome and Eth-HA-GC enhanced adhesion to negatively charged cell membranes, aiding skin permeation. In vivo results demonstrated that OVA@Eth-HA-GC/SF considerably boosted OVA-specific antibody levels in serum & the secretion of IL-2, IL-6, and IFN-γ by spleen cells, effectively stimulating both humoral and cellular immune responses [30].
Dextran, a natural branched glucose polymer, has a molecular weight range of 2 to 3000 kilodaltons and unique attributes like resistance to cell adsorption, low protein adsorption, biodegradability, and ease of modification [64]. Dextran sulfate (DS) is a ligand for scavenger class A receptors, offering excellent biocompatibility [65]. Zhao et al. employed DS to formulate and target the delivery of Dexamethasone (DEX) within flexible nano-liposomes. In vitro DEX release from conventional liposome hydrogel (DS-RLs/DEX) & (DS-FLs/DEX) DEX-loaded flexible liposome hydrogel showed similar profiles at pH 7.4, releasing approximately 20% in 48 h. At pH 6.5, an initial burst release occurred, releasing 40% (DS-RLs/DEX) & 33% (DS-FLs/DEX) in 5 h, followed by sustained release, reaching nearly 90% at 48 h. In vivo accumulation study revealed that treatment with [DIR: (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaineiodide)] DIR-DS-FLs/DEX hydrogel presented 1.7 times higher intensity of fluorescence than DIR-DS-RLs/DEX & 1.8 times higher than treatment with DIR hydrogel [49].
PEGylation, an extensively employed technique, involves the substantial use of PEG for surface modification of nanocarriers. PEG is a prominent drug-delivery polymer recognized for its versatility and biocompatibility [28]. PEGylation of lipid bilayers is widely used to improve lipid vesicle stability and prolong their circulation in bodily fluids. This process involves surface shielding, which reduces aggregation, opsonization, and phagocytosis, thus decreasing immunogenicity. Additionally, it finely regulates the release kinetics of encapsulated active pharmaceutical ingredients into the systemic circulation [50, 66]. Employing mixed monoterpenes (comprising limonene and citral) as edge activators with the incorporation of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt) (DSPE-PEG2000) for the modification of transfersomes (TFSs), results in the formation of Mixed Monoterpenes Edge Activated PEGylated TFSs (MMPTs) which was utilized by Zheng et al. for the sinomenine (SIN) delivery to improve percutaneous absorption. In vivo pharmacokinetics study in rabbits indicated that SIN concentrations and AUC0→t in joint cavities were 2.1 & 2.5-fold greater, respectively, with MMPTs compared with liposomes [50]. In another research conducted by Ma and colleagues, a novel complex consisting of polyethylenimine-modified ethosomes (Eth−PEI) & sodium cholate-modified ethosomes (Eth−SC), referred to as Eth−PEI/Eth−SC, was developed. The cellular uptake study against B16 cells indicated higher uptake of [Eth−PEI/Eth−SC(7:3)] complex with efficient s via the stratum corneum in ex vivo experiments. The anticancer potential of the Eth−PEI/Eth−SC complex by combining doxorubicin (DOX) and curcumin (CUR) [CUR@Eth−PEI/DOX@Eth−SC] was tested in vitro & in vivo against B16 melanoma cells & mice bearing melanoma tumors, respectively. The results consistently showed that the CUR@Eth−PEI/DOX@Eth−SC complex (7:3) effectively inhibited melanoma cell growth, indicating a synergistic anticancer effect between DOX@Eth−SC & CUR@Eth−PEI [51]. Albash et al. used PEGylated surfactant Brij® to develop and optimize FTN-loaded PEGylated cerosomes. Brij®, a single-chain surfactant with varied acyl chain and PEG components, enhances skin hydration by potential corneocyte and lipid changes and inducing SC swelling [67, 68]. In vivo dermatokinetic investigations demonstrated significantly higher skin drug concentrations at all time intervals, with a notably higher AUC0–10 (1120.61 ± 33.97 µgh/cm2) and Cmax (172.02 ± 22.86 µg/cm2) compared to FTN suspension. The histopathological assessment revealed no adverse effects on epidermal and dermal cells in the rat skin, affirming the safety & tolerance of topically administered PEGylated cerosomes in treating skin disorders. [19]. PEGylation of the vesicles altered their surface properties by prolonging their presence at the application site and improving topical drug delivery efficacy by maintaining desired drug concentrations over time [69]. A similar study by Albash et al. formulated pegylated bilosomes of Olmesartan medoxomil (OLM). The in vivo skin deposition study of OLM encapsulated transfersomes and OLM entrapped pegylated bilosomes reported 3.47- and 5.68-fold superior skin deposition than that of OLM suspension. In vivo pharmacokinetic study of the optimized OLM formulation showed a higher relative bioavailability of (235.04%) and 2.35 higher AUC0–∞ & AUC0–48 values than that of oral tablets, indicating effective transdermal delivery of developed and optimized formulation by bypassing the first pass liver metabolism [52].
Ligand-Modified Ultradeformable Vesicles
Lectin-Conjugated Ultradeformable Vesicles
Lectins, a structurally diverse group of glycoproteins and proteins, can bind to carbohydrate-exposed residues on epithelial cell membranes, functioning as efficient bioadhesive agents [70,71,72]. This characteristic arises from their high specificity in binding to sugar molecules, enabling precise recognition & reversible attachment to the carbohydrate motifs within intricate glycoconjugates without eliciting modifications to the covalent configuration of the known glycosyl ligands [73,74,75,76]. Given the widespread presence of cell surface carbohydrates, this property makes them potential candidates for enhancing targeted delivery at various biological sites [70]. Glycoproteins play a pivotal role in various cellular activities, including immune responses, cellular recognition, cellular differentiation & inflammation [27]. In cancer therapy, these lectins can be harnessed to identify specific carbohydrate moieties like glucose, mannose, dextrose, and their derivatives, thus serving as effective targeting agents [53, 77, 78]. Jangdey et al. designed apigenin-loaded nanotransfersomes using the rotary evaporation & sonication technique and subsequently conjugated the Concanavalin-A (Con-A) with carbodiimide chemistry. These nanotransfersomes were then incorporated into a carbopol 934 polymer to form a gel. The in vitro drug release showed an initial burst release of 10–15% within 2 h, followed by sustained release (54.46 ± 0.25%). Transdermal delivery through goat skin demonstrated higher flux (8.42 ± 0.73) and drug retention (0.91 ± 0.02%) than commercial products and pure suspension. Cytotoxicity results confirmed the specific targeting of cancer cells by the Con-A conjugated apigenin nano-transfersomal gel with lower cytotoxicity on A375 cells (human melanoma skin cell lines) in comparison to HaCaT cell lines (keratinocyte skin cell line) without affecting normal cells [35]. A study by parashar et al. achieved active targeting by surface modification of bilosomes using carbohydrates as targeting moieties. Dextrose, a carbohydrate moiety, was employed to coat the bilosome surface, facilitating binding to overexpressed lectins on cancer cells. Dextrose-modified bilosomes containing silymarin demonstrated significant intercellular uptake, as evidenced by high fluorescence intensity and a low IC50 value in Hep G2 cell lines. These findings were confirmed through in vitro cell line studies and cytotoxicity assessments.
Additionally, the modified bilosomes restored oxidative stress markers and normalized biochemical lipid parameters, demonstrating stability in gastrointestinal fluids, site-specific accumulation, and targeted drug delivery to tumor cells [53]. Another study by Verma et al. formulated mannosylated naringenin encapsulated transfersomes for macrophage-targeted drug delivery [54]. Mannose receptors, including the Macrophage Mannose Receptor, are significantly upregulated on the surfaces of macrophages. The other cell types, like endothelial & dendritic cells, also express mannose receptors, and several other lectins with mannose-binding capabilities have been identified [79, 80]. Exploiting these robust interactions between complex carbohydrates and lectins, glycotargeting becomes a valuable approach [81]. Mannose receptors, which are highly prevalent in immune cells, are a prime focus, achieved through mannosylation. This effectively allows the design and development of nanosystems for targeting mannose receptors [82]. Mannosylated naringenin encapsulated optimized transfersomes showed 62.03 ± 0.42% of in vitro drug release with 6.5 ± 3.07 μg/ch−2/h of higher ex vivo permeation flux and 0.76 ± 1.26% of drug retention compared to marketed formulation, drug dispersion and non-mannosylated naringenin encapsulated transfersomes. It also demonstrated effective cellular uptake through mannose receptor-mediated endocytosis [54].
Peptide-Conjugated Ultradeformable Vesicles
Peptides have also been studied for modifying UDVs for applications such as skin cancer, anti-inflammatory drug delivery, and enhancing skin penetration capabilities. Li et al. explored cell-penetrating peptide (CPPs) modified lycorine (LR) loaded transfersomes (Table I). CPPs constitute a category of small peptides characterized by cationic & amphiphilic features, enabling them to effectively traverse cell membranes, layers, and even tissues like the skin. These CPPs have the remarkable capacity to transport both macromolecules and minute particles [83, 84]. Among these, arginine oligomers, a subset of CPPs, stand out for their notable ability to enhance permeation. This is primarily attributed to the multiple positively charged arginine residues, which grant them a strong affinity for anionic cell membranes, facilitating the internalization of drug payloads [55, 85].
Furthermore, when CPPs incorporate arginine and histidine, they leverage arginine’s penetration-enhancing properties while promoting nanoparticle escape from endosomes. This escape is driven by the histidine-elicited proton sponge effect, allowing nanoparticle permeation through the skin and ultimately augmenting their anti-tumor effectiveness [86, 87]. This study aimed to alter transfersomes containing anticancer agents by incorporating the CPP-R5H3 peptide to increase intracellular and transdermal penetration, ultimately enhancing its anticancer efficacy. The lycorine-oleic acid ionic loaded CPP modified cationic transfersome gel showed effective drug permeation to the tumor cells and skin with high safety and anticancer properties [55]. Kim and his colleagues considered Palmitoyl pentapeptide (Pal-KTTKS or PPP), a peptide derivative with superior skin penetration properties, to enhance the transdermal delivery through transformer-ethosomes (TES) vesicles. PPP originates from collagen breakdown and stimulates collagen synthesis. This peptide readily permeates the skin, targeting dermal fibroblasts effectively. Enhancing collagen and glycosaminoglycan production boosts skin cell activity, counteracts aging effects, improves elasticity, and increases skin thickness. Notably, it possesses impressive skin-healing capabilities, addressing damage from UV exposure and external factors [88, 89]. Including capric acid & myristic acid (MA) in the TES enhanced flexibility compared to ethosomes and conventional liposomes. Skin permeation study in the skin-like artificial membrane (Strat-M™) demonstrated increased drug permeation of PP-TES-MA with cumulative permeation of PPP (481.8 ± 47.2 μg/cm2) over 24 h, permeability coefficient (4.84 ± 0.64 cm/h) & transdermal flux of (24.2 ± 3.2 μg/cm2/h) indicating effective TESs penetration ability by altering the membrane bilayer fluidity and enhancing the skin penetration [56]. Niu et al. study also worked on PPP (Lysine-threonine-threonine-lysine-serine), a short palmitoyl-pentapeptide (Pal-KTTKS), which is a derivative of KTTKS combined with palmitoyl was utilized for modifying indomethacin (IMC) loaded ethosomes (IMC-KTTKS-Es). In vitro drug release of IMC-KTTKS-Es showed 40–44% controlled release, indicating drug permeation through a modified aqueous boundary layer. The IMC-KTTKS-Es gel showed 96.35 ± 11.03 µg/cm2 of in vitro IMC skin permeation with 4.51 fold higher drug retention after topical administration with improved anti-inflammatory and analgesic activity compared to IMC-Eths [57].
Bioactive Molecule-Modified Ultradeformable Vesicles
Glucocorticoids, which are steroid hormones originating from the adrenal gland cortex, play a central role in regulating protein, glucose, & lipid metabolism within the body [90]. Glycyrrhetinic acid (GA), derived from Glycyrrhiza radix, acts as a glucocorticoid receptor with diverse pharmacological roles [91]. Notably, it is an adjuvant therapy for psoriasis due to its corticosteroid-like function (Table II). GA exhibits anti-inflammatory and immunomodulatory properties by regulating various factors, including interleukin-23, phosphorylated p38, vascular endothelial growth factor & block arachidonic acid metabolism [92, 93]. Surface modification techniques can be pivotal in enabling synergistic approaches for psoriasis management. Guo and colleagues developed (GA-TPGS) glycyrrhetinic acid-D-α-tocopherol acid polyethylene glycol succinate to improve psoriasis treatment via mitigating inflammation & protecting against lipid peroxidation-induced damage (Table I). This GA-TPGS was subsequently employed to modify the surface of curcumin-encapsulated ethosomes (Cur@GA-TPGS-ES) to exhibit multifunctional synergetic therapeutic intervention. In vitro skin permeation studies of Cur and GA in Cur@GA-TPGS-ES were 2.88 and 1.93-fold higher in multi-functionalized ethosomes than in ethanol solution, respectively. In Sprague − Dawley rat experiments, the surface-modified ethosomes exhibited substantially higher Cmax for Cur (4.09-fold) and GA (2.14-fold) than nonmodified ethosomes. TPGS was pivotal in augmenting deformability and penetration capabilities in enhancing cur and GA transdermal delivery [41].
Analytical Methodologies for Assessing Surface Modifications of Ultradeformable Vesicles
To analyze the surface of the vesicular systems and nanomaterials, recent advancements have led to the development of conventional and advanced techniques covering the critical aspects of ligand density, ligand conformation and structure, binding affinity, thickness, surface charge, and surface morphology. The methodologies for analytical estimation are essential for ensuring proper modifications to improve the efficacy and functionality of vesicles [103].
Surface area, porosity, pore volume, and surface morphology of vesicles undergo alterations due to surface modifications, and tracking these changes can confirm the modification process by utilization of microscopy and conventional-based methods and their associated techniques for monitoring surface changes [103]. The present review discusses the commonly employed analytical techniques to determine the surface changes of the UDVs.
-
a.
Electron microscopy (EM), particularly high-resolution transmission electron microscopy (HRTEM), is an imaging technique for analyzing nanoscale materials, providing detailed information on shape, uniformity, size, and atomic lattice structures. It is used to assess vesicles’ size and shape post-synthesis and modification and to analyze the layer thickness, defects, and surface morphology [103].
-
b.
Scanning Electron Microscopy (SEM), another electron microscopy method, is employed for surface observation and characterization of nanoscale materials [103, 104].
-
c.
Atomic force microscopy (AFM) is a suitable technique for examining structural characteristics of vesicular systems. It allows for analyzing mechanical properties and the shape of bilayers at the nanoscale range. AFM facilitates the gain of high-resolution surface topographic images and force-distance curves, achieving remarkable spatial resolution close to 1 Å and a detection limit approaching 10–12 N [105]. Force-distance (F-D) measurements using AFM track the cantilever deflection as it engages with the surface. This data interprets properties like stiffness characteristics of the ligand shell and surface functionalization of the vesicles [103].
-
d.
Surface charge, a critical parameter for assessing nanoparticle behavior in an aqueous environment, is a primary indicator for evaluating colloidal stability. This charge begins from the ligand shell’s functional groups or vesicular surfaces. In aqueous mediums, nanoparticles acquire charge through surface deprotonation/protonation or ion adsorption. Experimental characterization of surface charge involves electrophoretic light scattering, with results typically expressed as zeta potential. It is a qualitative assessment of the surface charge density used to indicate nanoparticle stability. High negative or positive zeta potentials contribute to stable colloidal dispersions via the electrical double layer (EDL). However, surface charge density may fluctuate based on biomolecular interactions or external factors potentially altering the local EDL upon binding [103]. Laser Doppler electrophoresis analyzes light scattering frequency/phase changes during electrophoresis and calculates zeta potential, offering insights into surface modifications with charged molecules [106].
-
e.
Infrared (IR) spectroscopy is a widely utilized analytical molecular and material characterization technique [106]. Fourier transform infrared spectroscopy (FTIR) instruments are commonly available in analytical labs and are readily available. IR spectroscopy depends on the vibrational behavior of molecules with unique spectroscopic characters. (Attenuated total reflectance) ATR-FTIR does not necessitate ultrahigh vacuum conditions, which makes it suitable for studying the liquid-nanoparticle interface. In surface functionalization analysis, IR spectroscopy primarily confirms functionalization by comparing spectra to the free ligand, providing perspectives into intermolecular interactions and covalent bonds [103, 107].
Patents on Surface Modified Ultradeformable Vesicles
UDV carriers are an innovative strategic approach representing a pioneering pharmaceutical innovation stride in transdermal and topical drug delivery. It intricately details the encapsulation of different therapeutic agents with specialized surface-modifying agents. Table III summarizes and presents the patents on different UDVs. This innovative methodology not only highlights the ingenuity of UDVs but also holds the potential to augment drug efficacy and permeation, thereby paving the way for novel avenues in targeted drug delivery.
In the investigation conducted by Rong L et al., a ceramide ethosome formulation was developed to transport both water-soluble and fat-soluble actives concurrently. The process entails: (a) combining water-soluble active ingredients (0.1–8% mass fraction), fat-soluble active ingredients (0.1–0.5%), phospholipid (up to 30%), ceramide (0.05–1%), ethanol (25–50%), cholesterol (up to 1%), and water to reach 100%. The process involved dissolving ceramide, phospholipid, fat-soluble actives, cholesterol, and ethanol to form an organic phase (A). Simultaneously, the water-soluble active substance was dissolved in water to form the water phase (B). Using the ethanol injection method, organic phase A was continuously added to water phase B, followed by magnetic stirring and hydration at 60°C for 30–50 min to obtain the ceramide-modified ethosome. The resulting ethosome appeared as an opaque or semi-transparent uniform liquid with a particle diameter ranging from 40 to 500 nm. Importantly, no significant changes in particle diameter were observed after storing the ethosome at 4, 25, and 45°C for 70 days [108].
Transcutaneous immunization represents a novel vaccination approach wherein antigen and adjuvant are applied topically to induce a systemic immune response, bypassing the limitations of conventional injection methods. However, the skin’s barrier function poses a challenge to TCI efficacy. Hongsheng W et al. ’s patented study presents a method for preparing galactose-modified ethosomes to enhance TCI efficiency without compromising skin integrity. The process involves lipid mixture preparation by rotatory evaporation, antigen protein encapsulation via ultrasonic dispersion to attain an immune ethosome containing the antigen protein cargo, and layer-by-layer self-assembly with galactosylated chitosan. The resulting ethosomes offer good cell compatibility, hypoimmunity, and hypotoxicity, facilitating transdermal drug delivery. The method is straightforward, with a short preparation time and mild conditions, suitable for scalable production [109].
Liver cancer is a significant global health concern, ranking third in cancer-related mortality worldwide, with over 35% of cases occurring in our country. To address this, targeted treatment strategies are crucial. Galactose ligands selectively bind to liver cancer cells, enhancing drug uptake. Polyethyleneimine (PEI) is a potent gene vector, but its cytotoxicity limits its use; however, galactosylated polyethyleneimine (Gal-PEI) shows reduced toxicity and targets liver cancer cells effectively. Ethosomes are efficient drug delivery systems with high cell compatibility, enabling direct drug release into cells. Utilizing layer-by-layer self-assembly techniques and ethosome preparation, nucleic acid drugs and Gal-PEI can be effectively loaded and delivered, improving targeted treatment for liver tumors [110].
Psoriasis is a complex inflammatory skin disorder characterized by inflammatory infiltration and abnormal epidermal proliferation, leading to organ and skin damage. Current treatments mainly rely on corticosteroid topical applications, but long-term use can result in adverse reactions and does not address inflammatory recurrence. Traditional Chinese medicine (TCM) offers a promising approach due to its reduced systemic toxicity and direct local treatment effect. Transdermal drug delivery of TCM ingredients, such as GA and CUR, has shown efficacy in psoriasis treatment. GA, a metabolite of glycyrrhizic acid, exhibits strong corticoid, anti-inflammatory-like effects. Combining GA and CUR enhances their synergistic therapeutic effects. The patented invention by Teng G introduces a GA-modified curcumin-loaded multifunctional ethosome, utilizing a core–shell structure with curcumin-loaded ethosomes as the core and GA-vitamin E polyethylene glycol succinate as the shell. This innovative delivery system promises enhanced efficacy and reduced side effects in psoriasis treatment [111].
The novel acupoint drug delivery system described in the invention by Yongtai Z et al. facilitates the targeted delivery of TCM to acupoints for dispelling cold and warming middle jiao. This system enhances drug absorption in acupoint regions, amplifying their therapeutic effects. The acupoint HA-ES transdermal drug delivery system demonstrates superior performance in deeply coating drugs into acupoint regions, preventing leakage during penetration through the epidermal layers, and facilitating drug transport to the dermis and subcutaneous tissues with rich microcirculation. It promotes drug absorption into systemic circulation, significantly improving drug bioavailability. Additionally, umbilical administration of HA-ES-carrying cinnamon volatile oil effectively reduces H2O2 (Hydrogen Peroxide) and NO (Nitric Oxide) levels in ulcerative colitis (UC) model rats while up-regulating Ach (acetylcholine) and ATP levels in UC murine colon tissue. This administration also modulates VIP (Vasoactive Intestinal Peptide) and SCF (Stem Cell Factor) expression in colon tissue, indicating its potential to alleviate ICC (Interstitial Cells of Cajal) injury and regulate gastrointestinal motility. The acupoint drug delivery system holds promise as a carrier for treating symptoms associated with cold syndrome, particularly in cases of cold syndrome UC or yang deficiency, which offers significant medicinal potential [112].
Diosgenin, also known as diosgenin aglycone, is predominantly found in the rhizomes of Dioscorea plants and exhibits a solid affinity for cellulose in the cell wall. Hui Z et al., in their patent, revealed a method for preparing transfersomes of diosgenin with chitosan. The method involves grinding the peltate yam rhizomes, followed by enzymatic fermentation. Subsequently, the mixture undergoes ultrasonic extraction utilizing magnesium chloride and sodium dodecyl sulfate to obtain an extract. Following hydrolysis with minimal sulfuric acid, the extract produces diosgenin hydrolysate. Post-foam separation and rinsing with a phosphate buffer salt solution, the hydrolysate is amalgamated with a lipidic dry membrane derived from cholesterol and egg yolk lecithin to form a liposome suspension, the microporous filtration and ultrasonic treatment with chitosan coating results in the formation of diosgenin chitosan transfersomes [113].
Clinical Trials on Surface Modified Ultradeformable Vesicles
Clinical trials represent the primary and most efficient approach for assessing the effectiveness of a pharmaceutical compound on a particular medical condition. They serve as a pivotal stage in the development of drugs. Consequently, examining clinical trials, particularly the analysis of registered clinical trials, has gained significant prominence in research endeavors, offering valuable insights for advancing future clinical practices. ClinicalTrials.gov, a publicly accessible trial registry, is jointly maintained by the US Food and Drug Administration & the US National Library of Medicine [114]. The clinical trial data available on the different surface-modified UDVs is represented in Table IV.
Fathalla et al. conducted a study to enhance anthralin efficacy against psoriasis by formulating liposomes via thin-film hydration and ethosomes via cold method. Optimized preparations were incorporated into various gel bases to study drug release kinetics. Clinical trials (NCT03348462) with 20 patients followed a randomized, prospective, single-blinded, controlled design. In vitro drug release profiles after 24 h showed liposome suspension > (hydroxyethyl cellulose) HEC gel > (Pluronic® F-127) PL-127 gel > (hydroxypropyl methylcellulose) HPMC gel for liposomes, and ethosome suspension > ethosome HPMC gel > ethosome PL-127 gel > ethosome HEC gel for ethosomes. Ethosomal gel exhibited 2.5- to 4.5-fold higher drug permeation than liposomal gel, drug solution, and hydroalcoholic solution. Short contact anthralin therapy in adults led to a significant -63.3% reduction in (Psoriasis Area and Severity Index) PASI score. Group II (ethosomes) showed significantly greater PASI reduction and patient satisfaction than Group I (liposomes). Both groups achieved maximum therapeutic effect below the irritation threshold, indicating effective side effect mitigation by nanocarriers. Both formulations demonstrated substantial effectiveness against psoriasis, with ethosomal gel showing superior outcomes in effectiveness, safety, and patient quality of life improvement [120].
The efficacy and safety of ketoprofen were assessed in an ultra-deformable vesicle gel formulation compared to a ketoprofen-free gel in individuals suffering from osteoarthritis (OA) knee pain. A randomized, prospective, double-blind, phase III clinical trial study (NCT00722852) was conducted at 39 centers throughout the United States. Eligible participants were subjected to randomization to administer either 100 mg of ketoprofen within a 4.4 g transfersome gel (IDEA-033) or a 4.4 g ketoprofen-free vehicle (TDT 064). The percentage of patients achieving a ≥ 50% reduction in (Western Ontario and McMaster Universities Osteoarthritis Index) WOMAC pain score from baseline at Week 12 was 50.5% (0.45–0.57; 95% CI) for TDT 064 and 41.2% (0.35–0.47; 95%CI) for IDEA-033. IDEA-033 exhibited inferiority relative to TDT 064 in mitigating moderate osteoarthritis (OA) knee pain and enhancing joint function [121].
Osteoarthritis (OA) significantly impacts the quality of life and healthcare resources due to the aging population. Oral (Nonsteroidal anti-inflammatory drugs) NSAIDs, commonly used for OA pain relief, pose systemic toxicity risks affecting cardiovascular, renal, and gastrointestinal systems. Therefore, their use is limited or cautioned against in individuals with concomitant and comorbidities medications. Topical NSAID therapy presents as a feasible substitution for oral NSAIDs. The topical application of ketoprofen encapsulated in an ultradeformable phospholipid vesicle, specifically a Transfersome (IDEA AG), and formulated into a Transfersome gel (IDEA-033), facilitates the ketoprofen biodistribution to a peripheral target. This method ensures both prolonged drug deposition and localization. The study (NCT00716547) aimed to evaluate the safety and efficacy of IDEA-003 of 12-week treatment in a controlled, randomized trial containing either 50 or 100 mg ketoprofen in patients suffering from knee pain due to OA.
Additionally, the study aimed to compare the efficacy of IDEA-033 with that of a matching oral celecoxib and ketoprofen-free vehicle (TDT 064). IDEA-033 showed no significant advantage over the ketoprofen-free vehicle TDT 064 for OA knee pain treatment. However, all topical treatments surpassed the oral placebo, comparable to celecoxib, suggesting non-inferiority. Further investigation into TDT 064’s efficacy in OA pain management is warranted [122].
Ketoprofen, a peripherally acting NSAID, can lead to dose-related GI bleeding. Topical application of NSAIDs to affected areas reduces these adverse effects. Diractin, containing ketoprofen in Transfersome® gel, has demonstrated pain reduction comparable to oral celecoxib in knee osteoarthritis. Transfersome carriers, lipid vesicles loaded with active substances, are applied topically. The study aimed to assess Diractin’s effect on pain induced by eccentric muscle contractions, a model for DOMS (Delayed Onset Muscle Soreness). It refers to the skeletal muscle discomfort that reaches its peak 24 to 48 h following exercise. A dose of 25 mg ketoprofen in Diractin was superior to a placebo in treating muscle pain and oral ketoprofen at some time points.
Conversely, 25 mg oral ketoprofen was not statistically different from placebo. Diractin was effective compared to untreated controls after multiple applications. Further investigation in larger trials is needed to confirm optimal dosing and regimen. DOMS proves suitable for evaluating Diractin’s therapeutic effect on acute muscle pain, suggesting it is a valuable option for musculoskeletal pain treatment [123].
Rother et al. conducted a double-blinded, randomized, multicenter clinical trial comparing epicutaneous ketoprofen in Transfersome (IDEA-033) with oral celecoxib and placebo for reducing signs and symptoms of knee osteoarthritis. IDEA-033 demonstrated safety and good tolerability. While IDEA-033 exhibited slightly more skin irritations than the placebo gel, erythema intensity was generally mild and reversible. Other adverse events were evenly distributed across all treatment groups. Gastrointestinal adverse events for IDEA-033 were comparable to placebo. Ketoprofen plasma concentrations in the IDEA-033 group ranged from 4.6 to 677 ng/ml, corresponding to 0.1–10% of maximum concentrations seen with a standard 200 mg ketoprofen therapeutic oral dose per day. Thus, systemic exposure to ketoprofen with IDEA-033 was markedly lower than oral administration [124].
Limitations/Learnings from Clinical Trials
Different UDVs, including ethosomes and transfersomes, were investigated and administered topically in the clinical trials outlined above. These formulations exhibited favorable safety profiles, with adverse events reported as mild, transient, and reversible with UDV-based treatments [124]. The clinical trials conducted thus far have not identified significant safety issues associated with UDVs. The multicentre double-blinded trials have been initiated further to enhance the clinical comprehension of UDVs [125]. The limitation of the study conducted by Fathalla et al. is the relatively small size of the treatment groups and the lack of inclusion of a conventional anthralin formulation as a comparative control. The sample size in the clinical trial aligns with the only published clinical trial focusing on anthralin liposomes encompassing 23 patients [126]. However, a conventional anthralin preparation such as ointment and creams were omitted from the study design as a control due to its adverse effects, including staining of both clothing and skin, cutaneous irritation, and limited efficacy in improving psoriasis lesions [120]. The clinical trial conducted by Rother et al. on ketoprofen delivery identified certain limitations in their findings, which included the absence of an actively established control group within the study and the potential impact of rescue medication on the assessment of treatment response [121]. The clinical trial study by Conghan et al. lacked a double-dummy blinding approach. However, it’s important to note that patients receiving topical treatment may have been less influenced by their expectations of pain relief, as they knew they were not receiving celecoxib. Furthermore, the study’s requirement for radiographically confirmed OA in patients under 45 years old could limit the broader applicability of the findings. Given the observed disparity between actual knee pain and radiographic evidence of knee OA, future studies may benefit from selecting patients based solely on clinical symptoms, thus improving the clinical trial’s application [122].
Conclusion and Future Perspectives
The surface modification of UDVs represents a pivotal advancement in transdermal/topical drug delivery. The current review article has unveiled novel insights into the potential of these carriers to revolutionize the delivery of drugs in medicine. Surface modification has emerged as the optimal strategy for addressing challenges related to the incapabilities of lipid-based nano vesicular carriers. Different surface modifying agents such as polymers, ligands, peptides, and bioactive molecules have been explored to efficiently advance the delivery of drugs by improving their targeting capacity, penetration, bioavailability, and entrapment efficiency. This review discussed the different surface modification strategies for surface functionalization of UDVs by covering an overview of applications through recent case studies with its patents and clinical trials related to surface-modified UDVs. Thus, this strategic approach demonstrates considerable and significant potential in drug delivery through the skin to treat various diseases.
Additional efforts are needed to surmount challenges intrinsic to optimizing formulations, developing economically viable nanocarriers, addressing toxicity concerns, and scaling up production processes. Moreover, there is a need for further in silico modeling research to elucidate the molecular interactions between the surfaces of nanocarriers and coating materials. Moreover, it is imperative to thoroughly understand the optimal physicochemical attributes, pharmacokinetic profiles, drug release kinetics, and clearance dynamics of these surface-modified UDVs before achieving successful clinical translation. Besides the potential therapeutic benefits of surface modifications, it is also essential to systematically explore their intricate interactions with the cellular microenvironment by considering their potential impact on human health and toxicity[28]. The mechanism behind successfully delivering these modified UDVs through skin remains unclear, which requires a detailed understanding. Not much exposure in the research related to other surface-modified UDV applications has been carried out, and there is a markedly limited availability of patents, which requires more focus. The long-term stability studies and scale-up considerations represent a significant challenge due to the inadequate availability of research or data in this field. The absence of pivotal clinical and preclinical trials on surface-modified UDVs poses complexities in elucidating and establishing the requisite safety profile for developing pharmaceuticals within the industrialized sector.
Abbreviations
- SC:
-
Stratum Corneum
- UDV:
-
Ultradeformable vesicles
- EDC:
-
(1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide),
- NHS:
-
N-hydroxysuccinimide,
- DMAP:
-
4-Dimethylaminopyridine,
- DOPE:
-
Dioleoyl phosphoethanolamine
- rpm:
-
Revolutions per minute
- HA:
-
Hyaluronic acid
- PEG:
-
Polyethylene glycol
- ODA:
-
Octadecylamine
- IND:
-
Indomethacin
- IND-TS:
-
Transfersomes
- OHAEC:
-
HA-enriched spironolactone-loaded cerosomes
- HA-ES:
-
Ethosome
- PGS:
-
Propylene glycol
- HPLC:
-
High Performance Liquid Chromatography
- TCI:
-
Transcutaneous immunization
- ESNFM:
-
Silk fibroin nanofibrous mats, encapsulated with ethosomes
- GC:
-
Galactosylated chitosan
- OVA:
-
Ovalbumin
- DC:
-
Dendritic cells
- DS:
-
Dextran sulphate
- DEX:
-
Dexamethasone
- DS-RLs:
-
Conventional liposome hydrogel
- FLs:
-
Flexible liposome hydrogel
- DIR:
-
1,1-Dioctadecyl-3,3,3,3- tetramethylindotricarbocyaineiodide
- DSPE-PEG-2000:
-
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol) 2000
- TFS:
-
Transfersomes
- MMPTS:
-
Mixed Monoterpenes Edge Activated PEGylated TFSs
- SIN:
-
Sinomenine
- Eth− PEI :
-
Polyethylenimine-modified ethosomes
- Eth− SC :
-
Sodium cholate-modified ethosomes
- DOX:
-
Doxorubicin
- OLM:
-
Olmesartan medoxomil
- Con-A:
-
Concanavalin-A
- HaCaT:
-
Keratinocyte skin cell line
- A375 cells:
-
Human melanoma skin cell lines
- CPP:
-
Cell-penetrating peptide
- LR:
-
Lycorine
- Pal-KTTKS or PPP:
-
Palmitoyl pentapeptide
- TES:
-
Transformer-ethosomes
- MA:
-
Myristic acid
- IMC:
-
Indomethacin
- IMC-KTTKS-Es:
-
Indomethacin loaded ethosomes
- GA:
-
Glycyrrhetinic acid
- GA-TPGS:
-
Glycyrrhetinic acid-D-α-tocopherol acid polyethylene glycol succinate
- CUR:
-
Curcumin
- ES:
-
Ethosomes
- EM:
-
Electron microscopy
- HRTEM:
-
High-Resolution Transmission Electron Microscopy
- SEM:
-
Scanning Electron Microscopy
- AFM:
-
Atomic force microscopy
- EDL:
-
Electrical Double Layer
- IR:
-
Infrared
- FTIR:
-
Fourier Transform Infrared Spectroscopy
- ATR-FTIR:
-
Attenuated total reflectance
- PEI:
-
Polyethyleneimine
- Gal-PEI:
-
Galactosylated polyethyleneimine
- TCM:
-
Traditional Chinese medicine
- H2O2 :
-
Hydrogen Peroxide
- NO:
-
Nitric Oxide
- UC:
-
Ulcerative Colitis
- Ach:
-
Acetylcholine
- ATP:
-
Adenosine triphosphate
- VIP:
-
Vasoactive Intestinal Peptide
- SCF:
-
Stem Cell Factor
- ICC:
-
Interstitial Cells of Cajal
- HEC:
-
Hydroxyethyl cellulose
- PL-127:
-
Pluronic® F-127
- HPMC:
-
Hydroxypropyl Methylcellulose
- PASI:
-
Psoriasis Area and Severity Index
- OA:
-
Osteoarthritis
- WOMAC:
-
Western Ontario and McMaster Universities Osteoarthritis Index
- NSAIDs:
-
Nonsteroidal anti-inflammatory drugs
- DOMS:
-
Delayed Onset Muscle Soreness
References
Mohammad Z, Zeeshan A, Faisal S, Suhail A, Sahar I, Mohd S, et al. Vesicular drug delivery system used for liver diseases. World J Pharm Sci. 2017;5:28–35. https://wjpsonline.com/index.php/wjps/article/view/vesicular-drug-delivery-system-liver-diseases.
Tewabe A, Abate A, Tamrie M, Seyfu A, Siraj EA. Targeted drug delivery — from magic bullet to nanomedicine: Principles, challenges, and future perspectives. J Multidiscip Healthc. 2021;14:1711–24.
Verma S, Utreja P. Vesicular nanocarrier based treatment of skin fungal infections: Potential and emerging trends in nanoscale pharmacotherapy. Asian J Pharm Sci. 2018;000:1–13.
Thakur A, Roy A, Chatterjee S, Chakraborty P, Bhattacharya K, Mahata PP. Recent Trends in Targeted Drug Delivery. In: Smgebooks. 2015.
Devi V, Jain N, Valli K. Importance of novel drug delivery systems in herbal medicines. Pharmacogn Rev. 2010;4:27–31.
Faheem AM, Abdelkader DH. Novel drug delivery systems. Eng. Drug Deliv. Syst. Elsevier LTD.; 2019. https://doi.org/10.1016/B978-0-08-102548-2.00001-9.
Benson HAE. Transfersomes for transdermal drug delivery. Expert Opin Drug Deliv. 2006;3:727–37. Available from: https://pubmed.ncbi.nlm.nih.gov/17076595/.
Garg V, Singh H, Bhatia A, Raza K, Singh SK, Singh B, et al. Systematic development of transethosomal gel system of piroxicam: formulation optimization, in vitro evaluation, and ex vivo assessment. AAPS PharmSciTech. 2017;18:58–71. Available from: http://link.springer.com/10.1208/s12249-016-0489-z.
Song CK, Balakrishnan P, Shim C, Chung S, Chong S, Kim D. A novel vesicular carrier, transethosome, for enhanced skin delivery of voriconazole: Characterization and in vitro / in vivo evaluation. Colloids Surf B Biointerfaces. 2012;92:299–304. https://doi.org/10.1016/j.colsurfb.2011.12.004.
Kumar L, Utreja P. Formulation and characterization of transethosomes for enhanced transdermal delivery of propranolol hydrochloride. Micro Nanosyst. 2019;12:38–47.
Tanner T, Marks R. Delivering drugs by the transdermal route: Review and comment. Ski Res Technol. 2008;14:249–60.
Ascenso A, Raposo S, Batista C, Cardoso P, Mendes T, Praça FG, et al. Development, characterization, and skin delivery studies of related ultradeformable vesicles: Transfersomes, ethosomes, and transethosomes. Int J Nanomedicine. 2015;10:5837–51.
Manosroi A, Jantrawut P, Khositsuntiwong N, Manosroi W, Manosroi J. Novel elastic nanovesicles for cosmeceutical and pharmaceutical applications. Chiang Mai J Sci. 2009;36:168–78.
Morilla MJ, Romero EL. Ultradeformable phospholipid vesicles as a drug delivery system: a review. Res Reports Transdermal Drug Deliv. 2015;4:55.
Ascenso A, Raposo S, Batista C, Cardoso P, Mendes T, Praça FG, et al. Development, characterization, and skin delivery studies of related ultradeformable vesicles: Transfersomes, ethosomes, and transethosomes. Int J Nanomedicine. 2015;10:5837–51.
Thadanki M, Babu AK. Review on ethosomes: a novel approach of liposomes. Int J Pharm Life Sci. 2015;6:4171–6.
Amnuaikit T, Limsuwan T, Khongkow P. Vesicular carriers containing phenylethyl resorcinol for topical delivery system; liposomes, transfersomes and invasomes. Asian J Pharm Sci. 2018;13:472–84.
Aziz DE, Abdelbary AA, Elassasy AI. Investigating superiority of novel bilosomes over niosomes in the transdermal delivery of diacerein: in vitro characterization, ex vivo permeation and in vivo skin deposition study. J Liposome Res. 2019;29:73–85. https://doi.org/10.1080/08982104.2018.1430831.
Albash R, Yousry C, Al-Mahallawi AM, Alaa-Eldin AA. Utilization of PEGylated cerosomes for effective topical delivery of fenticonazole nitrate: in-vitro characterization, statistical optimization, and in-vivo assessment. Drug Deliv. 2021;28:1–9. https://doi.org/10.1080/10717544.2020.1859000.
Albash R, Fahmy AM, Hamed MIA, Darwish KM, El-Dahmy RM. Spironolactone hyaluronic acid enriched cerosomes (HAECs) for topical management of hirsutism: in silico studies, statistical optimization, ex vivo, and in vivo studies. Drug Deliv. 2021;28:2289–300. https://doi.org/10.1080/10717544.2021.1989089.
Song YK, Kim CK. Topical delivery of low-molecular-weight heparin with surface-charged flexible liposomes. Biomaterials. 2006;27:271–80.
Duangjit S, Obata Y, Sano H, Kikuchi S, Onuki Y. Menthosomes, novel ultradeformable vesicles for transdermal drug delivery: optimization and characterization. Biol Pharm Bull. 2012;35:1720–8.
Lakshmi PK, Kalpana B, Prasanthi D. Invasomes-novel vesicular carriers for enhanced skin permeation. Syst Rev Pharm. 2014;4:26–30.
Shaji J, Bajaj R. Transethosomes: a new prospect for enhanced transdermal delivery. Int J Pharm Sci Res. 2018;9:2681–5.
Zhang Y, Xia Q, Li Y, He Z, Li Z, Guo T, et al. CD44 assists the topical anti-psoriatic efficacy of curcumin-loaded hyaluronan-modified ethosomes: A new strategy for clustering drug in inflammatory skin. Theranostics. 2019;9:48–64.
Priya S, Desai VM, Singhvi G. Surface modification of lipid-based nanocarriers: a potential approach to enhance targeted drug delivery. ACS Omega. 2022;8:74–86.
Khan AA, Allemailem KS, Almatroodi SA, Almatroudi A, Rahmani AH. Recent strategies towards the surface modification of liposomes: an innovative approach for different clinical applications. 3 Biotech. 2020;10. https://doi.org/10.1007/s13205-020-2144-3.
Osman N, Devnarain N, Omolo CA, Fasiku V, Jaglal Y, Govender T. Surface modification of nano-drug delivery systems for enhancing antibiotic. Nanomedicine and Nanobiotechnology. 2021;14:1–24.
Doane T, Burda C. Nanoparticle mediated non-covalent drug delivery. Adv Drug Deliv Rev. 2013;65:607–21. https://doi.org/10.1016/j.addr.2012.05.012.
Yang X, Wang X, Hong H, Elfawal G, Lin S, Wu J, et al. Galactosylated chitosan-modified ethosomes combined with silk fibroin nanofibers is useful in transcutaneous immunization. J Control Release. 2020;327:88–99.
Luiz MT, Viegas JSR, Abriata JP, Tofani LB, Vaidergorn M de M, Emery F da S, et al. Docetaxel-loaded folate-modified TPGS-transfersomes for glioblastoma multiforme treatment. Mater Sci Eng C. 2021;124:112033. https://doi.org/10.1016/j.msec.2021.112033.
Sanità G, Carrese B, Lamberti A. Nanoparticle surface functionalization: how to improve biocompatibility and cellular internalization. Front Mol Biosci. 2020;7:1–20.
Metkar SP, Fernandes G, Navti PD, Nikam AN, Kudarha R, Dhas N, et al. Nanoparticle drug delivery systems in hepatocellular carcinoma: A focus on targeting strategies and therapeutic applications. OpenNano. 2023;12:100159. https://doi.org/10.1016/j.onano.2023.100159.
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Cancer Nanotechnol. 2014;66:2–25.
Jangdey MS, Kaur CD, Saraf S. Efficacy of Concanavalin-A conjugated nanotransfersomal gel of apigenin for enhanced targeted delivery of UV induced skin malignant melanoma. Artif Cells Nanomed Biotechnol. 2019;47:904–16. https://doi.org/10.1080/21691401.2019.1578784.
Abualhasan MN, Al- Masri MY, Manasara R, Yadak L, Abu-Hasan NS. Anti-inflammatory and anticoagulant activities of synthesized NSAID prodrug esters. Scientifica (Cairo). 2020;2020:1–6.
Neises B, Steglich W. Simple method for the esterification of carboxylic acids. Angew Chemie Int Ed English. 1978;17:522–4.
Sheehan JC, Hess GP. A new method of forming peptide bonds. J Am Chem Soc. 1955;77:1067–8.
Baelo A, Levato R, Julián E, Crespo A, Astola J, Gavaldà J, et al. Disassembling bacterial extracellular matrix with DNase-coated nanoparticles to enhance antibiotic delivery in biofilm infections. J Control Release. 2015;209:150–8. https://doi.org/10.1016/j.jconrel.2015.04.028.
El Menshawe SF, Aboud HM, Elkomy MH, Kharshoum RM, Abdeltwab AM. A novel nanogel loaded with chitosan decorated bilosomes for transdermal delivery of terbutaline sulfate: artificial neural network optimization, in vitro characterization and in vivo evaluation. Drug Deliv Transl Res. 2020;10:471–85.
Guo T, Lu J, Fan Y, Zhang Y, Yin S, Sha X, et al. TPGS assists the percutaneous administration of curcumin and glycyrrhetinic acid coloaded functionalized ethosomes for the synergistic treatment of psoriasis. Int J Pharm. 2021;604:120762. https://doi.org/10.1016/j.ijpharm.2021.120762.
European Medicines Agency. ICH guideline Q3C (R6) on impurities: guideline for residual solvents. Int Conf Harmon Tech Requir Regist Pharm Hum Use. 2019;31:24.
Shah S, Dhawan V, Holm R, Nagarsenker MS, Perrie Y. Liposomes: Advancements and innovation in the manufacturing process. Adv Drug Deliv Rev. 2020;154–155:102–22.
Yuan M, Niu J, Xiao Q, Ya H, Zhang Y, Fan Y, et al. Hyaluronan-modified transfersomes based hydrogel for enhanced transdermal delivery of indomethacin. Drug Deliv. 2022;29:1232–42. https://doi.org/10.1080/10717544.2022.2053761.
Zhu J, Tang X, Jia Y, Ho CT, Huang Q. Applications and delivery mechanisms of hyaluronic acid used for topical/transdermal delivery – A review. Int J Pharm. 2020;578:119127. https://doi.org/10.1016/j.ijpharm.2020.119127.
Xie J, Ji Y, Xue W, Ma D, Hu Y. Hyaluronic acid-containing ethosomes as a potential carrier for transdermal drug delivery. Colloids Surf B Biointerfaces. 2018;172:323–9. https://doi.org/10.1016/j.colsurfb.2018.08.061.
Song L, Pan Z, Zhang H, Li Y, Zhang Y, Lin J, et al. Dually folate/CD44 receptor-targeted self-assembled hyaluronic acid nanoparticles for dual-drug delivery and combination cancer therapy. J Mater Chem B. 2017;5:6835–46.
Yang JA, Kim ES, Kwon JH, Kim H, Shin JH, Yun SH, et al. Transdermal delivery of hyaluronic acid - Human growth hormone conjugate. Biomaterials. 2012;33:5947–54. https://doi.org/10.1016/j.biomaterials.2012.05.003.
Zhao YP, Han JF, Zhang FY, Liao TT, Na R, Yuan XF, et al. Flexible nano-liposomes-based transdermal hydrogel for targeted delivery of dexamethasone for rheumatoid arthritis therapy. Drug Deliv. 2022;29:2269–82. https://doi.org/10.1080/10717544.2022.2096718.
Zheng H, Xu C, Fei Y, Wang J, Yang M, Fang L, et al. Monoterpenes-containing PEGylated transfersomes for enhancing joint cavity drug delivery evidenced by CLSM and double-sited microdialysis. Mater Sci Eng C. 2020;113:110929. https://doi.org/10.1016/j.msec.2020.110929.
Ma L, Wang X, Wu J, Zhang D, Zhang L, Song X, et al. Polyethylenimine and sodium cholate-modified ethosomes complex as multidrug carriers for the treatment of melanoma through transdermal delivery. Nanomedicine. 2019;14:2395–408.
Albash R, Refai H, Abdelbary AA. Tailoring of PEGylated bilosomes for promoting the transdermal delivery of olmesartan medoxomil: in- vitro characterization, ex-vivo permeation and in-vivo assessment. Int J Nanomedicine. 2019;14:6555–74.
Parashar P, Rana P, Dwivedi M, Saraf SA. Dextrose modified bilosomes for peroral delivery: improved therapeutic potential and stability of silymarin in diethylnitrosamine-induced hepatic carcinoma in rats. J Liposome Res. 2019;29:251–63.
Verma N, Saraf S. Development and optimization of mannosylated naringenin loaded transfersomes using response surface methodology for skin carcinoma. Int J Appl Pharm. 2021;13:235–41.
Li Y, Tai Z, Ma J, Miao F, Xin R, Shen C, et al. Lycorine transfersomes modified with cell-penetrating peptides for topical treatment of cutaneous squamous cell carcinoma. J Nanobiotechnology. 2023;21:1–18. https://doi.org/10.1186/s12951-023-01877-4.
Kim JE, Oh GH, Jang GH, Kim YM, Park YJ. Transformer-ethosomes with palmitoyl pentapeptide for improved transdermal delivery. J Drug Deliv Sci Technol. 2019;52:460–7. https://doi.org/10.1016/j.jddst.2019.04.039.
Niu J, Yuan M, Li H, Liu Y, Wang L, Fan Y, et al. Pentapeptide modified ethosomes for enhanced skin retention and topical efficacy activity of indomethacin. Drug Deliv. 2022;29:1800–10. https://doi.org/10.1080/10717544.2022.2081739.
Engelke L, Winter G, Hook S, Engert J. Recent insights into cutaneous immunization: How to vaccinate via the skin. Vaccine. 2015;33:4663–74. https://doi.org/10.1016/j.vaccine.2015.05.012.
Ita K. Transdermal delivery of vaccines – Recent progress and critical issues. Biomed Pharmacother. 2016;83:1080–8. https://doi.org/10.1016/j.biopha.2016.08.026.
Villablanca EJ, Mora JR. A two-step model for Langerhans cell migration to skin-draining LN. Eur J Immunol. 2008;38:2975–80.
Zhang D, Fan L, Ma L, Liu J, Zhou K, Song X, et al. Helicobacter pylori ribosomal protein-A2 peptide/ silk fibroin nanofibrous composites as potential wound dressing. J Biomed Nanotechnol. 2019;15:507–17.
Song DW, Kim SH, Kim HH, Lee KH, Ki CS, Park YH. Multi-biofunction of antimicrobial peptide-immobilized silk fibroin nanofiber membrane: Implications for wound healing. Acta Biomater. 2016;39:146–55. https://doi.org/10.1016/j.actbio.2016.05.008.
Fan L, Li JL, Cai Z, Wang X. Creating biomimetic anisotropic architectures with co-aligned nanofibers and macrochannels by manipulating ice crystallization. ACS Nano. 2018;12:5780–90.
Diaz-montes E. Dextran: sources, structures, and properties. Polysaccharides. 2021;2:554–65.
Yang M, Ding J, Feng X, Chang F, Wang Y, Gao Z, et al. Scavenger receptor-mediated targeted treatment of collagen-induced arthritis by dextran sulfate-methotrexate prodrug. Theranostics. 2017;7:97–105.
Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev. 2016;99:28–51. https://doi.org/10.1016/j.addr.2015.09.012.
Tagami T, Ernsting MJ, Li SD. Optimization of a novel and improved thermosensitive liposome formulated with DPPC and a Brij surfactant using a robust in vitro system. J Control Release. 2011;154:290–7. https://doi.org/10.1016/j.jconrel.2011.05.020.
Rangsimawong W, Opanasopit P, Rojanarata T, Ngawhirunpat T. Terpene-containing PEGylated liposomes as transdermal carriers of a hydrophilic compound. Biol Pharm Bull. 2014;37:1936–43.
Vega E, Egea MA, Garduño-Ramírez ML, García ML, Sánchez E, Espina M, et al. Flurbiprofen PLGA-PEG nanospheres: Role of hydroxy-β-cyclodextrin on ex vivo human skin permeation and in vivo topical anti-inflammatory efficacy. Colloids Surfaces B Biointerfaces. 2013;110:339–46.
Jepson MA, Clark MA, Hirst BH. M cell targeting by lectins: A strategy for mucosal vaccination and drug delivery. Adv Drug Deliv Rev. 2004;56:511–25.
Sharon N. Lectin-carbohydrate complexes of plants and animals: an atomic view. Trends Biochem Sci. 1993;18:221–6.
Clark MA, Hirst BH, Jepson MA. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev. 2000;43:207–23.
Mishra N, Tiwari S, Vaidya B, Agrawal GP, Vyas SP. Lectin anchored PLGA nanoparticles for oral mucosal immunization against hepatitis B. J Drug Target. 2011;19:67–78.
Kilpatrick DC, Pusztai A, Grant G, Graham C, Ewen SWB. Tomato lectin resists digestion in the mammalian alimentary canal and binds to intestinal villi without deleterious effects. FEBS Lett. 1985;185:299–305.
Lis H. Lectins as Molecules and as Tools. Annu Rev Biochem. 1986;55:35–67.
Lehr CM, Bouwstra JA, Kok W, Noach ABJ, de Boer AG, Junginger HE. Bioadhesion by Means of Specific Binding of Tomato Lectin. Pharm Res. 1992;9:547–53.
Lammers T, Hennink WE, Storm G. Tumour-targeted nanomedicines: Principles and practice. Br J Cancer. 2008;99:392–7.
Yan H, Kamiya T, Suabjakyong P, Tsuji NM. Targeting C-type lectin receptors for cancer immunity. Front Immunol. 2015;6:1–9.
Irache JM, Salman HH, Gamazo C, Espuelas S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin Drug Deliv. 2008;5:703–24.
Kerrigan AM, Brown GD. C-type lectins and phagocytosis. Immunobiology. 2009;214:562–75. https://doi.org/10.1016/j.imbio.2008.11.003.
Chen P, Zhang X, Jia L, Prud’homme RK, Zoltan S, Sinko PJ. Optimal Structural Design of Mannosylated Nanocarriers for Macrophage Targeting. J Control release. 2014;194:341–549.
Gupta A, Deep KC. Comparative Evaluation of Two Different Novel Formulations of Quercetin Against Non Melanoma Skin Cancer in Human Subjects. J Clin Exp Dermatol Res. 2016;07:3–7.
Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES. Cell-penetrating Peptides as a Novel Transdermal Drug Delivery System. Chem Biol Drug Des. 2012;80:639–46.
Abdelhamid HN, Dowaidar M, Hällbrink M, Langel Ü. Gene delivery using cell penetrating peptides-zeolitic imidazolate frameworks. Microporous Mesoporous Mater. 2020;300:1–10.
Rothbard JB, Garlington S, Lin Q, Kirschberg T, Kreider E, McGrane PL, et al. Conjugation of arginine oligomers to cyclosporin A facilitates topical delivery and inhibition of inflammation. Nat Med. 2000;6:1253–7.
Jiang T, Wang T, Li T, Ma Y, Shen S, He B, et al. Enhanced transdermal drug delivery by transfersome-embedded oligopeptide hydrogel for topical chemotherapy of melanoma. ACS Nano. 2018;12:9693–701.
Jiang T, Mo R, Bellotti A, Zhou J, Gu Z. Gel-liposome-mediated co-delivery of anticancer membrane-associated proteins and small-molecule drugs for enhanced therapeutic efficacy. Adv Funct Mater. 2014;24:2295–304.
Dumont C, Bourgeois S, Fessi H, Jannin V. Lipid-based nanosuspensions for oral delivery of peptides, a critical review. Int J Pharm. 2018;541:117–35. https://doi.org/10.1016/j.ijpharm.2018.02.038.
Robinson LR, Fitzgerald NC, Doughty DG, Dawes NC, Berge CA, Bissett DL. Topical palmitoyl pentapeptide provides improvement in photoaged human facial skin. Int J Cosmet Sci. 2005;27:155–60.
Chourpiliadis C, Aeddula NR. Physiology, glucocorticoids. StatPearls Publishing; 2023. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560897/.
Cheng X, Qiu L, Wang F. 18α-Glycyrrhetinic acid (GA) ameliorates fructose-induced nephropathy in mice by suppressing oxidative stress, dyslipidemia and inflammation. Biomed Pharmacother. 2020;125:1–14.
Murck H. Symptomatic protective action of glycyrrhizin (Licorice) in COVID-19 infection? Front Immunol. 2020;11:1–5.
Shi J, Li J, Li J, Li R, Wu X, Gao F, et al. Synergistic breast cancer suppression efficacy of doxorubicin by combination with glycyrrhetinic acid as an angiogenesis inhibitor. Phytomedicine. 2021;81:153408. https://doi.org/10.1016/j.phymed.2020.153408.
Mishra A, Behura A, Mawatwal S, Kumar A, Naik L, Mohanty SS, et al. Structure-function and application of plant lectins in disease biology and immunity. Food Chem Toxicol. 2019;134:110827. https://doi.org/10.1016/j.fct.2019.110827.
Alavi M, Asare-Addo K, Nokhodchi A. Lectin protein as a promising component to functionalize micelles, liposomes and lipid nps against coronavirus. Biomedicines. 2020;8:1–16.
Wei X, Li L. Comparative glycoproteomics: Approaches and applications. Brief Funct Genom Proteom. 2009;8:104–13.
Silva MLS. Lectin biosensors in cancer glycan biomarker detection. Adv Clin Chem. 2019;1–61.
Del Genio V, Bellavita R, Falanga A, Hervé-Aubert K, Chourpa I, Galdiero S. Peptides to Overcome the Limitations of Current Anticancer and Antimicrobial Nanotherapies. Pharmaceutics. 2022;14:1–21.
Khairkhah N, Namvar A, Bolhassani A. Application of cell penetrating peptides as a promising drug carrier to combat viral infections. Mol Biotechnol. 2023;65:1387–402. https://doi.org/10.1007/s12033-023-00679-1.
Dinh CT, Vu HT, Phan QTH, Nguyen LP, Tran TQ, Van Tran D, et al. Synthesis of glycyrrhetinic acid-modified liposomes to deliver Murrayafoline A for treatment of hepatocellular carcinoma. J Mater Sci Mater Med. 2022;33:1–14.
Timmermans S, Souffriau J, Libert C. A General Introduction to Glucocorticoid Biology. Front Immunol. 2019;10:1–17.
Lühder F, Reichardt HM. Novel drug delivery systems tailored for improved administration of glucocorticoids. Int J Mol Sci. 2017;18:1–19.
Jayawardena HSN, Liyanage SH, Rathnayake K, Patel U, Yan M. Analytical Methods for Characterization of Nanomaterial Surfaces. Anal Chem. 2021;93:1889–911.
Bhosale SS, Avachat AM. Design and development of ethosomal transdermal drug delivery system of valsartan with preclinical assessment in Wistar albino rats. J Liposome Res. 2013;23:119–25.
Morilla M, Romero E. Carrier deformability in drug delivery. Curr Pharm Des. 2016;22:1118–34. Available from: http://www.eurekaselect.com/openurl/content.php?genre=article&issn=1381-6128&volume=22&issue=9&spage=1118.
Takechi-Haraya Y, Ohgita T, Demizu Y, Saito H, Izutsu K ichi, Sakai-Kato K. Current status and challenges of analytical methods for evaluation of size and surface modification of nanoparticle-based drug formulations. AAPS PharmSciTech. 2022;23. https://doi.org/10.1208/s12249-022-02303-y.
Yin Y, Lu Y. Handbook Of Synthetic Methodologies And Protocols Of Nanomaterials. World Sci Ser Nanosci Nanotechnol. 2019.
Rong L, Liping J, Cheng Y. Preparation method and application of ceramide ethosome carrying water-solubleliposoluble active substances. China: Jiangnan University; 2023.
Hongsheng W, Xingxing Y, Siyufan L, Linlin M, Gejie L, Xiaohan P. Preparation method of galactosylated chitosan modified immune ethosome. China: Donghua University; 2016.
Hongsheng W, Linlin M, Xingxing Y, Dongdong Z, Fan Y, Xiaohan P, et al. Preparation method of drug-loaded ethosome modified by galactosylated polyethyleneimine. China: Donghua University; 2017.
Teng G, Nianping F, Chunyun Z, Wei L, Yunlong F, Jianying L. Glycyrrhetinic acid-modified curcumin-loaded multifunctional ethosome as well as preparation method and application thereof. China: Shanghai University of Traditional Chinese Medicine; 2020.
Yongtai Z, Nianping F, Hongyu Z, Zhi W, Teng G, Xiaolin H, et al. Hyaluronic acid modified ethosome, acupoint drug delivery system containing same and application of system. China: Shanghai University of Traditional Chinese Medicine; 2021.
Hui Z, Tao L, Xuening Y, Bingquan W. Preparation method of diosgenin chitosan transfersome. China: Jiangxi Yongtong Technology Co ltd; 2021.
Feng Z, Gu Y, Yuan M, Xiao R, Fei Z. Clinical trials of liposomes in children’s anticancer therapy: a comprehensive analysis of trials registered on ClinicalTrials.gov. Int J Nanomedicine. 2022;17:1843–50.
Formulation and clinical evaluation of ethosomal and liposomal preparations of anthralin in psoriasis. Available from: https://clinicaltrials.gov/study/NCT03348462?term=NCT03348462&rank=1&tab=table. Accessed 6 Nov 2023.
Study of safety and efficacy of Diractin® for the treatment of Osteoarthritis (OA) of the knee. Available from: https://clinicaltrials.gov/study/NCT00722852?term=NCT00722852&rank=1. Accessed 6 Nov 2023.
Safety and efficacy of two dosages of Diractin® in Osteoarthritis (OA). Available from: https://clinicaltrials.gov/study/NCT00716547?term=NCT00716547&rank=1&tab=table. Accessed 6 Nov 2023.
Study of epicutaneously applied Ketoprofen Transfersome® Gel with or without combination with oral celecoxib for the treatment of muscle pain induced by eccentric exercise. Available from: https://clinicaltrials.gov/study/NCT01020279?term=NCT01020279&rank=1. Accessed 6 Nov 2023.
Ketoprofen in Transfersome Compared to Oral Celecoxib and Placebo for Pain Associated With Osteoarthritis of the Knee. Available from: https://clinicaltrials.gov/study/NCT00317733?term=NCT00317733&rank=1. Accessed 6 Nov 2023.
Fathalla D, Youssef EMK, Soliman GM. Liposomal and ethosomal gels for the topical delivery of anthralin: Preparation, comparative evaluation and clinical assessment in psoriatic patients. Pharmaceutics. 2020;12:1–24.
Rother M, Conaghan PG. A randomized, double-blind, phase III trial in moderate osteoarthritis knee pain comparing topical ketoprofen gel with ketoprofen-free gel. J Rheumatol. 2013;40:1742–8.
Conaghan PG, Dickson J, Bolten W, Cevc G, Rother M. A multicentre, randomized, placebo- and active-controlled trial comparing the efficacy and safety of topical ketoprofen in Transfersome gel (IDEA-033) with ketoprofen-free vehicle (TDT 064) and oral celecoxib for knee pain associated with osteoarthritis. Rheumatol (United Kingdom). 2013;52:1303–12.
Rother M, Seidel EJ, Clarkson PM, Mazgareanu S, Vierl U, Rother I. Efficacy of epicutaneous Diractin® (ketoprofen in Transfersome® gel) for the treatment of pain related to eccentric muscle contractions. Drug Des Devel Ther. 2009;143–9.
Rother M, Lavins BJ, Kneer W, Lehnhardt K, Seidel EJ, Mazgareanu S. Efficacy and safety of epicutaneous ketoprofen in Transfersome (IDEA-033) versus oral cefecoxib and placebo in osteoarthritis of the knee: Multicentre randomised controlled trial. Ann Rheum Dis. 2007;66:1178–83.
Yao L, Bojic D, Liu M. Applications and safety of gold nanoparticles as therapeutic devices in clinical trials. J Pharm Anal. 2023;13:960–7. https://doi.org/10.1016/j.jpha.2023.06.001.
Saraswat A, Agarwal R, Katare OP, Kaur I, Kumar B. A randomized, double-blind, vehicle-controlled study of a novel liposomal dithranol formulation in psoriasis. J Dermatolog Treat. 2007;18:40–5.
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal Nil.
Author information
Authors and Affiliations
Contributions
Planning of original draft, writing, conceptualization, literature search, and editing: DN.
Guidance, editing, review, conceptualization, & supervision: Dr. VKT & Dr MR.
Corresponding author
Ethics declarations
Conflict of Interest
No conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nayak, D., Rathnanand, M. & Tippavajhala, V.K. Navigating Skin Delivery Horizon: An Innovative Approach in Pioneering Surface Modification of Ultradeformable Vesicles. AAPS PharmSciTech 25, 126 (2024). https://doi.org/10.1208/s12249-024-02847-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-024-02847-1