
Abstract
The present study investigates the impact of the solid-state disorder of vortioxetine hydrobromide (HBr) on oxidative degradation under accelerated conditions. A range of solid-state disorders was generated via cryogenic ball milling. The solid-state properties were evaluated by calorimetry, infrared-, and Raman spectroscopies. While salt disproportionation occurred upon milling, no chemical degradation occurred by milling. The amorphous fraction remained physically intact under ambient storage conditions. Subsequently, samples with representative disordered fractions were mixed with a solid oxidative stressor (PVP-H2O2 complex) and were compressed to compacts. The compacts were exposed to 40°C/75% RH for up to 6 h. The sample was periodically withdrawn and analyzed for the physical transformations and degradation. Two oxidative degradation products (DPs) were found to be formed, for which dissimilar relations to the degree of disorder and kinetics of formation were observed. The degradation rate of the major DP formation obtained by fitting the exponential model to the experimental data was found to increase up to a certain degree of disorder and decrease with a further increase in the disordered fraction. In contrast, the minor DP formation kinetics was found to increase monotonically with the increase in the disorder content. For the similar crystallinity level, the degradation trend (rate and extent) differed between the single-phase disorder generated by milling and physically mixed two-phase systems. Overall, the study demonstrates the importance of evaluating the physical and chemical (in)stabilities of the disordered solid state of a salt form of a drug substance, generated through mechano-activation.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Diverse solid forms (salt, co-crystal, amorphous) are explored to enhance the stability, solubility, and dissolution of the active pharmaceutical ingredients (APIs) in early development stages [1,2,3,4]. Predicting the formation of degradation products (DPs) and the drug’s stability aids in setting shelf-life and storage specifications, thereby guarantying safety and efficacy. The high energy disordered crystal or amorphous state is reactive towards diverse physical and chemical instabilities [5]. While recrystallization of an amorphous API is commonly studied [6,7,8], a relationship between the physical and chemical stabilities of the disordered solid states is less often explored. Drug degradation can compromise safety, efficacy, tolerability, and use [9]. Forced degradation is performed in the early stages of drug development to identify intrinsic reactivity to different stress conditions [10, 11]. Conventional methods to predict the solid drug’s degradation and shelf-life are empirical, and do not necessarily consider the effects of crystal disorder [12]. Drug oxidation is a common degradation reaction [13, 14] and a risk factor to stability during manufacturing and upon storage, especially in the disordered solid state of susceptible APIs. The presence of reactive oxidative species in excipient(s) creates a high instability risk. To screen solid drug oxidation propensity, some methods have been reported in the literature [15,16,17,18,19], including some recent ones [20, 21]. Although the finished product contains a uniform quantity of drug, diverse solid states such as polymorphs, disorders, morphology, and crystallinity can alter the chemical reactivity [22].
Solid dosage processing such as milling, sieving, compaction, granulation, drying, and extrusion can generate surface or bulk crystal disorder [23]. Micronization is performed for API crystal size reduction and for improving solubility [24]. Cryo-milling is increasingly recognized as a green and environment-friendly route for the generation of amorphous and nanomaterials for drug delivery [25]. Also, prolonged milling is the process applied to generate fine grades of excipients such as lactose to use as inhaled carrier [26]. The mechanical stresses exerted by milling can trigger the physical transformation [27]. Cryo-milling is a preferred route to generate the disordered forms [28]. Feng et al. showed that cryogenic milling of griseofulvin leads to a reduction in crystallinity and accumulation of crystal defects [29]. Mechano-activation is reported to induce Maillard reaction between metoclopramide HCl and lactose [30]. Sagud et al. reported drug degradation during co-milling with excipients [31]. We have demonstrated the impact of solid-state disorder in simvastatin, nifedipine, and mifepristone on their oxidative instabilities [20, 32,33,34]. Our group previously reported the application of mechano-activation as a tool to assess autoxidation of olanzapine and mifepristone [23, 35]. In addition to amorphization, salt disproportionation has been reported to occur during drug product process such as compaction [36, 37]. Yet, it is seldom reported under milling [38]. The disproportionation of a salt form loses the immediate benefit associated with drug solubility. The degradation kinetics can compete with physical transitions. While the physical stability of such solid drugs subjected to mechanically induced disorders (types/states) is well-studied [39], their effects in the chemical reactivity have been rarely addressed.
In this study, we assess the role of solid-state disorder on the chemical reactivity of an antidepressant drug, vortioxetine (VOR), in its commercially available hydrobromide salt form (VOR-HBr) [40]. The strong basic character of VOR (pKa = 9.1) makes it prone to form salts with the weak acids. The acidic proton is transferred to the secondary N-atom on the piperazine ring of vortioxetine, forming the charge-assisted hydrogen bond (N+–H…Br−) [41]. The oxidative susceptibility of VOR-HBr is reported in the literature [42, 43]. The current work aims at evaluating the (in)stability of short- and long-range solid disorders. The disproportionation of hydrobromide counter-ion is also investigated herein. The powder compacts were prepared of VOR-HBr pre-mixed with a PVP-hydrogen peroxide (PHP) powder as a solid stressor and exposed to 40°C/75% RH. The samples were evaluated for the physical change using Raman spectroscopy, and chemical change using LC–MS, respectively. Also, a quantitative relationship is established between the content of the oxidative stressor and the total drug degradation. The present work emphasizes the relation between the degree/types of disorder and the extent of chemical degradation, which could be useful in the development of predictive stability models.
Materials and Methods
Materials
The crystalline materials, salt form vortioxetine hydrobromide (Mw, 379.37 g mol−1; molecular formula, C18H22N2S.HBr) and vortioxetine free base (molecular weight, 298.45 g mol−1; molecular formula, C18H22N2S) were purchased from Shenzhen Nexconn Pharmatechs Ltd. (Shenzhen, China). PVP-hydrogen peroxide stressor (PHP), a well-characterized and safe-to-handle solid-state oxidative reagent was prepared using the in-house procedure as reported in a previous study [18]. All other chemicals and buffer salts were of analytical reagent grade.
Generation of Solid-State Disorder Using Cryo-milling
Approximately, 2 g of VOR-HBr crystalline powder sample was milled in a 50 mL jar with a 2.0-cm stainless steel ball at cryogenic temperature (liquid nitrogen) using the milling frequency of 25 Hz (Retsch Mill, Germany). The milling was carried out separately for 2 min, 5 min, 10 min, 30 min, and 60 min. For each milling time point, using a thermocouple, the temperature of the powder was measured immediately after opening the jar.
The milled samples were handled under the controlled environment in the laboratory with RH < 55%. Immediately after milling, the collected powder samples were transferred to glass vials with air-tight closure. The glass vials were placed in the desiccator containing the silica-gel as desiccant.
For simplicity, the milling-induced disorders (termed as single phases) are referred to as defective crystalline (short-time-milled), partially crystalline (intermediate milled comprises crystalline and amorphous fractions), and fully amorphous (longest-milled with short-range order) and abbreviated as DC, PC, and FA, respectively. The unmilled VOR-HBr is designated as a fully crystalline (FC). Further, within PC, the different time-milled fractions are abbreviated as PC5, PC10, and PC30. In parallel, a two-phase system was prepared by physically mixing (PM) the amorphous and highly crystalline fractions (using an appropriate ratio of 2- and 60-min milled powders) as having expected crystallinity theoretically equivalent to a 10-min milled sample.
Characterization of Solid States of Milled Samples
Fourier Transform Infrared (FTIR) Spectroscopy
The spectroscopic measurements of the VOR-HBr powder samples were performed using an FTIR spectrometer, VERTEX 70 (Bruker Optik GMBH, Germany) with an attenuated total internal reflectance (ATR) sampling unit. The background was collected with 32 scans, and the spectra were collected as the average of 32 scans acquired in the spectral range from 600 to 4000 cm−1 with a spectral resolution of 4 cm−1. Using these same parameters, the vortioxetine free base powder was also measured for comparing it to the HBr salt form.
Differential Scanning Calorimetry (DSC)
The thermal properties of the milled VOR-HBr samples were evaluated using the DSC 204 F1 Phoenix (NETZSCH, Germany) equipped with an autosampler and intra-cooler. Adamantane, indium, zinc, and bismuth were used as references for temperature and enthalpy calibration. Calibration of heat capacity (Cp) was carried out using a Sapphire. Approximately, 10–15 mg of a powder sample was placed in an aluminum pan and covered with a lid with a pinhole. The samples were heated from 20 to 250°C at a heating rate of 10 K/min. The data was analyzed with NETZSCH Proteus Thermal Analysis Software version 6.1.0. The VOR-HBr and VOR free base were evaluated using the same DSC method, as controls.
Particle Size Distribution Analysis
At the end of each milling cycle, the particle size distribution was evaluated by using laser diffraction (HELOS/KR, Sympatec GmbH, Clausthal-Zellerfeld, Germany) with a dry dispersing system (Rodos/L, Sympatec) and a vibrating chute (Vibri, Sympatec). A dispersion pressure of 2.0 bar was applied with a 30-s sampling time. The data evaluation was conducted using Windox 5.6.0.0 (Sympatec) software.
Preparation and Characterization of VOR-HBr-PHP Compacts
Preparation and Exposure to Storage Conditions
An equal amount of PHP complex (a PVP-H2O2 reagent) and drug powder was weighed accurately and mixed together [18]. The powder mixture was compressed (using a compression force of 50 kN for 30 s) into a compact disc employing a hydraulic hand press (PerkinElmer, Waltham, MA, USA). Of all the generated milled samples, an accelerated oxidation study was performed on 2-min milled (defective crystalline), 10-min milled (partial crystalline), and 60-min milled (amorphous) samples and was abbreviated as DC-PHP, PC-PHP, and FA-PHP. The mixture of unmilled crystalline sample is termed as FC-PHP. The physically mixed two-phase system (theoretically equivalent to 10 min milled) is referred to as PM-PHP and treated similarly.
The solid compacts were filled into the glass vial exposed open to 40°C/75% RH in controlled stability cabinets (WTC Binder, Tuttlingen, Germany). Within the 6 h of total exposure, the samples were taken at intermittent intervals 0, 0.5, 1, 2, 4, and 6 h. The PHP-only compact was also exposed to identical conditions. The solid compacts were subjected to physical and chemical evaluation after exposure. The degradation extent of the drug in these compacts was first converted to a fraction degraded (dividing the value by 100) and was plotted against exposure time using Origin software (OriginLab®, Northampton, USA).
Analysis of Solid Compacts by Raman Spectroscopy
The solid compacts were analyzed by a RamanRXN Systems analyzer to evaluate the solid form. The spectra were acquired using a Kaiser PhAT Probe with a 6-mm large-area laser spot size providing a non-contact sample analysis. The emission power of the laser beam was kept below 500 mW at 830 nm and scans ranged from 150 to 2000 cm−1. The exposure time was kept 2 s, and triplicate samples were analyzed at each time point. Prior to Raman analysis, the morphology (physical form and color of compact) was visually noted.
Liquid Chromatography Photodiode Array/Mass Spectrometry (LC-PDA/MS) Analysis
As per our previous study [18], the LC method employed a reversed-phase separation. Gradient elution was used for the separation of drug and degradation products (DPs) in a Waters Acquity UPLC instrument using HSS T3 2.1 × 100 mm, 1.8 µm, column. The stability-indicating ability of the developed method was proven to separate the drug from oxidative DPs. After each stability exposure, solid compacts were individually removed and dissolved in a diluent mixture of acetonitrile and water (50:50 v/v). The final drug concentration was kept at 500 ppm, and the injection volume was kept at 1 µL. Detection of the drug was carried out using an ultraviolet-photo diode array detector (UV-PDA) at 230 nm. The degradation of FC-PHP, DC-PHP, PC-PHP, FA-PHP, and PM-PHP samples was expressed as an area percentage, i.e., based on the relative area under the curve (AUC) of the drug with respect to the DPs.
The solid compacts with the highest levels of DP-I and DP-II (after storing at 40°C/75% RH) were analyzed via LC–MS using a QExactiveTM Orbitrap (Thermo Fisher ScientificTM, USA) and electrospray ionization (ESI) operating in a positive mode (scans range from 150 to 2000 m/z). Data analysis was carried out using XcaliburTM 3.0. The line spectrum was derived from the total ion chromatogram (TIC) and extracted ion chromatogram (EIC) plots. The molecular ion peaks were compared with the literature reports for postulating possible chemical structures of DPs.
Estimation of Peroxide Content Using Amplex Kit and UV–Vis Spectrophotometry
The peroxide content in the PHP complex was determined using an Amplex® Red Hydrogen Peroxide/Peroxidase assay procedure. A calibration curve was established in the range of 1–25 µM from a 20 mM H2O2 working standard solution dissolved in 1 × Reaction Buffer (PBS). The detection range chosen for UV–visible spectrophotometry was kept between 590 and 560 nm. The sample solution (powder compact dissolved in 10 mL of phosphate buffer solution) was treated similarly to the standard using the same dilution regime. The content of hydrogen peroxide (H2O2) was estimated for FC-PHP, DC-PHP, PC-PHP and FA-PHP, and PHP alone (used as a control). The peroxide content of stability samples (n = 3) was also analyzed at 0.5, 1, and 6 h.
Results
Physical Characterization of Solid-State Disorder
Thermal Characterization
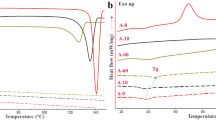
The milled samples were analyzed using the DSC method to evaluate the change in the thermal properties upon milling. The material properties related to the thermal event are tabulated in Table S1. The thermal events observed in each milled sample along with the unmilled (control) sample were integrated and reported. The unmilled VOR-HBr crystal demonstrated an onset of melting (Tm) at 230.60 ± 0.28°C having a melting enthalpy (ΔHm) of 110.75 ± 1.34 J g−1. For the intermediate and longer milled samples at 5 min, 10 min, 30 min, and 60 min, a glass transition event followed by recrystallization was clearly visible, while they were absent in unmilled (control) and were not clear in the profile of short-time milled (2 min) sample (Fig. 1).

Overlaid DSC thermograms of milled and unmilled vortioxetine samples; FC (unmilled), DC (2 min milled), PC5 (5 min milled), PC10 (10 min milled), PC30 (30 min milled), and FA (60 min milled). A zoomed inset of glass transition event is represented indicating the Tg onset
Also mentioned in Table S1 (supplementary information) is the heat capacity change (∆Cp) related to the glass transition temperatue (Tg) for each of the milled samples. Figure 2a depicts the change in the Tg over milling time. Both Tg and onset of crystallization decreased for the sample with increased milling time. The Tg profile was distinctly higher for the lower milling time (lower amorphous fraction) and that was higher with the increasing milling time/amorphous content. Since the moisture content values obtained of the representative milled samples using Karl Fischer titration were below the quantification limit (< 0.10%), we assume there is no or trivial contribution of varying moisture content among disordered samples to their observed Tg. Next, the DSC data was used to estimate the degree of crystallinity of milled VOR-HBr (shown in supplement Table S1). Assuming that the amorphous fraction undergoes recrystallization during DSC analysis, the degree of crystallinity was derived using Eq. 1 as follows:
where ∆Hc and ∆Hm are the enthalpy of crystallization and heat of fusion, respectively.

The plot of Tg (a) and the degree of crystallinity (b) of VOR-HBr as a function of milling time
The plot of the degree of crystallinity remaining over milling time is depicted in Fig. 2b. The crystallization enthalpy systematically increased upon milling for extended durations suggesting an increased formation of amorphous content upon milling. Although no clear glass transition and recrystallization exotherm signal were evident for the 2-min milled sample, we believe that surface disordering (formation of the local defects and crystal imperfections) would have already been initiated. The overall loss of crystallinity showed an exponential decay profile as a function of milling time. We applied here the assumption of a complete crystallization of the amorphous fraction present during DSC analysis, and thus the degree of amorphicity is simply the ratio of area under the crystallization exotherm to that under the melting endotherm. However, as these two events occur far apart at different temperatures, there would be the need for correction for the temperature-dependent enthalpy. We are aware of the method applied by Grisedale et al. including the enthalpy correction [44]. While the actual crystallinity across the milled samples might differ, we speculate that the values obtained by enthalpy-corrected methods will be comparable as the crystallization regions are within a narrow temperature zone.
In addition to HBr salt form, the melting onset of VOR free base was 116.90 ± 0.17°C, and the corresponding melting enthalpy (ΔHm) was 30.27 ± 3.60 J g−1 (see supplement Figure S1). These values are substantially lower than the HBr salt form.
The extent of recrystallization in the milled samples stored at 2–8°C (a temperature well below the Tg) was obtained after 30 days and 60 days. For a 10-min milled sample, the complete recrystallization was noted at the end of 30 days, and the fully amorphous sample (60-min milled) remained non-crystalline unit 30 days and fully crystallized after 60 days.
The WAXS measurements were performed on selected samples to verify the presence of a disordered phase formed upon milling. The intensity of Bragg peaks reduced upon milling for intermediate time (2–10 min), the intensity further decreased, and the peaks completely disappeared upon milling for 60 min (shown in supplement Figure S2). For these milled samples, UPLC analysis depicts no degradation product peaks confirming the absence of chemical degradation occurring during milling (shown in supplement Figure S3).
ATR-FTIR Spectroscopic Analysis of Disordered Samples
As depicted in Fig. 3, the spectral profiles of the milled samples comprise a distinct change in the 3100–2400 cm−1 region. As the milling progressed (with increased amorphization), a systematic broadening of the vibrational bands with a corresponding decrease in the peak intensity was evident. In the N–H stretching region (3100–2950 cm−1), a broadening of the peaks and a concomitant drop in the relative peak intensity at with the peak maximum of 2915 cm−1 were observed. The extent of broadening (owing to intermolecular H-bond disruptions) and drop in intensity follows the order of increasing amorphicity such that FA (fully amorphous) > PC30 (partially crystalline) > PC10 (partially crystalline) > PC5 (partially crystalline) > DC (defective crystalline) > FC (fully crystalline). The peak with a maximum at 2700 cm−1 (Ar–C–H region) also followed a similar decreasing trend over milling. A feeble, yet a clear change is observed for the shortest milled samples (2-min DC) as compared to the unmilled samples (crystalline). Amorphization appears to disrupt the H-bonds in the between molecules in the crystal lattices.

Overlays of partial and normalized IR spectra of milled and unmilled VOR-HBr powders in the 3100–2400 cm−1 region; FC (0 min milled), DC (2 min milled), PC5 (5 min milled), PC10 (10 min milled), PC30 (30 min milled), and FA (60 min milled)
For the peak with a maximum of 2450 cm−1 (a typical region of interest for the HBr) of the longest-milled samples (fully amorphous), a reduction in the peak intensity was observed (Supplementary Figure S4). This peculiar observation was indicating the possibility of disproportionation of HBr counter-ion from the salt form (VOR-HBr) during milling. In order to confirm the disproportionation, we recorded the spectrum for the VOR free base. Clearly, the peak with the maximum at 2450 cm−1 was absent in the free base sample. Relatively, the drop in intensity for the milled sample (fully amorphous) form was about 75–80% of the crystalline salt form. A complete absence of this peak in the free base and a slight decrease in intensity in the fully amorphous (60 min milled) VOR-HBr signify an amorphization occurring concomitantly with an extent of salt disproportionation. However, the exact extent of disproportionation was not quantified.
Particle Size and Specific Surface Area as a Function of Milling
Particle size distribution (PSD) of the sample was determined by laser diffraction. The measured particle size (mean ± standard deviation (SD), n = 3) of the unmilled VOR-HBr (FC) powder was 372.41 ± 6.64 µm (d50) with the specific surface area (SSA) of 0.92 ± 0.02 m2 g−1 (calculated using the particle size and powder true density). After milling, the d50 decreased to a specific value and remained comparable to all the milled samples over time. The surface area of the powder was calculated using the volumetric particle size d50 obtained from laser diffraction. The d50 (and corresponding SA) for the various fraction was determined as 14.13 ± 1.15 µm (2.29 ± 0.12 m2 g−1), 3.28 ± 0.29 µm (3.79 ± 0.05 m2 g−1), 5.29 ± 0.10 µm (3.07 ± 0.03 m2 g−1), 4.77 ± 0.21 µm (3.23 ± 0.31 m2 g−1), and 3.09 ± 0.55 µm (3.89 ± 0.07 m2 g−1) for the 2-min (DC), 5-min (PC5), 10-min (PC10), 30-min (PC30) and 60-min (FA) milled samples, respectively.
Physical and Chemical Transformation under Accelerated Storage Conditions
Physical Appearance
The physical appearance of the solid compacts after 6 h exposure to 40°C/75% RH was distinctive. The coloration (off-white to browning of the tablet) was noted which could have resulted from oxidation of VOR and from the disproportionation of HBr acid from the salt (Fig. 4). An intense color change was evident for the samples with a lower crystallinity (higher amorphous content). The enriched color shades reflect the chemical change which was further confirmed with chromatographic and mass spectroscopic analyses. Among all, for the defective crystalline state (originates from shortest milling), a sticky and slight shrinking mass was noted. This could have resulted from the high hygroscopicity of the PVP in addition to the enhanced surface area.

Physical appearance for representative solid compacts of VOR-HBr and PHP exposed to 40°C/75% RH; a 0 min milled, b 2 min milled, c 10 min milled, and d 60 min milled. The upper panel demonstrates unexposed samples at time zero (T0), while the lower panel is for exposed samples withdrawn after 6 h
Physical Transformation Evaluated Using Raman Spectroscopy
In order to confirm the solid-state property over the storage, the solid compacts were immediately subjected to the Raman analysis. The benefit of using a PhAT probe was the wide coverage of the sample area, good spectral resolution, and reduced variations associated with sub-sampling. In addition, the same sample was subsequently used for the chemical analysis via UPLC. For the given solid compacts containing the crystalline to amorphous range of disorders, the distinct spectral region was identified (Fig. 5).

Raman spectra of solid compacts of VOR-HBr with PHP exposed to 40°C/75% RH recorded after different time points (h) of exposure: a 0 min milled, b 2 min milled, c 10 min milled, and d 60 min milled
The ratio of the peak intensity of the Raman band at 1580 cm−1 to that at 1605 cm−1 in the initial powder compact was found to systematically decrease with the decrease in the amorphous content. This Raman band ratio in the stability samples overall remained unaltered over the exposure period (seen in supplement Table S2). Moreover, the lack of interference of the PHP signal in this spectral region was confirmed by comparing that with the pure PHP spectrum (data not shown). In general, as seen in Fig. 6, the relative trend of crystallinity in VOR-HBr powder compacts with PHP remained largely unchanged during the exposure period. Our objective of this ratiometric representation of the Raman spectral data is to provide an indication of the change in the relative crystallinity or not. At this moment, we are unable to present the exact spectral assignment of the selected peak, even though they are more likely attributable to the Raman polarizability of double bond systems. Even so, we found a systematic response of the ratio of these two peaks that was useful to provide insight into the relative crystallinity of the sample. The signal ratio was selective, yet we admit that the values are not fully quantitative as we did not develop any calibration curve to quantify crystallinity using this method nor we verify this to other methods.

Plots of the peak intensity ratio of Raman bands for solid compacts of VOR-HBr and PHP exposed to 40°C/75% RH for 6 h; 0 min milled (light blue square), 2 min milled (purple diamond), 10 min milled (red triangle) and 60 min milled (black circle). Vertical bars in each point represent s.d. among triplicate measurements
Degradation Kinetics in the Solid State
As illustrated in our previous study [18], by using the solution state peroxide stress, a specific and precise stability-indicating UPLC method was developed for the separation of the drug from its oxidative degradation products. The same chromatographic method was employed in this study for the assessment of stability samples. The separation of the drug peak from its degradation products is evident (as shown in supplement Figure S5). The calculated relative retention time (RRT) and observed UV λmax for DP-I were 0.68 and 240 nm. For DP-II, these values were 1.08 and 234 nm, respectively. The UV spectral maxima show a bathochromic shift in λmax by 13 nm for DP-I and 7 nm for DP-II relative to the parent drug (227 nm). Unlike the solid samples, the liquid stress degradation study was found to form only DP-I.
The relative percentage of degradation was reported in the area percentage of the drug peak with respect to that of the DPs. The compacts containing varying amounts of disorder showed a faster VOR-HBr degradation as compared to the compacts with pure crystalline powder. For the studied powder solid compacts, both DPs formed to a different extent. DP-I was assigned as a major degradation product and the extent of degradation ranged from 2 to 20% among the samples. DP-II was assigned as a minor DP, and the extent was overall less than 2% among the stored samples. The formation of DP-I in the crystalline (FC-PHP) and fully amorphous (FA-PHP) samples was within the 5%, while for the defective crystalline (DC-PHP), partially crystalline (PC-PHP), and two-phase disorder (PM-PHP) samples, the amount exceeded 5% level. However, DP-II in FC-PHP and DC-PHP was formed below 0.5% and slightly able 0.5% in PC-PHP, FA-PHP, and PM-PHP samples. The exact amounts formed at each stability time point are presented in supplement Table S3. For the formation of DP-I, the order of degradation upon exposure was fully crystalline (FC-PHP) < fully amorphous (FA-PHP) < defective crystalline (DC-PHP) < partially crystalline (PC-PHP), while for DP-II formed upon exposure increased systematically with the initial amorphous content, crystalline (PC-PHP) < defective crystalline (DC-PHP) < partially crystalline (PC-PHP) < fully amorphous (FA-PHP). The two-phase disordered solid compact (PM) (with equivalent crystallinity) differs from respective the single-phase with comparable crystallinity.
Using the degradation data of each time point (0–6 h), the kinetics of each DP was plotted and compared as depicted in Fig. 7. The resulting experimental degradation kinetics were fitted by modified Avrami’s model [45], as expressed in Eq. 2.
where k is the reaction rate constant (h−1), t is the exposure time (h), and n is the reaction order. In all the cases, the value of n obtained was below 0.5, suggesting the diffusion-controlled degradation reaction. The diffusion of peroxide to the disordered and reactive solid surface can be considered as the rate-determining step in this case.

Degradation kinetic profile for a DP-I (major DP) and b DP-II (minor DP); unmilled (black), 2 min milled (blue), 10 min milled (red), 60 min milled (green), and physically mixed two-phase system theoretically equivalent to 10 min milled (purple and dotted fit). The data points were fitted using a modified Avrami model [45]. The vertical error bars in each point represent the standard deviation of three independent measurements, and the lines represent the best fit to the presented data points
The extracted k for the disordered samples and the goodness of fit (R2) data are listed in supplement Table S4. As evident from Fig. 7, the degradation followed non-linear kinetics to form the major (DP-I) and the minor (DP-II) degradation product. The reaction rates were found to generally increase with the increase in the milling time; hence, the degradation depends on the levels of disorder. However, the rate of formation of DP-I is lower for the fully amorphous sample as compared to the partially crystalline counterparts. The latter perhaps suggests that the rate-limiting step of the peroxidation in fully amorphous systems might differ from that in the one-phase system.
For the physical mixture (PM-PHP), i.e., a two-phase system comprising of equivalent crystallinity to a 10-min milled sample, the degradation (extent and profile) was not similar to the single-phase system suggesting the reactive role of defects present within the crystal lattice. This result indicates an important aspect of the relation between disorder type and degradation. Thus, the same level of disorder present as a continuum in a crystalline particle versus that present as a separate phase in the powder mixture can result in a differing reactivity.
Furthermore, using the observed MS data and literature information, the chemical structures were postulated for both DPs. The molecular ion peaks [M + H]+ in the ESI positive mode appeared at m/z 299.1601, 315.1544, and 377.0682 corresponding to the drug, DP-I, and DP-II, respectively. The presence of N–H and O–H functionalities in the drug structure allows the use of ESI positive mode leading to better ionization efficiency during MS analysis. The MS line spectrum for DP-I showed an addition of 16 Da unit mass (equivalent to an oxygen atom) and for DP-II by 78 Da. Interestingly, for DP-II, the presence of isotopic abundance pattern in MS spectra was clearly attributing to bromination, as evident by its characteristic isotopic pattern (1:1) of 50.50% and 49.50% corresponding to 79Br (M + 1) and 80Br (M + 2), respectively. The EIC corresponding to the m/z of drug and DPs and TIC plots along with line spectra are provided in supplement Figure S6.
For DP-I, the most probable structures derived and correlated to the reported one was 3-methyl-4([2-(piperazin–1-yl)phenyl]sulfanyl)phenyl)methanol [42]. However, the other structural possibility of DP-I carrying similar m/z also cannot be ruled out [43]. The obtained MS data and isotopic abundance enabled to propose the structure for DP-II to be a known impurity of vortioxetine, 1-(2[(3-bromo-2,4dimethylphenyl)-sulfanyl] phenyl)piperazine (Fig. 8) [46]. These results indicate that the oxidation of the drug in the solid state occurred mainly at the methyl group attached to the phenyl ring (para position to the sulfur atom) as was reported in our prior study [18]. In addition, a minor product was formed by bromination occurring at the carbon in the phenyl ring (meta position to the sulfur atom). The FTIR spectral data also reveals the disproportionation tendency of the salt form (liberating bromine) upon milling. The peroxide/free radical catalyzed anti-Markovnikov bromination reaction is a widely reported mechanism. The favorable conditions including elevated temperature, accessible peroxide species, and bromine from the salt disproportionation seem to be contributing to this chemical reaction [46, 47].

Chemical structures of a drug (VOR-HBr), b DP-I (major DP), and c DP-II (minor DP)
Estimation of Peroxide Content
In order to quantify the amount of peroxide in the samples, the Amplex Red Hydrogen Peroxide/Peroxidase Assay method was implemented. As highlighted in Figure S7 (supplementary), the obtained amount of 3.99 ± 0.30 mmol H2O2 per g (average ± s.d., n = 3) in PHP correlates with the theoretical amount. Interestingly, over the entire exposure period (6 h), the level of peroxide remains constant and matches the value at T0 for pure PHP. While for the drug and PHP complex, the peroxide levels were found to decrease with the time (compared to its starting value). This observation indicates the consumption of peroxide by the API oxidation.
Discussion
VOR-HBr is known to be susceptible to exclusive peroxide-mediated degradation. The cryo-milling was chosen for amorphization via mechano-activation, thereby generating the samples with controlled disorder content. The PHP stressor was utilized to perform oxidative reactions in solid compact form. Furthermore, the exposure to 40 °C/75% RH yields the kinetic profiles of the physical (recrystallization) and chemical (degradation) transformation for the selected disorder states, when exposed in the form of solid compacts (drug + PHP).
Generation of Crystal Disorder
As reported, the crystal structure of VOR-HBr contains one molecule of each ion in the asymmetric unit, specifically, NH2+ and Br− ions are bonded through ionic interactions [41]. The two phenyl rings bridged by the S-atom are perpendicular to each other, while the piperazine ring adopts a chair conformation. Upon varying milling times, a sufficient solid-state disorder was generated. The shortest time-point milled solid (2 min) led to a defective crystal (with no sign of amorphous fraction with Tg in DSC). Milling for 5–30 min resulted in a partial crystallinity, and the longest time-point milling generated a fully amorphous phase. The DSC profiles confirmed that milling was able to progressively amorphize the drug. The exponential relationship of milling-induced amorphization kinetics of dexamethasone was described previously by Oliveira et al. [48] wherein an empirical model was fitted to the experimental profile to obtain average relaxation time. In VOR-HBr case, the model did not converge to fit the experimental profile. This is presumably due to the sudden change in the structural relaxation dynamics from the glassy to super-cooled liquid state as the temperature of the milled solid exceeds the Tg of amorphous phase. Another important feature was the concomitant salt disproportionation observed with disordering during milling. As notable in Fig. 2b, the Tg values of earlier milling time points were notably higher than that of later time points. The Tg drop over the increase of the milling period reflects the contribution of the amorphous state of free base version of vortioxetine. The first two milling times led to trivial salt disproportionation, while last two milling time led to prominent salt form conversion to free base as noted clearly in infrared red spectra. While we did not measure the Tg of free base form of drug due to the limited availability of the same, we had measured and thus reported its melting point which is 100 °C lower than salt form (Fig. S1). Therefore, we expect free base would have lower Tg than the salt form contributing to the lower overall Tg of highly milled samples. Moreover, the solids generated upon milling were not degraded and physically stable (no polymorphic or solid-state form changes were observed). For the fully amorphous form, the nucleation induction time was sufficiently longer (> 30 days). The recrystallized form was similar to the original polymorphic form of VOR-HBr (matching Tc and Tm).
Disordering during milling followed an interesting manifestation on the PSD behavior. As can be noted, there is an abrupt reduction (from 370 to 14 µm) in particle size just by 2 min intensive milling, which is an indication of a sharp mechano-active brittle fracture of the crystals. From 2 to 5 min, the particles go from 14 to 3 µm, less drastic, yet brittle deformation seems to still occur. Interestingly, from 5 min onward, the overall range of d50 values was between 3 and 5 µm. Although the replicate measurements are reproducible (low RSD), these values indicate that from 5 min onward, particles exhibit plastic deformation to mechanical response as they turn increasingly amorphous and get smaller. A slight increase in PSD between 5 and 30 min milling could be due to the agglomeration of high energy particles that again tend to slightly de-agglomerate upon longer milling (30 min). To duly factor the effect of these particle changes and thus account only to amorphization, we thus expressed degradation as the rate constant per unit surface area of the powder (derived from the measured PSD).
The FTIR spectral signatures revealed the salt disproportionation of VOR-HBr. In the literature, ample cases of salt disproportionation have been reported. However, to the best of our knowledge, this is the first time mechano-activated disproportionation is shown. High volatility of the HBr can enhance salt disproportionation. Comparison of the FTIR spectral signatures with that of the free base form enabled confirming this phenomenon. The LC–MS analysis of the stability samples of disordered forms supports the formation of brominated DP. The elucidated structure matches the known brominated impurity of the drug.
The well-characterized PHP solid prudently allowed to study the degradation behavior of the selected disorder systems. For DP-I, the defective crystalline state was more reactive as compared to other forms. For DP-II, the amorphization order follows the chemical reactivity. For the 2-min milled sample, we believe that surface disorder would have higher interfacial energy enabling higher reactivity over the amorphous forms. Interestingly, the physical appearance of solid compacts upon exposure to high humidity suggested the formation of a sticky, shrink mass (could be due to higher surface moisture absorption). As was reported earlier [18], the measured microenvironment pH of the oxidative reagent (PHP complex) was neutral (around pH 6.6). Also, the crystallinity level of the stability samples of VOR-PHP remained nearly the same as the initial values. In our previous study, the higher oxidative reactivity of a partially crystalline state (having the highest nanoscale powder heterogeneity) over its crystalline and fully amorphous counterpart was demonstrated for a simvastatin [32].
Relationship of Drug Crystallinity Versus Degradation Potential
A visualization of the oxidation degradation rates in the powder compacts as a function of the physical transformation is shown in Fig. 9. A peculiar relation among the degree of disorder to the types of degradation is notable. The rate of DP-I formation increased with the increase in the disorder up to 75% disorder (10-min milled sample) and abruptly decreased in the case of fully amorphous sample (60 min). Whereas, in the case of DP-II formation kinetics, a monotonic increase in the DP is observed with the decrease in the crystallinity.

The plots of the rate constants of formation of DP I (KDP-1) and DP-II (KDP-2) in the solid compacts of VOR-HBr and PHP exposed to 40 °C/75% RH for 6 h versus the degree of crystallinity of the samples obtained by DSC. Vertical bars in each point represent S.D. among replicate measurements (n = 3)
DP-I was found to form both in the peroxide solution and in the solid state. In contrast, DP-II formed only in the solid state. If we hypothesize that DP-I is formed mainly in a liquid layer of the solid compacts (via dissolved drug and peroxide fraction in the surface water layer), it would be sensible that with the increase in amorphous content to a certain level (up to 10 min milling), the rate of dissolution increases, thereby the availability of reacting drug in liquid is higher. However, this does not directly explain the drop in DP-I formation for a completely amorphous sample (60 min milled). In this case, it can be expected that due to the highest amorphicity and smaller particle size, a drastic increase in particle growth and agglomeration could take place. This can lead to a decrease in the available surface for dissolution. Since DP-II was only formed in the solid state, this reaction appears to commence on a viscous solid surface with a few molecular layers of water present. A higher surface energy of an amorphous state could favor the reaction, despite an intense agglomeration at the mesoscopic scale. Also, DP-II formation will depend on the rate of HBr formation from the parent compound. DP-I results from direct peroxidation, while DP-II via bromination with peroxide plays a catalytic role. Since this anomalous relation of the relative crystallinity to the two different types of degradation reaction of the same molecule is firstly observed, our hypothesis further needs a carefully designed experiment to (dis)prove them.
Similarly, the reduction in peroxide content is due to the consumption of peroxide species. As illustrated in Fig. 10, the consumption of peroxide and reduction in drug content (based on the chemical degradation) show a good linear relationship (R2 = 0.99). This observation clearly links and confirms the oxidative susceptibility of drug in the presence of reactive oxidative species.

Plot showing the relationship between the amounts of hydrogen peroxide consumed versus the decrease in drug content in the solid compacts. Bars represent standard deviation among replicate measurements (n = 3)
Another important factor that can contribute to this anomalous relation of initial crystallinity to the degradation rate would be the moisture sorption behavior of the samples. While one would expect that the higher the amorphicity, the higher the moisture sorption propensity, we have found in our earlier study on the disordered simvastatin solid that moisture sorption extent can have a complex relation to the crystallinity [32]. Therefore, future attempt to clarify this unique degradation profile of disordered solid needs to duly report and interpret the role of moisture content.
Conclusion
Through this study, we report a differential oxidative reactivity of the vortioxetine hydrobromide solid state containing varying degrees of disorders. The formation kinetics of a minor brominated product catalyzed by peroxide was found to increase with the amorphous content of the sample. In contrast, the major oxidative degradation product formed via direct peroxidation was found to have a lower rate of formation in a fully amorphous sample than the partially crystalline sample. This highlights the importance of monitoring both degradation kinetics as well as degradation pathway and product types as a function of the solid state of the reactant. The thermal and spectral (infrared, Raman, and mass) signatures helped in interpreting the transformations. For an equivalent crystallinity level, the outcome for the oxidative chemical reactivity was different between the milled single-phase and the physically mixed two-phase disordered system. Also, mechano-activated salt disproportionation was evidenced. This led to the involvement of bromine counter-ion in drug degradation reaction catalyzed by peroxide. A wider case study involving diverse salt forms of drugs is needed to further explore these findings and to build a quantitative relationship between disordered–disproportionated–degraded forms of the drug. In general, the traditional accelerated stability designs do not account for or measure the implications of solid-state disorder, salt disproportionation, etc. on the chemical degradation of drugs. Hence, the outcome of this study suggests the criticality of solid-state properties such as molecular mobility, Tg, and crystallization rate constant to better predict the chemical stability of pharmaceuticals.
References
Newman A, Wenslow R. Solid form changes during drug development: good, bad, and ugly case studies. AAPS Open. 2016;2. https://doi.org/10.1186/s41120-016-0003-4.
Ng LH, Ling JKU, Hadinoto K. Formulation strategies to improve the stability and handling of oral solid dosage forms of highly hygroscopic pharmaceuticals and nutraceuticals. Pharmaceutics. 2022;14:2015.
Shi Q, Moinuddin SM, Cai T. Advances in coamorphous drug delivery systems. Acta Pharm Sin B. 2019;9:19–35. https://doi.org/10.1016/j.apsb.2018.08.002.
Paudel A, Meeus J, Mooter G Van den. Structural characterization of amorphous solid dispersions. Amorph Solid Dispersions. Springer, New York, NY; 2014. p. 421–85.
Schammé B, Monnier X, Couvrat N, Delbreilh L, Dupray V, Dargent É, et al. Insights on the physical state reached by an active pharmaceutical ingredient upon high-energy milling. J Phys Chem B. 2017;121:5142–50. https://doi.org/10.1021/acs.jpcb.7b02247.
Mehta M, Ragoonanan V, McKenna GB, Suryanarayanan R. Correlation between molecular mobility and physical stability in pharmaceutical glasses. Mol Pharm. 2016;13:1267–77. https://doi.org/10.1021/acs.molpharmaceut.5b00853.
Newman A, Zografi G. What are the important factors that influence API crystallization in miscible amorphous API–excipient mixtures during long-term storage in the glassy state? Mol Pharm. 2022;19:378–91. https://doi.org/10.1021/acs.molpharmaceut.1c00519.
Newman A, Zografi G. [ASAP] What we need to know about solid-state isothermal crystallization of organic molecules from the amorphous state below the glass transition temperature. Mol Pharm. Available from: http://feedproxy.google.com/~r/acs/mpohbp/~3/EdaGsuzSQOM/acs.molpharmaceut.0c00181?utm_source=researcher_app&utm_medium=referral&utm_campaign=RESR_MRKT_Researcher_inbound.
Qiu F, Scrivens G, editors. Accelerated predictive stability (APS): fundamentals and pharmaceutical industry practices. London: Elsevier; 2018.
Singh S, Junwal M, Modhe G, Tiwari H, Kurmi M, Parashar N, et al. Forced degradation studies to assess the stability of drugs and products. TrAC Trends Anal Chem. 2013;49:71–88 (Available from: http://www.sciencedirect.com/science/article/pii/S0165993613001313).
Hotha KK, Roychowdhury S, Subramanian V. Drug-excipient interactions: case studies and overview of drug degradation pathways. Am J Anal Chem. 2016;07:107–40. https://doi.org/10.4236/ajac.2016.71011.
Williams HE, Bright J, Roddy E, Poulton A, Cosgrove SD, Turner F, et al. A comparison of drug substance predicted chemical stability with ICH compliant stability studies. Drug Dev Ind Pharm. 2019;45:379–86. https://doi.org/10.1080/03639045.2018.1542707.
Gabrič A, Hodnik Ž, Pajk S. Oxidation of drugs during drug product development: problems and solutions. Pharmaceutics. 2022;14:325 (Available from: https://www.mdpi.com/1999-4923/14/2/325).
Hovorka SW, Schöneich C. Oxidative degradation of pharmaceuticals: theory, mechanisms and inhibition. J Pharm Sci. 2001;90:253–69 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022354916307286).
Kawaguchi-Murakami Y, Fukutsu N, Kajiro T, Araki T, Murakami T, Kawasaki T, et al. A prediction system of oxidation reaction as a solid-state stress condition: applied to a pyrrole-containing pharmaceutical compound. J Pharm Biomed Anal. 2009;50:328–35 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0731708509002714).
Ueyama E, Tamura K, Mizukawa K, Kano K. Realistic prediction of solid pharmaceutical oxidation products by using a novel forced oxidation system. J Pharm Sci. 2014;103:1184–93 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022354915306365).
Zhu DA, Zhang GGZ, George KLST, Zhou D. A novel accelerated oxidative stability screening method for pharmaceutical solids. J Pharm Sci. 2011;100:3529–38 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022354915320256).
Modhave D, Barrios B, Paudel A. PVP-H2O2 complex as a new stressor for the accelerated oxidation study of pharmaceutical solids. Pharmaceutics. 2019;11:457 (Available from: https://www.mdpi.com/1999-4923/11/9/457).
Iyer J, Karn A, Brunsteiner M, Ray A, Davis A, Saraf I, et al. Screening autoxidation propensities of drugs in the solid-state using PVP and in the solution state using N-Methyl pyrrolidone. Pharmaceutics. 2023;15:848 (Available from: https://www.mdpi.com/1999-4923/15/3/848).
Saraf I, Modhave D, Kushwah V, Neshchadin D, Gescheidt G, Trausinger G, et al. Feasibility of rapidly assessing reactive impurities mediated excipient incompatibility using a new method: a case study of famotidine-PEG system. J Pharm Biomed Anal. 2020;178: 112893. https://doi.org/10.1016/j.jpba.2019.112893.
Iyer J, Brunsteiner M, Ray A, Davis A, Saraf I, Paudel A. Theoretical and experimental investigation of autoxidation propensity of selected drugs in solution state. Mol Pharm. 2023;20:1768–78. https://doi.org/10.1021/acs.molpharmaceut.2c00967.
Byrn SR, Sally T, Alice M. Solid-state chemistry of drugs. 1st ed. New York: Academic Press, INC.; 1982.
Iyer J, Brunsteiner M, Modhave D, Paudel A. Role of crystal disorder and mechanoactivation in solid-state stability of pharmaceuticals. J Pharm Sci. 2023; Available from: https://www.sciencedirect.com/science/article/pii/S0022354923000709.
Loh ZH, Samanta AK, SiaHeng PW. Overview of milling techniques for improving the solubility of poorly water-soluble drugs. Asian J Pharm Sci. 2014;10:255–74. https://doi.org/10.1016/j.ajps.2014.12.006.
Nirmal Kumar Katiyar KB, Tiwary CS. Cryomilling as environmentally friendly synthesis route to prepare nanomaterials. Int Mater Rev. 2021;66:493–532. https://doi.org/10.1080/09506608.2020.1825175.
Stankovic-Brandl M, Zellnitz S, Wirnsberger P, Kobler M, Paudel A. The influence of relative humidity and storage conditions on the physico-chemical properties of inhalation grade fine lactose. AAPS PharmSciTech. 2021;23:1. https://doi.org/10.1208/s12249-021-02159-8.
Vasa DM, Wildfong PLD. Solid-state transformations of ribavirin as a result of high-shear mechanical processing. Int J Pharm. 2017;524:339–50. https://doi.org/10.1016/j.ijpharm.2017.04.002.
Einfalt T, Planinšek O, Hrovat K. Methods of amorphization and investigation of the amorphous state. Acta Pharm. 2013;63:305–34. https://doi.org/10.2478/acph-2013-0026.
Feng T, Pinal R, Carvajal MT. Process induced disorder in crystalline materials:differentiating defective crystals from the amorphous form of griseofulvin. J Pharm Sci. 2008;97:3207–21 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0022354916326454).
Qiu Z, Stowell JG, Morris KR, Byrn SR, Pinal R. Kinetic study of the Maillard reaction between metoclopramide hydrochloride and lactose. Int J Pharm. 2005;303:20–30 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0378517305004448).
Šagud I, Zanolla D, Perissutti B, Passerini N, Škorić I. Identification of degradation products of praziquantel during the mechanochemical activation. J Pharm Biomed Anal. 2018;159:291–5 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0731708518313839).
Modhave D, Laggner P, Brunsteiner M, Paudel A. Solid-state reactivity of mechano-activated simvastatin: atypical relation to powder crystallinity. J Pharm Sci. 2019;108:3272–80. https://doi.org/10.1016/j.xphs.2019.05.032.
Modhave D, Saraf I, Karn A, Paudel A. Understanding concomitant physical and chemical transformations of simvastatin during dry ball milling. AAPS PharmSciTech. 2020;21:1–10. https://doi.org/10.1208/s12249-020-01687-z.
Iyer J, Morgan LM, Harrison P, Davis A, Ray A, Mitsche S, et al. Applying material science principles to chemical stability: modelling solid state autoxidation in mifepristone containing different degrees of crystal disorder. J Pharm Sci. 2023;000:1–20. https://doi.org/10.1016/j.xphs.2023.03.020.
Iyer J, Barbosa M, Saraf I, Pinto JF, Paudel A. Mechanoactivation as a tool to assess the autoxidation propensity of amorphous drugs. Mol Pharm. 2023;20:1112–28. https://doi.org/10.1021/acs.molpharmaceut.2c00841.
Thakral NK, Kelly RC. Salt disproportionation: a material science perspective. Int J Pharm. 2017;520:228–40. https://doi.org/10.1016/j.ijpharm.2017.02.001.
Christensen NPA, Rantanen J, Cornett C, Taylor LS. Disproportionation of the calcium salt of atorvastatin in the presence of acidic excipients. Eur J Pharm Biopharm. 2012;82:410–6. https://doi.org/10.1016/j.ejpb.2012.07.003.
Hsieh Y-L, Merritt JM, Yu W, Taylor LS. Salt stability – the effect of pHmax on salt to free base conversion. Pharm Res. 2015;32:3110–8. https://doi.org/10.1007/s11095-015-1691-5.
Guerrieri P, Taylor LS. Role of salt and excipient properties on disproportionation in the solid-state. Pharm Res. 2009;26:2015–26.
Vortioxetine hydrobromide [Internet]. [cited 2023 Feb 15]. Available from: https://www.axios-research.com/products/vortioxetine-hbr.
Zhou X, Hu X, Wu S, Ye J, Sun M, Gu J, et al. Structures and physicochemical properties of vortioxetine salts. Acta Crystallogr Sect B Struct Sci Cryst Eng Mater. 2016;72:723–32 (Available from: https://scripts.iucr.org/cgi-bin/paper?S2052520616010556).
de Diego M, Correa D, Mennickent S, Godoy R, Vergara C. Determination of vortioxetine and its degradation product in bulk and tablets, by LC-DAD and MS/MS methods. Biomed Chromatogr. 2018;32:1–6.
Ragi NC, Velma GR, Pallerla PK, Siddiqua S, Alugonda V, Rachamalla HKR, et al. Identification and characterization of forced degradation products of vortioxetine by LC/MS/MS and NMR. J Pharm Biomed Anal. 2020;188:113442 (Available from: https://www.sciencedirect.com/science/article/pii/S0731708520313285).
Grisedale LC, Jamieson MJ, Belton PS, Barker SA, Craig DQM. Characterization and quantification of amorphous material in milled and spray-dried Salbutamol Sulfate: a comparison of thermal, spectroscopic, and water vapor sorption approaches. J Pharm Sci. 2011;100:3114–29.
Yang J, Grey K, Doney J. An improved kinetics approach to describe the physical stability of amorphous solid dispersions. Int J Pharm. 2010;384:24–31 (Available from: https://linkinghub.elsevier.com/retrieve/pii/S0378517309006966).
Amati A, Dosualdo G, Zhao L, Bravo A, Fontana F, Minisci F, et al. Catalytic processes of oxidation by hydrogen peroxide in the presence of Br 2 or HBr. Mechanism and synthetic applications. Org Process Res Dev. 1998;2:261–9.
Radical additions: anti-Markovnikov product formation [Internet]. [cited 2023 Feb 12]. Available from: https://chem.libretexts.org/Bookshelves/Organic_Chemistry/Supplemental_Modules_(Organic_Chemistry)/Alkenes/Reactivity_of_Alkenes/Free_Radical_Reactions_of_Alkenes/Radical_Additions%3A_Anti-Markovnikov_Product_Formation.
Oliveira PFM, Willart J-F, Siepmann J, Siepmann F, Descamps M. Using milling to explore physical states: the amorphous and polymorphic forms of dexamethasone. Cryst Growth Des. 2018;18:1748–57. https://doi.org/10.1021/acs.cgd.7b01664.
Funding
Open access funding provided by Graz University of Technology. This work was funded through the Austrian COMET–Competence Centres for Excellent Technologies program. The COMET program is operated by the Austrian Research Promotion Agency (FFG) on behalf of the Federal Ministry for Transport, Innovation, and Technology (BMVIT) and the Federal Ministry for Digital and Economic Affairs (BMDW). Our projects are also funded by Land Steiermark and the Styrian Business Development Agency (SFG). Open access funding provided by Graz University of Technology Open Access Publishing Fund.
Author information
Authors and Affiliations
Contributions
DM: conceptualization, investigation, resources, supervision, visualization, writing–original draft, writing–review and editing. BB: conceptualization, methodology, investigation, writing–original draft, writing–review and editing. JI: data curation, investigation, writing–review and editing. AP: conceptualization, funding acquisition, project administration, resources, writing–review and editing.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Modhave, D., Barrios, B., Iyer, J. et al. Influence of Crystal Disorder on the Forced Oxidative Degradation of Vortioxetine HBr. AAPS PharmSciTech 25, 10 (2024). https://doi.org/10.1208/s12249-023-02721-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-023-02721-6