
Abstract
The manufacturing of biopharmaceutical drug solutions can involve close contact with various polymeric components, including common filter membranes. Potential leachable substances from filters may interact with the protein and thereby increase the structural damage and aggregation. The main aim of the study deals with the assessment of extractable and leachable (E/L) from different filters and the potential effect of E/Ls on protein (human granulocyte-colony stimulating factor (rh-GCSF) stability. The present study examines the E/L profile of five different polymeric filter membranes using various chromatographic techniques including LC–MS and GC–MS. In order to investigate their effect on protein stability, G-CSF (human granulocyte colony-stimulating factor) formulations were spiked with filter leachable stock solutions at two different pH levels. The spiked formulations were further analyzed with respect to their aggregation behavior. The results demonstrated a higher E/L content in the case of polyamide (PA), polycarbonate (PC), and polyethersulfone (PES) filters as compared to the polytetrafluoroethylene (PTFE) and regenerative cellulose (RC) filter materials. The E/L from RC and PES was found surface-active, whereas E/L from PA and RC significantly altered the particle size/structure resulting in the aggregation of proteins. Furthermore, bisphenol A was found to be one of the E/L substances from PC filters and can impose significant health problems when administered along with pharmaceutical products. The present study reports a qualitative rank ordering of the filter membranes in terms of their propensity to generate E/Ls and thus can be helpful in selecting a suitable membrane filter.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sterilization is an essential step for the development of injectable protein formulations to remove microorganisms. Owing to heat labile property of the proteins, such products are commonly sterilized via sterile filtration, also called “cold” sterilization [1,2,3,4]. Preferably, the filter membranes used for this purpose need to be inert so that no release of membrane material into pharmaceutical products can take place [4, 5]. However, there are reports of filter membranes used in sterilization releasing extractables and leachables into the final products, which can interact and thus alter the protein conformation and structure [1, 6].
Extractables are defined as organic and inorganic chemical compounds that can be released from a contact material under laboratory conditions (accelerate or exaggerate the normal conditions of storage) such as solvent, temperature, and stoichiometry. Extractables have the potential to leach into a drug product under normal conditions of storage. Whereas, leachables are organic and inorganic chemical compounds that migrate from the contact material (primary and secondary packaging) into the product solution under conditions of clinical use, recommended storage, and during accelerated drug product stability studies [6,7,8,9]. For better processability and stability, additives are added to the filter polymers. These additives are generally non-covalently attached with the polymer, and thus are readily available to release into the contacting solutions. The leaching propensity of the filter membrane in contact with a formulation could possibly increase when it contains solubilizing agents, like surfactants, organic solvents, etc. Even though, during sterile filtration, the filtration area is often very small, the contact surface that actually interacts with the protein compound and the contact time are markedly higher owing to porous structure of the filter. In addition, auxiliary parts of the filter, like O-rings, filter housings, or other components (such as wetting agent, lubricants, plasticizers, and antioxidants) can also act as a source of extractables and leachables (E/L). E/L can cause serious health risk for the patient either due to their own toxicity or via affecting the potency, efficacy, and stability of the pharmaceutical product [7, 8]. The health hazards includes dermal sensitization, genetic toxicity, carcinogenicity, reproductive toxicity, and developmental toxicity. Furthermore, the E/L can change the absorption, distribution, metabolism, and excretion (ADME) of the biological active moiety, resulting in altered pharmacokinetics [10,11,12]. Thus, it is vital to perform the qualitative and/or quantitative analysis of the potential E/L from the filters.
In the present study, five different types of filter membranes (PA, polyamide; PC, polycarbonate; PES, polyethersulfone; PTFE, polytetrafluoroethylene; RC, regenerative cellulose) were screened for their E/Ls profile and their effect on protein stability. PA filters are naturally hydrophilic and are chemically resistant to organic solvents and alkaline solutions [13, 14]. The application of PA is limited due to high non-specific adsorption resulting in lower filtration yield of small volume samples [15]. Whereas, PC filter membranes possess improved thermal stability, lower adsorption, and are highly resistant towards chemicals, thus they can be used for a broad range of samples [16,17,18]. PES filter membranes have a high filterable volume thus allow the use of high flow speeds [19]. PES is hydrophilic in nature and is acid and base resistant [20]. PTFE (known as Teflon) consists of repeated units of tetrafluoroethene monomers [21]. PTFE filter membranes are hydrophobic in nature and mainly used for air/gas-filtration [22]. They have an excellent chemical resistance that allowed the use for filtration of aggressive bases, acids, and solvents that are incompatible with other filter membranes [23]. RC filter membranes are hydrophilic and highly resistant towards aqueous and organic solvents. RC are primarily used for filtration of biological solutions owing to their low non-specific protein adsorption [24,25,26].
The effects of E/L from the filters were evaluated on the model protein human granulocyte-colony stimulating factor (G-CSF). The G-CSF is a therapeutically relevant glycoprotein that belongs to the four-α-helix-bundle class of cytokines, having molecular weight of 19.6 kDa [27]. G-CSF protein is mainly produced by macrophages and is involved in the proliferation of neutrophils, differentiation of precursor cells for neutrophil production, and stimulates the activity of full-fledged neutrophil granulocytes. Because of its diverse functions, it has various fields of application [28,29,30]. The prevention of protein aggregation, denaturation, and unfolding, respectively, owing to heat, mechanical stress, radiation, strong acids or bases, detergents, and organic solvents is a major challenge [31, 32] in pharmaceutical engineering. Protein aggregates are reported to cause adverse immunogenic responses or lead to plugging during parenteral delivery, and thus can be harmful for the patients.
Thus, the present study deals with the E/L screening from filter materials and to evaluate the effect of E/L on protein stability. Initially, the extractables were extracted from filters using solvents, followed with assessment of leachables from the filters and their impact on protein aggregation. The E/L materials were extracted and analyzed using various chromatographic methods. Thereafter, the extracts were spiked with the protein formulation and evaluated for any change in protein structure. Finally, the protein aggregation and/or denaturation due to E/L were evaluated by using size exclusion chromatography, electrophoretic light scattering, flow microscopy, differential scanning calorimetry, and Raman spectroscopy. In terms of the stability of the product, the results demonstrated PTFE and RC as the best and the worst case scenario, respectively, among all tested filter membranes (PA, PC, PES, PTFE, and RC).
Material and Methods
Materials
PA, polyamide; PC, polycarbonate; PES, polyethersulfone; PTFE, polytetrafluoroethylene; and RC, regenerative cellulose filter membranes with a diameter of 47 mm and a pore size of 0.2 µm were kindly provided by Sartorius Stedim Biotech GmbH (Göttingen, Germany). Human granulocyte colony-stimulating factor (G-CSF) from Sandoz (Kundl, Austria) was utilized as the model protein drug. All other chemicals were of analytical reagent grade.
Preparation of the Extractable Solutions for the Controlled Extraction Study
For the controlled extraction study, solvents of different polarities, namely ethanol (gradient grade for LC, Merck Millipore, Billerica, MA, USA), hexane (Rotisolv HPLC, Carl Roth GmbH + Co. KG, Karlsruhe, Germany), and purified water (TKA MicroPure system, TKA GmbH, Niederelbert, Germany) were used. The filter membranes were treated with 10 mL of extraction solvent at a temperature slightly below the solvent’s boiling point (Table I). The ethanol and hexane extracts were agitated in an orbital incubator shaker operated at 100 rpm for 48 h. Whereas, the water extractable stocks were kept in a compartment dryer at 90°C. Because of technical limitations (90°C with shaker), the water extracts were not agitated (Table I).
Preparation of the Leachable Stock for the Protein Stability Study
The leachable stocks for the protein stability study were prepared by extraction of PA, PC, PES, PTFE, and RC filters in citrate–phosphate buffer (10 mM) at pH 4 and pH 8. The different filter membranes were pre-wetted in purified water for 5 min and subsequently autoclaved by three cycles at 121°C for 30 min, without any solvent. Thereafter, the filter membranes were extracted in 10 mL CP-buffer at pH 4 and pH 8 in an orbital incubator operated at 100 rpm for 66 h. The extraction conditions were selected based on previous studies (see Table II) [6]. The leachable stocks for each type of filter were treated at 30°C or 50°C.
Preparation of the Protein Formulations for the Protein Stability Study
Human granulocyte colony-stimulating factor was obtained in acetate buffer with a concentration of 1 mg/mL and was stored at − 20°C, until used. The acetate buffer was exchanged with the 10 mM CP-buffer using spin columns (Vivaspin 20 mL, Sartorius Stedim, Göttingen, Germany) with a molecular weight cut-off membrane of 5000 Dalton. Briefly, the frozen protein was thawed to liquid, thereafter the liquid was centrifuged to concentrate using a spin column with 5000 g at 4°C and the buffer was exchanged two times. The concentrated protein was then re-suspended in buffer of pH 4 and 8 and the protein concentration was determined via UV at 280 nm. Thereafter, the G-CSF solutions were diluted with different leachable stocks to achieve a final protein concentration of 0.6 mg/mL in buffers (pH 4 and 8). As control, G-CSF-formulation without leachable stock was prepared in an analogous manner as the formulations with leachable stocks.
Analytical Methods
Solid Phase Micro Extraction of Headspace Volatiles and Add-On GC/MS
To profile volatile compounds in the filters, solid phase micro extraction (SPME) of headspace volatiles was performed. Initially, the HS-GC vials were pre-heated at 200°C for 30 min. The membranes were sliced in small pieces and 0.5 g of different membranes was preheated for 5 min at 50°C using a Combi Pal autosampler system and placed into 20 mL headspace-GC (HS-GC) vials. The vials containing membrane pieces were then sealed with a PTFE lined silicone septum. The resulting headspace volatiles were extracted at 50°C for 20 min onto a 2 cm Stableflex 50/30 µm DVB/Carboxen/PDMS SPME fiber (Supelco, Bellefonte, USA). Thereafter, the headspace volatiles were analyzed via 7890A gas chromatograph (Agilent Technologies, Santa Clara, USA) coupled to a 5975C mass spectrometer (Agilent Technologies, Santa Clara, USA) and using a CTC Combi Pal autosampler (CTC Analytics AG, Zwingen, Switzerland) with SPME option. ZB-5MSi column with a film thickness of 0.25 µm, an inner diameter of 0.25 mm, and a length of 30 m was used for the analysis. The oven program was as follows: initial hold — 35°C for 1 min, heating up to 230°C with a heating rate of 5.5°C per min, final hold — 230°C for 1 min. Helium was used as carrier gas with a flow rate of 0.86 mL/min. The MS interface temperature was set to 280°C and mass detection was operated in scan mode from 35 to 300 m/z with a delay time of 5 min after injection. Volatile compounds were identified via comparison of their mass spectra with the NIST mass spectral library. Compounds with a match quality higher that 90% were considered identified.
GC/MS
Gas chromatography was applied to analyze the semi-volatile compounds in the extracts. The hexane and ethanol extractable stocks were analyzed by a Shimadzu GC-2010 Plus gas chromatograph coupled to a GCMS-QP2010SE mass spectrometer (Shimadzu, Kyōto, Japan). An HP5MS column with a film thickness of 0.25 µm, a diameter of 0.25 mm, and a length of 30 m was used as stationary phase. The following temperature program was applied: initial hold — 50°C for 1 min, heating up to 310°C with a heating rate of 10°C per min, final hold — 310°C for 3 min. Helium was used as carrier gas at a flow rate of 0.8 mL/min. Injection of 1 µl of sample solution was performed automatically by an AOC-20I auto injector (Shimadzu, Kyōto, Japan). The interface temperature of the mass spectrometer was set to 250°C and a mass range from 30 to 600 m/z was scanned starting 3.5 min after the injection. Detected compounds were identified by the comparison of their mass spectra with the NIST mass spectral library.
RP-HPLC/UV/MS
HPLC/UV and HPLC/MS measurements were performed for the extracts of the controlled extraction study and for the extracts of the protein stability study. Beside the hexane extracts, all other extracts were injected directly after extraction. Hexane extracts from the CES were treated as follows: 1 mL of each extract, as well as a blank, was filled in a HPLC vial and evaporated to dryness under a gentle stream of nitrogen. The residue was dissolved in 1 mL mobile phase (10% acetonitrile, 90% purified water). Forty microliters of each vial was injected and analyzed by an Alliance 2695 Separations Module (Waters Corp., Milford, MA, USA) coupled with a 2996 Photodiode Array Detector (Waters Corp., Milford, MA, USA) and a Micromass® Quattro micro™ API mass spectrometer (Waters Corp., Milford, MA, USA). A C-18 reversed phase column (Purospher® STAR, Merck Millipore, Billerica, MA, USA) was used as stationary phase at a temperature of 20°C. Acetonitrile (A) (Sigma Aldrich, St.Louis, MO, USA) and water (B) were used as mobile phase using the following gradient program: 0–40 min linear gradient from 10% acetonitrile (ACN) to 100% ACN, 40–45 min isocratic hold 100% ACN, 45–45.1 min from 100% ACN to 10% ACN, and 45.1–55 min isocratic 10% ACN. The flow rate was set to 1 mL/min and UV detection was performed at 220 nm. In order to enable appropriate electrospray ionization, the flow towards the MS detector was split in a 1:1 ration resulting in a 0.5 mL/min flowrate. To improve ionization, a 30 mM ammonium acetate solution in water was infused post column via a “T” connector into the mobile phase at a flow rate of 0.1 mL/min using an external pump (LC10AD, Shimadzu, Kyōto, Japan). The operation parameters for the mass spectrometer measurements are shown in Table III. A mass range from 110 to 1200 m/z was scanned. Data analysis was performed with the corresponding MassLynx 4.1 software (Waters Corp., Milford, MA, USA).
SEC-HPLC
For the size exclusion chromatography measurements, a Merck Hitachi LaChrom liquid chromatography system with an L-7400 UV detector was used (Hitachi, Darmstadt, Germany). The separation was accomplished by a TSK-GEL G3000SWXL column that was equipped with a guard column from Tosoh Bioscience (Tosoh Biosience, Tokyo, Japan). Citrate–phosphate buffer at pH 4 and pH 8 was used as mobile phases with a flow rate of 0.5 mL/min. The column temperature was set to 30°C [33]. Before the measurements, the column was saturated with G-CSF at a concentration of 1.71 mg/mL.
Density of Sample Solutions
Prior to surface tension measurements, density needed to be determined. The solvents from the controlled extraction study and the protein stability study were analyzed using a DSA 5000 M density and sound velocity meter (Anton Paar GmbH, Graz, Austria).
Surface Tension Analysis
The surface tension of the ethanol and water extracts obtained from the controlled extraction study as well as the leachable stocks of the protein stability study was determined using the EasyDrop System (Krüss, Hamburg, Germany). The measurements were performed with the pendant drop method using a 500 µL Hamilton syringe with a 0.8 mm needle diameter (Hamilton, Reno, NV, USA). A ten-fold determination was executed for each sample and subsequently averaged.
Zeta Potential Measurement
Electrophoretic light scattering was applied to analyze the zeta potential of the protein formulations using a Malvern ZetaSizer Nano-ZS (Malvern Instruments, UK) equipped with a 532 nm laser. A sample of approximately 1 mL was filled in a folded capillary cell (Malvern Instruments, UK), equilibrated at 25°C. The analysis was performed in triplicate with three different samples. Data analysis was conducted by the Malvern Zetasizer software.
Microflow Imaging
The protein aggregate formation (for the acquisition of protein particles between 2 and 300 µm) of the different protein formulations was analyzed using micro-flow imaging (MFI™ 5100 flow microscope, from ProteinSimple, Santa Clara, CA, USA). Milli-Q-water was flushed through the MFI device before and after each measurement in order to retain a clean base line. The sample purge volume was set at 0.20 ml/min and approx. 330 μL of the different protein formulations were analyzed. The particles of aspect ratio of ≥ 0.80 were considered for the analysis.
Raman Spectroscopy
To analyze the stability of the protein formulations, Raman spectroscopy was applied. For this, a RamanRXN2™ Hybrid Spectrometer (Kaiser Optical systems, Ann Arbor, MI, USA) with a 785 nm laser was used. Samples of 3.5 mL were taken from the different protein formulations and stirred with 100 rpm. During the agitation, the samples were scanned between a Raman shift of 100 cm−1 and 1900 cm−1 six times for 45 s using an MR probe, which was immersed into the formulation. The corresponding iC Raman 4.1™ software was used to record the spectra. For the following data analysis, SIMCA 13.0 software (Umetrics, Umeå, Sweden) was utilized and an SNV correction was performed.
Results and Discussion
The first part of this section deals with the evaluation of extractables and leachables from filter materials using SPME-GC/MS, GC/MS and LC/UV/MS. Whereas, the second part covers the protein stability study (PSS) comprising the study on the effect of leachable stock solutions on human granulocyte-colony stimulating factor (rh-GCSF) (Fig. 1).

Flowchart for the evaluation of E/L and the following protein stability study
Evaluation of Extractables and Leachables
Solid Phase Micro Extraction (SPME) of Headspace Volatiles and Add-On GC/MS
The volatile compounds in filters were analyzed using solid phase micro extraction, followed by GC/MS. The total ion chromatogram and list of volatile compounds extracted from the PA filter membrane is shown in Fig. S1 (please refer supplementary information) and Table IV. Five main peaks at RT (RT) of 5.97 min, 8.39 min, 12.32 min, 22.00 min, and 27.00 min were observed corresponding to hexanal, styrene, 2-ethylhexanol, tetradecane, and isopropyl laurate, respectively. In addition, few volatile compounds were also detected in lower concentration and were identified as hexamethylcyclotrisiloxane, nonanal, N,N-dimethylformamide, and several branched and unbranched aliphatic hydrocarbons of short chain length. Furthermore, a large number of low intensity peaks were detected and were not identified.
The identified compounds (such as styrene, 2-ethylhexanol (2-EH), and N, N-dimethylformamide) are reported to be used for polymer fabrication. Styrene is a toxic liquid and is known to be used in the manufacturing of rubbers and polymers [34]. 2-EH is a high-boiling solvent and is widely used as an esterification component for the production of plasticizers [35]. Whereas, N,N-dimethylformamide is used as a common solvent during the manufacturing of diverse polymers like polyamides, polyurethane, etc. [36].
In the case of PC membrane filters, lower numbers of peaks were observed as compared to the PA samples (Fig. S2). The most prominent peak was detected at a RT of 7.19 min and was identified as chlorobenzene. Peaks of lower intensity were detected at an RT of 5.97 min, 8.39 min, and refer to hexanal and styrene, as already seen in PA samples. In addition, traces of toluene, butylacetate, xylene, siloxanes, and carene were also detected (Table IV). The chromatogram also shows several small peaks as in the case of PA, which were not identifiable.
Some of the identified compounds were associated with polymer manufacturing such as chlorobenzene, toluene, and xylene. Chlorobenzene, either alone or mixed with other suitable solvents, used in the manufacturing of PC and for dissolving chain terminator or branching agents. It is also used as the organic phase for interphase polycondensation reaction. Toluene is another solvent that could be used during the manufacturing of PCs [37, 38]. Pure o-xylene is used as a source material for the production of phthalic acid or phthalic anhydride, an intermediate for the manufacturing of plasticizers or polyesters. As the use of plasticizers is very common in PC, the appearance of xylene as an extractable from PC filters was not surprising [37, 39]. p-xylene is primarily used for the production of terephthalic acid, which further converts to polyesters. Whereas, meta-xylene is often isomerized to its ortho- or para isomer [39].
In general, the intensity of the peaks in case of PES was found to be lower as compared to PA, PC, and RC samples (Fig. S3), implying lower amounts of volatile components. Five branched saturated and unsaturated aliphatic hydrocarbons, one siloxane, hexanal, styrene, α-pinene, and longifolene were identified as the volatile compounds, which are mainly used for the manufacturing of polyethersulfonate filter membranes (Table IV).
Like PES, the chromatogram of the PTFE sample showed a lower abundance of volatile compounds as compared to PA, PC, and RC samples (Fig. S4 and Table IV). Two siloxanes, two aliphatic hydrocarbons, verebenone, 4-butylnonane, and isopropyl laurate were successfully determined and were present in higher quantity as compared to other compounds.
In RC samples, the most prominent peak was detected at an RT of 9.83 min and was identified as 6-methyl-3-heptanone. Furthermore, the peaks at RT of 5.96 min, 8.38 min, 8.43 min, 12.32 min, 14.43 min, 17.02 min, and 21.99 min were also found and were identified as hexanal, styrene, cyclohexanone, 2-ethylhexanol, nonanal, dodecane, and tetradecane, respectively. Cyclohexanone plays an important role in RC manufacturing (via hydrolysis of cellulose triacetate (CTA) films). CTA was dissolved in a solvent mixture of cyclohexanone and methylene chloride is prepared. Thereafter, the solution is casted and the membrane is precipitated through the immersion of the cast film into a methanol bath. The recovered CTA membrane was then hydrolyzed to obtain RC membranes [40].
GC/MS Analysis
Gas chromatography coupled to mass spectrometry was applied to detect and identify semi-volatiles in the hexane and ethanol extracts from the CES. Chromatograms of the hexane extracts showed no additional peaks as compared to the blank. Whereas, in the case of ethanol extracts, only PA and PC showed significant peaks compared to the corresponding ethanol blank (Figs. S6 and S7).
In the PA-ethanol extract, two intensive peaks, at a RT of 11.35 min and 22.50 min, and one peak of lower intensity at a RT of 23.48 min were detected. The first and second peak at 11.35 and 22.50 min was identified as caprolactam and bisphenol A, respectively. However, the third peak could not be identified (Table V). As caprolactam is the base material for the production of polyamides via polycondensation, it was expected to be an extractable from PA filters [41].
The chromatogram of the PC-ethanol extract showed two intensive peaks at a RT of 6.72 min and 22.50 min, which were identified as phenol and bisphenol A. In addition, various smaller peaks, which were not determinable, were also detected. Bisphenol A was present in both membrane filters. Bisphenol A is mainly used as the base material for PC membrane production and as an antioxidants for plasticizers (Table VI) [42].
HPLC/UV/MS Analysis for the Controlled Extraction Study
The non-volatile compounds in the membranes were analyzed using liquid chromatography coupled with an UV detector and a mass spectrometer. HPLC/UV chromatograms of each filter extractable stock are shown in Figs. S8 to S12.
All the extracts of water, ethanol, and hexane showed significant peaks in UV chromatograms of PA samples (Fig. S8). In the PA water extract, peaks were detected at a RT of 8.33, 9.07, 9.58, and 10.00 min. Similar peaks were also detected in the PA ethanol extract along with an additional peak at 10.30 min. Whereas, in case of PA hexane extract, low intensity peaks were observed at a RT of 30.38 and 39.05 min.
The area under the curve (AUC) of different compounds was calculated at a wavelength of 220 nm. As mentioned in Table VII, the concentration of the peaks at RT 8.33 min, 9.07 min, and 9.58 min was found to be comparable in the case of water and ethanol extracts. However, higher concentration of peak at RT 10.00 min was found in the case of ethanol extract as compared to water extract. The total ion chromatograms of water and ethanol extract in ESI positive mode showed peaks at the same RTs, similar to UV chromatograms. The mass to charge (m/z) ratios for all peaks were found to be 566 (RT 8.33 min), 679 (RT 9.07 min), 792 (RT 9.58 min), 906 (RT 10.00 min), and 1019 (RT 10.30 min) (Table VII and Fig. S13). As evident from the m/z ratios, the subsequent m/z value was found to be 113 higher. The increase in m/z ratio with 113 was found to be due to the addition of a single caprolactam monomer (molecular weight is approximately 113 g/mol), to the caprolactam oligomers. Similar trend was also documented by Jenke et al. [43]. Thus, m/z of 566, 679, 792, 906, and 1019 represents the caprolactam pentamer (5 × 113 + H+), hexamer, pentamer, octamer, and nonamer, respectively. As mentioned above, the concentration of the caprolactam octamer was found to be higher in ethanol extracts compared to water extracts. The increase in octamer concentration in ethanol extracts was due to decrease in polarity of the caprolactam oligomer with increase in length of hydrocarbon chain.
In the case of PC water extract, two peaks with low (RT of 10.88 min) and high intensity (RT of 19.05 min) were observed. These peaks were found exclusively in case of water extract and thus indicating hydrophilic nature. Whereas, abundant peaks were observed in case of ethanol extract of PC (Table VII and Fig. S14). Apart from the peaks at RT of 34.90 and 39.15 min, which were also present in the PC hexane extract, all the peaks were found exclusively in ethanol extract. However, the AUC values of the peaks at RT of 34.90 and 39.15 min were found to be significantly higher in case of ethanol extract. Furthermore, the m/z ratios in ESI positive mode were found to be 438 (RT 33.30 min), 486 (RT 34.90 min), 740 (RT 39.15 min), 780 (RT 40.85 min), and 995 (RT 41.18 min).
The chromatograms of the PES samples showed the presence of two peaks of relatively low intensity in case of water extract. Whereas, multiple peaks were found in the ethanol extract and detected at the RTs of 26.58, 29.92, 32.98, 34.90, 36.31, and 37.30 min. Furthermore, the PES hexane extract showed low intensity peaks at 21.07, 25.57, and 30.38 min. The total ion chromatograms of all PES extracts showed no significant peaks, indicating presence of poorly ionizable compounds under the applied conditions.
The UV chromatograms of PTFE extracts showed peaks only in the hexane extract. The low intensity peaks were detected at a RT of 30.25, 38.26, 42.98, 43.38, and 43.83 min. Whereas, no peaks were detected in the total ion chromatograms of all extracts.
The RC extracts showed peaks in case of water and hexane extracts. In the water extract, one high intensity peak at RT 4.82 min and three smaller peaks at RT 2.38, 5.07 and 5.62 min were detected. Whereas, the RC hexane extract demonstrated a significant peak at RT 30.23 min. For the RC samples, no significant peaks were observed via MS-detection.
In general, UV detection showed many significant peaks in the different filter extracts compared to the sample blanks. As evident from the results (demonstrating higher number of compounds in ethanol), the application of ethanol as an extraction solvent seemed to mimic the worst-case scenario in this study. The detection via mass spectrometry showed just a few significant peaks which means that most extractables, detected in the controlled extraction study, were not ionizable under applied conditions. Interestingly, mass detection was exclusively possible in positive ion mode. The total ion chromatograms in negative ion mode showed no significant peaks despite the use of ammonium acetate as ionization source. Regarding the compounds detected via mass spectrometry, only the peaks occurring in PA samples were successfully identified. Furthermore, detected m/z ratios were not further analyzed in this study.
HPLC/UV/MS Analysis for the Leachable Study
For the leachable study filter, extracts at pH 4 and pH 8 were examined and evaluated using UV chromatograms (Figs. S15 and S16). The UV chromatogram of the PA pH 4 extract showed four peaks at an RT of 8.32, 9.06, 9.60, and 10.00 min. Whereas, three peaks at RT 8.33, 9.08, and 9.62 min were observed in case of PA buffer pH 8 (Table VIII). In accordance with the mass spectrometry results of the “HPLC/UV/MS Analysis for the Controlled Extraction Study” section (controlled extraction study), the peaks were found to be caprolactam oligomers (Fig. S17). The detected extractables in the CES actually became leachables, thus, extractable/leachable correlation was demonstrated. The only exception was found to be caprolactam monomer, which was detected only in case of CES study. Moreover, it is apparent that higher acidic pH (pH 4) seems to leach more compounds out of PA filters.
In case of PC pH 4 samples, only one peak at RT of 19.10 min was found, whereas, two peaks at RT of 10.83 and 19.08 min were observed in pH 8 samples. As evident from the AUC results (Table VIII), the peak at RT of 19.10 min was found to be substantial higher in buffer at pH 8 as compared to buffer at pH 4. Interestingly, the peak occurring at 10.83 min was exclusively present in pH 8 samples. The peaks were also found in case of water extract of the PC samples of CES. Thus, extractable/leachable correlation was demonstrated.
The UV chromatograms of the PES buffer extracts showed only one peak which was detected in the pH 4 buffer at a RT of 5.13 min. The same peak was also observed in the water extracts of the CES, which indicates that the corresponding substance is an extractable that became a leachable. No m/z ratio could be determined for the observed peak. The PTFE samples showed no significant peaks, which was found to be in accordance with the water extract of the controlled extraction study. Whereas, the UV chromatograms of the RC buffer extracts demonstrated two peaks at both pH values. The intense first peak was detected at 2.38 min, whereas, the other peak was detected at 4.77 min. Both peaks were also detected in the controlled extraction study.
In general, no leachables were detected, which were not already identified as extractables, due to the fact that leachables are typically a subset of extractables. In addition, it was observed that the pH of the solvents has a significant impact on the amount of extractables and leachables leached from the contact materials.
Solution Colloidal State Properties of Proteins in the Presence of E/L
Surface Tension Analysis
Surface tension is a critical quality attribute in the formulation development of biopharmaceuticals. Surface active agents are added to protein formulation to prevent accumulation of protein molecules at solid–liquid or air–liquid interfaces during product development. Surfactants interact with the protein surface and inhibits the interaction of protein surface with the primary packing surface, thereby preventing protein aggregation and denaturation. Despite the addition of surfactants critical for the protein formulations, accidental introduction of surface active leachables is considered an adulteration and have significant potential to alter the quality, purity, or safety of the drug product [6].
The surface tension was measured for each filter material in CP-buffer at pH 4 and pH 8. The samples were treated at temperature of 30°C and 50°C to mimic routine and slightly aggressive scenario during sterile filtration, respectively [44]. As illustrated in Fig. 2, the surface tension decreased in the presence of leachables from RC membranes at both extraction conditions. A clearly higher concentration of leachables were observed in case of pH 4 as compared to pH 8.

Results of surface tension measurements from filter leachable stocks which were extracted at 30°C (a) and 50°C (b)
As evident from Table VIII, higher concentration of compound at RT 2.38 min was observed in CP-buffer of pH 4. When comparing the leachable and surface tension results, the compound at RT 2.38 was found to be surface active. In addition, PES and PA extracts showed comparable surface tension value at both extraction conditions at pH 4 and pH 8. PA extracts demonstrated marginally lower surface tension at pH of 4 and 8 at temperature 50°C only. This might be due to the fact that the filter membrane at higher temperature could produce a higher amount of surface active leachable. However, the decrease in surface tension due to the surface active leachables needs further verification. In PC and PTFE extracts, no notable decrease of surface tension was observed.
Micro Flow Imaging
Micro flow imaging was used to evaluate the protein aggregation, which is a major problem in biopharmaceutical formulations due to protein instability and can lead to an increase in particulate size and number of particle/volume, and also trigger severe allergic reaction, etc. [45].
As evident from Fig. 3, higher particle concentration was observed in samples at pH 4 as compared to pH 8. Furthermore, the G-CSF protein formulations at pH 4 showed an extremely high particle concentration when spiked with RC extracts. In addition, the G-CSF samples treated with PA pH 4 extracts also demonstrated significantly higher particle concentration as compared to control. Whereas, no significantly higher concentration was observed in case of formulations containing PC, PES, and PTFE filter solutions.

Results (total number of protein particles between 2 and 300 µm) of MFI measurements from filter leachable stocks which were extracted at 30°C (a) and 50°C (b)
Even though G-CSF has higher stability at pH 4, the G-CSF formulations at pH 4 showed a higher particle concentration as compared to pH 8 [12]. According to Arakawa et al., lower stability at pH 8, as compared to pH 4, is due higher propensity towards oxidation and disulfide exchange reactions of the free cysteinyl residue at position 17 [10,11,12]. The increase in particle concentration could be due to the presence of higher concentration of compounds leached out at pH 4, adversely affecting the protein stability.
In case of RC filters, significantly higher particle concentration in formulations at pH 4 was observed. The increase in particle concentration was found to be in accordance with the surface tension results. This implies that RC stocks at pH 4 comprised of significantly higher surface active compounds, altering the protein conformation and thus reducing the stability. In addition, the PA extracts also demonstrated alteration in the protein aggregate concentration, however, to lower extend as compared to the RC extracts. In contrary, the formulations with leachables from PTFE filters extracted at 50°C demonstrated stability ameliorating properties of G-CSF formulations leading to decrease of particle concentration.
Size Exclusion Chromatography
As control, G-CSF formulation without filter extracts was used. The G-CSF protein in its native form (control group) showed RTs of approximately 24 and 18.5 min in case of pH 4 and pH 8, respectively. Thus, other peaks in the chromatograms with lower and higher RTs could be due to protein aggregates and protein degradation, respectively [46].
The pH 4-treated samples demonstrated no significant alteration in the protein size. Whereas, in case of pH 8 samples, significantly lower protein aggregation was observed in case of only RC treated samples (Fig. 4). The decrease in the degree of protein aggregation could be due to the unfolding and subsequently degradation of protein into smaller subunits. The degradation of the protein into smaller subunits could be due to the presence of surface-active compounds (as evident from the surface tension analysis), leached out from the RC filters. Furthermore, on comparing the results of micro flow imaging, it can be concluded that the extracts of RC filters resulted in formation of higher amount of protein aggregates, most probably of lower size below the detection limit of MFI.

Decrease in molecular weight levels (total low molecular weight species) in G-CSF formulations at pH 8 analyzed by SEC-HPLC after spiking with filter leachable stocks, extracted at 30°C (a) and 50°C (b)
Furthermore, a shoulder at the monomer peak was observed in case of PA treated samples. The shoulder could be due to minor aggregation of the monomer unit (Fig. 5) [33]. The results of protein aggregation in case of PA extract treated samples were found to be in accordance with the micro flow imaging results (Fig. 5).

SEC chromatograms of the control and the PA samples at pH 8
Zeta Potential
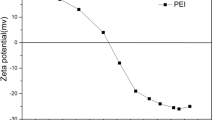
By the use of electrophoretic light scattering, the zeta potential of charged molecules like protein complexes can be determined. When charged particles are associated with an electric field, its motion vector is the sum of its charge dependent motion in the direction of the oppositely charged electrode and its random motion [31].
As evident from the Fig. 6, spiking G-CSF-formulations at pH 4 with filter extracts (extracted at 30°C) showed insignificant decrease in zeta potential except samples spiked with RC filter extract. This implies that RC containing samples were less stable than the control, and therefore shows a higher propensity to form aggregates. The results were in accordance with the micro flow imaging and size exclusion analysis (Fig. 3) where RC containing samples at pH 4 showed an extremely higher particle concentration of lower particle size. Whereas, in case of pH 8 samples, higher variability in zeta potential was observed as compared to pH 4 samples. Compared to the control PA, PC and RC samples showed a significant drop in zeta potential and are therefore more likely to form aggregates. The results were found to be in accordance with the extractable and leachables analysis (“Results and Discussion” section), demonstrating higher amounts of impurities in case of PA, RC, and PC samples as compared to the PES and PTFE samples. Thus, affirming extractable and leachables mediated alteration in the protein structure and thus zeta potential, which could be due to the ionic interaction between the impurities and the carboxylic and/or amine group of the proteins.

Results of zeta potential analysis from filter leachable stocks which were extracted at 30°C (a) and 50°C (b)
As compared to the 30°C samples, on spiking the pH 4 samples with the filter extracts at 50°C demonstrated decrease in zeta potential only in case of RC samples. Whereas, in case of pH 8 samples, all the protein samples spiked with filter extracts at 50°C showed a lower zeta potential as compared to the control. The decrease in zeta potential at 50°C could be due to combined effect of higher particle collision or interaction between the extracts and proteins and lower stability at higher temperature.
Raman Spectroscopy
Raman spectroscopy was applied to detect potential changes in confirmation when G-CSF formulations were spiked with filter leachables. As evident from Figs. S18 and S19, the Raman spectra demonstrated characteristic peaks at wavenumber, 1300, 1550, and 1650 cm−1 corresponding to amide III, II, and I, respectively [47]. Interestingly, no shift in the wavenumber was observed. However, significant changes in the relative intensity of the peaks were observed in case of protein spiked with extracts at pH 4 and 8. A marked decrease in the peak intensity of amide I band at 1650 cm−1 was observed in case of samples spiked with the PA filter extracts. Similarly, PA filter extracts demonstrated significant decrease in the amide II signals as compared to control samples. Interestingly, all the filter extracts, i.e., from PA, RC, PC, PES, and PTFE demonstrated significant decrease in the amide III signals. The results were found to be in accordance with the results of other protein stability studies, suggesting significant effect of filter extracts on the protein denaturation and aggregation [6].
Relevance and Limitations of the Study
In the present investigation, the volatile, semi-volatile, and non-volatile E/L were analyzed using HS-GC, GC/MS, and HPLC/UV/MS, respectively. Diverse volatile and non-volatile E/L were observed in the case of all the filter samples. However, only PA and PC showed some semi-volatile E/L. The E/L was found to be mainly the monomers, reagents used for polymerization reaction, reaction intermediate, and solvents used for the filter preparation. The E/L were qualitatively and semi-quantitatively analyzed using various analytical methods. However, the analytical methods used in this study lack a comprehensive verification steps such as extraction stoichiometry, which could be critical in absolute quantification of E/L. Therefore, the study design and the results therefrom are by no means quantitative and validated, which is often the case of common E/L screening studies reported [43]. Nevertheless, the present study deals with comparative effects of various E/L, from five different filter membranes, on the stability of a model protein formulation using various analytical techniques and methodologies. Thus, overall, the results provided potential insight into the effect of E/L on the protein formulation and can be vital in rank ordering the materials which are in contact during aseptic manufacturing of sterile biologics products.
Conclusion and Outlook
In the present manuscript, various E/Ls were extracted from different filters and evaluated using state-of-the art analytical techniques. Some of the identified extractables obtained from specific filter materials, such as bisphenol A from PC filters, are known to be harmful for patients. Other detected extractables were not found in literature so far and were found to be associated with polymer production processes. Remarkably, PA, PC, and PES filters showed the highest extractable content, whereas PTFE and RC membranes contained a low level of extractables. By applying less aggressive extraction conditions, in order to mimic actual process or storage conditions, leachables were obtained and identified. As typical, all the detected leachables were found to be a subset of extractables. Since so far it was also not possible to discern and exactly chemically identify the several E/Ls detected, a follow up study with advanced characterization would be encouraging.
The stability of the selected model protein was evaluated upon spiking with E/Ls. As evident from the results, E/Ls from RC membranes were found to be surface active. Whereas, E/Ls from PES slightly decreased the surface tension. Furthermore, E/Ls from PA and RC filters showed a distinct change in the protein structure. In this context, the evaluation of the effect of monomers of PA and RC leachables on G-CSF formulations, for example, effect of caprolactam monomers on G-CSF could additionally corroborate the present findings.
Abbreviations
- 2-EH:
-
2-Ethylhexanol
- ACN:
-
Acetonitrile
- AUC:
-
Area under the curve
- CES:
-
Controlled extraction study
- CP-buffer:
-
Citrate-phosphate buffer
- CTA:
-
Cellulose triacetate
- DSC:
-
Differential scanning calorimetry
- EIC:
-
Extracted ion chromatogram
- E/L:
-
Extractables and leachables
- ESI:
-
Electrospray ionization
- E-stock:
-
Extractable stock solution
- GC:
-
Gas chromatography
- HPLC:
-
High performance liquid chromatography
- HS-GC:
-
Gas chromatography of headspace volatiles
- kDa:
-
Kilo Dalton
- LC:
-
Liquid chromatography
- m/z:
-
Mass-to-charge ratio
- MFI:
-
Micro flow imaging
- MS:
-
Mass spectrometry
- n.d.:
-
Not determinable
- PA:
-
Polyamide
- PC:
-
Polycarbonate
- PES:
-
Polyethersulfone
- PSS:
-
Protein stability study
- PTFE:
-
Polytetrafluoroethylene
- RC:
-
Regenerated cellulose
- RP:
-
Reversed phase
- RT:
-
Room temperature
- SEC:
-
Size exclusion chromatography
- SPME:
-
Solid phase micro extraction
- TIC:
-
Total ion current
- UV:
-
Ultraviolet
References
Jenke DR, et al. Accumulation of extractables in buffer solutions from a polyolefin plastic container. Int J Pharm. Jun. 2005;297(1–2):120–33. https://doi.org/10.1016/j.ijpharm.2005.03.010.
Behme S. Manufacturing of pharmaceutical proteins: from technology to economy. John Wiley & Sons; 2021. https://www.wiley.com/en-us/Manufacturing+of+Pharmaceutical+Proteins%3A+From+Technology+to+Economy%2C+3rd+Edition-p-9783527833801.
Akers MJ. Sterile drug products: formulation, packaging, manufacturing and quality. CRC Press; 2016. https://www.taylorfrancis.com/books/mono/10.3109/9781420020564/sterile-drug-products-michael-akers.
The European Agency for the Evaluation of Medicinal Products Evaluation of Medicines for Human Use Committee for Proprietary Medicinal Products (cpmp) Decision Trees for the Selection of Sterilisation Methods Decision Trees for the Selection of Sterilisat. 1997. Available: http://www.eudra.org/emea.html. Accessed 16 July 2021.
Committee for Medicinal Products for Human use (CHMP) Committee for Medicinal Products for Veterinary use (CVMP) Guideline on the sterilisation of the medicinal product, active substance, excipient and primary container. 2019, Available: www.ema.europa.eu/contactTelephone+31. Accessed 16 July 2021.
Huang M, Horwitz TS, Zweiben C, Singh SK. Impact of extractables/leachables from filters on stability of protein formulations. J Pharm Sci. 2011;100(11):4617–30.
Colton RH, Bestwick DG. Extractables and leachables evaluation for filters. In: Jornitz M, Meltzer T, editors. Filtration and Purification in the Biopharmaceutical Industry. 2nd ed. USA: Oregon; 2008. p. 389–411.
Riley CM, Rosanske TW, Reid GL, editors. Specification of drug substances and products: development and validation of analytical methods. Elsevier; 2020. https://www.elsevier.com/books/specification-of-drug-substances-and-products/riley/978-0-08-102824-7
Peri P. Regulatory Perspectives on Extractables and Leachables Structures of potential leachables. Bethesda (MD). 2011. https://pqri.org/wp-content/uploads/2015/11/PQRI_Extractables_and_Leachables_Talk_Prasad_2011.pdf. Accessed 8 Aug 2022
T. Arakawa, S. J. Prestrelski, L. O. Narhi, T. C. Boone, and W. C. Kenney, “Cysteine 17 of recombinant human granulocyte-colony stimulating factor is partially solvent-exposed,” J. Protein Chem. 1993 125, vol. 12, no. 5, pp. 525–531, Oct. 1993, https://doi.org/10.1007/BF01025117
B. H. Do et al., “Granulocyte colony-stimulating factor (GCSF) fused with Fc domain produced from E. coli is less effective than polyethylene glycol-conjugated GCSF,” Sci. Reports 2017 71, vol. 7, no. 1, pp. 1–9, Jul. 2017, https://doi.org/10.1038/s41598-017-06726-7
Oh-Eda M, et al. O-linked sugar chain of human granulocyte colony-stimulating factor protects it against polymerization and denaturation allowing it to retain its biological activity*. J Biol Chem. 1990;265(20):11432–5. https://doi.org/10.1016/S0021-9258(19)38416-9.
Caseri W. Polyamide, RD-16-03046. RÖMPP [Online], Stuttgart, Georg Thieme Verlag, 2022. https://roempp.thieme.de/lexicon/RD-16-03046. Accessed 8 Aug 2022.
Nylon membranes, Sterlitech Corporation. https://www.sterlitech.com/nylon-membranes.html. Accessed 8 Aug 2022
Nylon membrane filter 25007-293-N, Sartorius AG. https://www.sartorius.com/shop/ww/de/eur/applications-laboratory-filtration/polyamide-membrane-filters-type-25007/p/25007-293------N. Accessed 8 Aug 2022.
Caseri W, Polycarbonate, RD-16-03130. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-16-03130. Accessed 8 Aug 2022.
Polycarbonate 23007-50-N, Sartorius AG. https://www.sartorius.com/shop/ww/de/eur/applications-laboratory-filtration/polycarbonate-track-etched-membrane-filters-0-2-%c2%b5m-50-mm-discs/p/23007--50------N. Accessed 8 Aug 2022.
Whatman® Nuclepore Track-Etched Membranes - Analysis and Detection of Microorganisms, Sigma-Aldrich. https://www.sigmaaldrich.com/AT/de/substance/whatmannucleporetracketchedmembranes1234598765. Accessed 8 Aug 2022.
Polyethersulfone 15407-25-N, Sartorius AG. https://www.sartorius.com/shop/ww/de/eur/applications-laboratoryiltration/polyethersulfone-membrane-filters---type-15407--0-2-%c2%b5m-pore-size--25-mm-diameter--100-pieces-per-pack/p/15407--25----MIN. Accessed 8 Aug 2022.
Polyethersulfone, Sterlitech Corporation. https://www.sterlitech.com/polyethersulfone-pes-membrane-filters.html. Accessed 8 Aug 2022
Brüggemann O, Polytetrafluoroethylene, RD-16-03604. RÖMPP [Online], Stuttgart, Georg Thieme Verlag, 2022. https://roempp.thieme.de/lexicon/RD-16-03604. Accessed 8 Aug 2022.
Polytetrafluorethylene, Sartorius AG. https://www.sartorius.com/shop/ww/de/eur/applications-laboratory-filtration-sterile-filtration/minisart-syringe-filter%2c-polytetrafluorethylene-%28ptfe%29%2c-pore-size-0-2-%c2%b5m%2c-non-sterile%2c-female-luer-lock%2c-male-luer-slip%2c-pack-size-500/p/1751A----------Q. Accessed 8 Aug 2022
PTFE Membrane Filters, Sterlitech Corporation. https://www.sterlitech.com/0ql8222005.html. Accessed 8 Aug 2022
Türk W. Regenerated cellulose, RD-18-00636. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-18-00636. Accessed 8 Aug 2022.
Regenerated Cellulose, Sartorius AG. https://www.sartorius.com/shop/ww/de/eur/applications-laboratory-filtration/regenerated-cellulose-membrane-filters-type-18406%2c-0-45-%c2%b5m-pore-size%2c-25-mm-diameter%2c-100-pieces-per-pack/p/18406--25------N. Accessed 8 Aug 2022.
DIAFIL RC syringe filter Membrane material: Regenerated cellulose. http://www.filtershop.cc/contents/de/d2.html. Accessed 8 Aug 2022
Raso SW, et al. Aggregation of granulocyte-colony stimulating factor in vitro involves a conformationally altered monomeric state. Protein Sci. 2005;14(9):2246–57. https://doi.org/10.1110/ps.051489405.
Hübel K, Engert A. Clinical applications of granulocyte colony-stimulating factor: an update and summary. Ann Hematol. 2003;82(4):207–13. https://doi.org/10.1007/s00277-003-0628-y.
Hill CP, Osslund TD, Eisenberg D. The structure of granulocyte-colony-stimulating factor and its relationship to other growth factors. Proc Natl Acad Sci U S A. 1993;90(11):5167–71. https://doi.org/10.1073/pnas.90.11.5167.
Prakash A, Medhi B, Chopra K. Granulocyte colony stimulating factor (GCSF) improves memory and neurobehavior in an amyloid-β induced experimental model of Alzheimer’s disease. Pharmacol Biochem Behav. 2013;110:46–57. https://doi.org/10.1016/j.pbb.2013.05.015.
Jiskoot W, Crommelin D, editors. Methods for structural analysis of protein pharmaceuticals. Springer Science & Business Media. 2005. https://link.springer.com/book/9780971176720?utm_medium=referral&utm_source=google_books&utm_campaign=3_pier05_buy_print&utm_content=en_08082017
Stoker HS. Biological chemistry. In: General, Organic and Biological Chemistry. 6th ed. Belmont: CA, USA; 2013. p. 733.
Roessl U, Wiesbauer J, Leitgeb S, Birner-Gruenberger R, Nidetzky B. Non-native aggregation of recombinant human granulocyte-colony stimulating factor under simulated process stress conditions. Biotechnol J. 2012;7(8):1014–24. https://doi.org/10.1002/biot.201100436.
National Center for Biotechnology Information. PubChem Compound Summary for CID 88025, Styrene-d8, PubChem, https://pubchem.ncbi.nlm.nih.gov/compound/Styrene-d8. Accessed 8 Aug 2022.
RÖMPP-Redaktion, 2-Ethylhexan-1-ol, RD-05-02039. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-05-02039. Accessed 8 Aug 2022.
RÖMPP-Redaktion, dimethylformamide, RD-04-01717. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-04-01717. Accessed 8 Aug 2022.
Dujardin R, Meyer RV, Siebourg W. Polycarbonates with aralkyl phenyl end groups, their preparation and their use. EP0327951B1, 1988. https://patents.google.com/patent/EP0327951B1/en.
Herrig W, Kadelka J, Hucks U. Process for the production of polycarbonates. DE4241619 A1. 1994. https://patents.google.com/patent/DE4241619A1/pt-PT.
Caseri W. Xylene Formaldehyde Resins, RD-24-00156. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-24-00156. Accessed 8 Aug 2022.
Johnson BM. Production of permeable cellulose triacetate membranes. US 4631157 A. 1986. https://patents.google.com/patent/US4631157A/en.
Türk W, Perlon®, RD-16-01098. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-16-01098. Accessed 8 Aug 2022.
Sefkow M. RÖMPP-Redaktion, Bisphenols, RD-02-01737. RÖMPP [Online], Stuttgart, Georg Thieme Verlag. 2022. https://roempp.thieme.de/lexicon/RD-02-01737. Accessed 8 Aug 2022.
Jenke D. Evaluation of the chemical compatibility of plastic contact materials and pharmaceutical products; safety considerations related to extractables and leachables. J Pharm Sci. 2007;96(10):2566–81. https://doi.org/10.1002/jps.
Nema S, Ludwig JD, editors. Pharmaceutical dosage forms: parenteral medications, volume 3 set. CRC Press; 2010. https://www.routledge.com/Pharmaceutical-Dosage-Forms-Parenteral-Medications-Third-Edition-3-Volume/author/p/book/9781482281781.
Zhang A, Singh S, Shirts M. Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze/thaw-stresses revealed by hydrogen exchange. Pharm Res. 2012;29(1):236–50.
Uversky V. Use of fast protein size-exclusion liquid chromatography to study the unfolding of proteins which denature through the molten globule. Biochemistry. 1993;32:13288–98.
Lewis IR, Edwards H. Handbook of Raman spectroscopy: from the research laboratory to the process line. CRC Press; 2001. https://www.routledge.com/Handbook-of-Raman-Spectroscopy-From-the-Research-Laboratory-to-the-Process/Lewis-Edwards/p/book/978082470557.
Acknowledgements
The authors duly acknowledge RCPE laboratory team for their support during the execution of this study. We thank Steven A. Zdravkovic (Baxter, USA) for his expert suggestion and for careful revision of the work.
Funding
Open access funding provided by Graz University of Technology. This study was funded by the Austrian COMET Programme under the auspices of the Austrian Federal Ministry of Transport, Innovation and Technology (BMVIT), the Austrian Federal Ministry of Economy, Family and Youth (BMWFJ) and the State of Styria (Styrian Funding Agency [SFG]). COMET is managed by the Austrian Research Promotion Agency (FFG).
Author information
Authors and Affiliations
Contributions
Varun Kushwah: methodology, software, formal analysis, writing — original draft, writing — review and editing, and supervision.
Juliana Münzer: investigation, methodology, and writing — original draft.
Verena Feenstra: supervision and writing — review and editing.
Stefan Mohr: supervision and writing — review and editing.
Amrit Paudel: conceptualization, supervision, resources, project administration, and writing — review and editing.
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kushwah, V., Münzer, J., Feenstra, V. et al. Impact of Extractables/Leachables from Filter Materials on the Stability of Protein-Based Pharmaceutical Products. AAPS PharmSciTech 23, 233 (2022). https://doi.org/10.1208/s12249-022-02374-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02374-x