
ABSTRACT
The purpose of this study was to evaluate the surface mineralization activity and in vitro drug behavior potential of two forms of mesoporous silica: powder and granulate. Ordered mesoporous SiO2 powder was synthesized by surfactant-assisted sol-gel process using tetraethoxysilane as a silica precursor and hexadecyltrimethylammonium bromide as the structure-directing agent. The granulate was prepared using silica powder and ethyl cellulose as a binding agent. Metronidazole (MT)—an anti-inflammatory substance and doxorubicin hydrochloride (ChD)—an anti-cancer drug were chosen as drug models for delivery studies. The results of structural characteristic studies, utilizing transmission electron microscope (TEM) and scanning electron microscope (SEM) images, powder X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), and nitrogen adsorption–desorption (BET) measurements, show that obtained materials have two-dimensional hexagonal p6mm symmetry, high specific surface area, narrow pore size, and a satisfactory mineralization behavior in the simulated body solution (SBF, pH = 7.4). The release rate of drugs depends upon the structural features of the drug molecules and the form of the carrier material. Of both the drugs analyzed, faster release was observed for small MT molecules characterized by weaker interactions with the carrier. In addition, the slower drug release was observed with granulate form due to increased diffusion barrier for drugs. Obtained results prove that the MT/ChD-loaded silica formulations could be attractive materials for filling bone defects and for local delivery systems.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Some of the problems faced in the field of reconstructive medicine were the creation and successful application of biomaterials whose properties need to be as close to the damaged, diseased, or resected bone tissue. It is also desirable to stimulate the growth of the natural tissue, helping in its replacement or complementation. The desired biomaterial should demonstrate appropriate physicochemical stability and good adaptation in in vivo conditions.
Ceramics play a very important role among the broad range of biomaterials used in the reconstructive medicine (1). They have been in use in various fields of medicine such as orthopedics, stomatology, traumatology, and cardiology. These biomaterials demonstrate high chemical stability, degradation-resistance properties, and in vivo biocompatibility. The porosity of the materials is varied, which enables ingrowth of vascularized soft tissue and osteocytes into the pores (2), thus reinforcing the bond between the implant and the bone tissue.
Among the ceramic oxide materials, ordered mesoporous silica-based materials are the leading candidates in the tissue reconstruction technology. Their characteristic feature is low susceptibility to bacterial colonization and absolute resistance to corrosion, unlike metallic implants. In addition, because of their uniform porosity and pore size, which is similar in magnitude to the molecular size of the drugs, they may be used as carrier designated for the controlled release of therapeutic drugs (3–5).
Many ordered types of mesoporous silica have been tested as drug carriers. First, Vallet-Regi discovered the new property of MCM-41 silica in drug release, for the sustained release of ibuprofen (6). Since that time, many related materials, especially SBA-15 and the organic modification of MCM-41, have been used to control the release of various active substances such as famotidine, amoxicillin, itraconazole, and methotrexate (7–10).
There is large amount of literature available on mesoporous materials. These studies focused on the synthesis and functionalization procedures in addition to the descriptions on the influence of the chemical interaction between silica and the drug molecule with respect to adsorption and delivery rate of drugs. However, there is less information on the forms of mesoporous materials as implantable carriers of drugs. In general, these are tested either in the form of powder or in the form of compressed disks. These forms have some disadvantage: powder forms will be too dusty and the compression of powder to disks can destroy their ordered structure (11–13).
During granulation of the material, small particles are gathered into larger, permanent masses where the original particle can still be identified. In addition to this, granulated material results in reduced dustiness and sustained drug release (14). Therefore, in this study, we selected ethyl cellulose (EC) as the binding agent due to its widespread use as a binder in wet granulation and the use of cellulose derivatives as components of bioactive materials (15).
In this study, we investigated the possibility of using granulated mesoporous silica (in comparison to the MCM-41 powder) in the construction of bioactive implant to reconstruct the bone structure and at the same time as the model of therapeutic system—matrix to the targeted, controlled release of drug. Another objective of this study was to introduce a combination of two mutually complementary drugs acting in the diseased bone area. Introduction of a bioactive implant containing a cytostatic—doxorubicin hydrochloride (ChD) complemented with an anti-inflammatory substance—metronidazole (MT) into the defect left upon resection of cancerous bone tissue can substantially improve the effectiveness of the therapeutic process.
The choice of the aforementioned substances is dictated by our earlier results of the dissolution of the gel of the oxide carrier (15,16). We found that due to the construction and interaction with the matrix oxide, ChD was released much slower than MT. Based on these preliminary results, we hypothesize that ChD-MT-MCM-41 formulation can be a promising system for (i) the immediate protection against inflammation after implantation due to the rapid release of the anti-inflammatory substance—MT and (ii) targeted treatment of the cancerous diseased bone tissue in controlled, prolonged manner due to the slow release of ChD.
MATERIALS AND METHODS
Tetraethoxysilane (TEOS), n-hexadecyltrimethylammonium bromide (CTAB), EC, and drugs—MT, 2-methyl-5-nitroimidazole-1-ethanol, and ChD (Sigma-Aldrich Co., Poznań, Poland) were used without further purification. Ethanol and 25% aqueous ammonia (POCh Co., Gliwice, Poland) were of analytical grade purity.
Simulated body fluid (SBF) as the dissolution medium for release analysis and bioactivity test was prepared according to Kokubo (17).
Synthesis of Mesoporous Silica MCM-41
MCM-41 powder was prepared according to the procedure reported by Grün, Lauer, and Unger (18). Approximately 2.39 g of CTAB was dissolved in a mixture of de-ionized water (125 g), 25% aqueous ammonia (9.18 g), and ethanol (12.5 g) and stirred in a polypropylene beaker on a hot plate magnetic stirrer (40°C) for 1 h (200 rpm). Then, 10.03 g of TEOS was added dropwise with continuous stirring. After 2 h of stirring, the white precipitate was aged at 100°C for 5 days under static conditions. The resulting solid product was recovered by filtration on a Buchner funnel and washed with absolute ethanol, dried at 50°C for 24 h, and calcined at 550°C for 5 h (Muffle Furnace, M-525, USA, heating rate 1 K/min) to remove the CTAB surfactant.
The final powder samples with the fraction size ranging from 100 to 200 μm were obtained by sieving for 10 min with a nest of sieves (100 and 200 μm) mounted on a sieve shaker (Retsch-Haan, Germany).
Drug-loading Efficiency
Drugs were loaded into MCM-41 using the incipient wetness method (19). In this method, drug substance in the form of a solution is diffused into the carrier pore by capillary force. Then, the carrier is dried to evaporate the solvent, thereby depositing the drug on the carrier surface.
The procedure for loading solutions was as follows: 50 mL of water solution containing ChD (200 μg/mL) was added to a flask containing 1 g of MCM-41. This mixture was shaken for 24 h at room temperature (25 ± 0.5°C) under light-sealed conditions. After this, the supernatant was separated from the mixture by centrifugation, and the residual ChD content was determined using the calibration curve of ChD standard solution in SBF by reading the absorbance at 480 nm. The % drug adsorption versus initial concentration was plotted, which allowed to determine the adsorption capacity of the silica samples. The results were obtained from data groups of n ≥ 3 and are expressed as mean ± standard deviation. The loading efficiency obtained was 10 mgChD/gMCM-41 (98.2 ± 1.8%).
The loading of MT into MCM-41 was in a similar way except that the concentration of MT was 1 mg/mL, and the residual MT content was determined using the calibration curve of MT standard solution in SBF by reading the absorbance at 320 nm. The loading efficiency obtained was 18 mgMT/gMCM-41 (36.3 ± 0.4%).
Manufacture of Powder with Drugs
ChD-MCM-41 powder was physically combined with MT-MCM-41 at 1:1 mass ratios using tumble mixing for 3 min to achieve a homogeneous solid mixture.
Manufacture of Granules with Drugs
First, the granulation liquid (5% ethanolic solution of EC, 15 mL) was added dropwise with a pipette to the dry ChD-MT-MCM-41 (~5 g) powders and mixed using the ceramic mortar. After the wetting process, the obtained mixture was pressed through a sieve (Retsch 800 μm). The wet granulate was dried at 37°C for 2 h and again screened through a sieve. For further analysis, the 500–800-μm fraction of granules was selected.
Characterization of Silica Carriers
The molecular structure of samples was determined using a Fourier transform infrared (FT/IR) spectrometer (410 Spectrometer, Jasco), using the potassium bromide (KBr) disk technique. The spectra were collected over a range of 4000–400 cm−1 (64 scans, resolution 4 cm−1).
Surface area, pore volume, and pore diameter of the powder were determined by N2 adsorption using a Micromeritics ASAP 2405N instrument and the Barret–Joyner–Halenda (BJH) methods.
The X-ray diffraction (XRD) spectra of samples were taken with an Empyrean/PANalytical XRPD diffractometer using CuKα1 radiation, operating at 20 kV and 40 mA.
The morphology of the silica was characterized using transmission electron microscope (TEM) (Tecnai G2 20X-TWIN, FEI Company) and scanning electron microscope (SEM) equipped with an energy dispersive spectroscope (EDS) and a mapping detector (Quanta 3D FEG/FEI).
Assessment of In Vitro Mineralization
In vitro mineralization—in vitro bioactivity (ability to apatite formation on the silica surface) was performed as follows: 0.5 g of sample (powder or granulate) was soaked in 50 mL of SBF (solution with inorganic ion concentrations close to that of human blood plasma) and was shaken in the thermostated shaking water bath (Julaba, Germany, 50 rpm) at 37°C (±0.5°C) for 1 month. The samples were examined daily using FTIR, XRD, and SEM-EDS techniques to detect apatite.
Dissolution Studies
The dissolution behavior was determined using the Flow-Through-Cell USP Apparatus 4 (closed loop) (Erweka, DZT-770, Germany). For the drug release study, 100 mg of the carrier samples (powder and granulate) with drugs was placed on glass beads in a 22.6-mm flow-through cell. The SBF solution was used as an acceptor fluid. The experiment was conducted at 37°C with a flow rate of fluid of 4 mL/min and 100 mL of acceptor fluid. At certain time intervals, a portion of the eluate (3 mL) was drawn to measure the concentration of the released drug and then fresh SBF (3 mL) was added to keep the volume constant. The concentration of ChD and MT was determined using a UV–VIS spectrophotometer (Shimadzu UV-1800 spectrometer), at λmax of 480 and 320 nm, respectively. Drug elution data were plotted as the cumulative mass amount of drug released as a function of time. The results were obtained from data groups of n ≥ 3 and are expressed as mean ± standard deviation.
The Peppas power law model (Eq. (1)) (20) was used as a mathematical description of drug release:
where M t and M ∞ are the absolute cumulative amounts of drug released at time t and infinite time, respectively, k is a constant rate, and n is the release exponent indicative of the mechanism of drug release. When the exponent n is close to 1, the drug release rate is time-independent. This case corresponds to zero-order release kinetics. When n is close to 0.5, the model predicts a diffusion-controlled drug release; for n between 0.5 and 1, the drug release is considered as anomalous. The Peppas model is valid up to 60% drug release.
RESULTS AND DISCUSSION
Characterization of Silica Carriers
FTIR spectroscopy provided qualitative information about the chemical structure of the synthesized drug carriers. It allowed the assessment of complete reaction of substrates and also the complete removal of surfactant from the pores of the synthesized MCM-41 material. The resulting FTIR spectrum for the powder material showed bands derived only from bonds present in the silica (Fig. 1). As seen in Fig. 1, strong bands were caused by the vibrations of Si–O–Si groups at 1076, 924, and 800 cm−1, which is indicative of a high degree of polymerization. The lack of bands derived from the vibrations of CH stretching in the region of 2800 cm−1 indicates complete hydrolysis of the alkoxy groups (–OC2H5) derived from TEOS. Two broad bands, one at 3450 cm−1 and the other at 1631 cm−1, were caused by O–H vibrations of Si–OH groups and the water molecule retained in the pores of the oxide network. The granulates’ FTIR spectrum was slightly different from the spectrum of the MCM-41 powder. Additional bands were visible in the 2900–2800 cm−1 region. These bands originated from the C–H vibrations of the alkyl group derived from EC used as a binder (21).

FTIR spectra of a MCM-41 in the powder form and b MCM-41 in the granulate form
Figure 2a shows nitrogen adsorption–desorption isotherms of the MCM-41. According to the IUPAC classification (22), the isotherm obtained for the powder belongs to type IV with a pronounced increase of capillary condensation and a narrow hysteresis loop. A rapid increase in nitrogen adsorption at a relative pressure within the range of 0.35–0.45 is characteristic for such type of materials and corresponds to capillary condensation of nitrogen in regular mesopores. This fragment of the adsorption–desorption isotherm allows to calculate the pore diameter of the tested material.

a Nitrogen adsorption–desorption isotherms plots. b Barrett–Joyner–Halenda pore size distribution of the MCM-41
The graph in Fig. 2b shows that the MCM-41 had a narrow pore distribution.
Table I presents the calculated parameters of the porous structure of materials tested based on nitrogen adsorption isotherms using BJH method. Analysis of morphology of MCM-41 confirmed the ordered pores’ structure. Although the walls of MCM-41 are amorphous, it is possible to conduct the diffraction tests due to long-range order of the pore system.
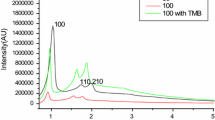
Figure 3 shows the XRD pattern measured for the MCM-41 material. The obtained diffraction pattern of the MCM-41 exhibited three well-resolved peaks, which were assigned to reflections from the (100), (110), and (200) planes of a hexagonal well-ordered structure with p6mm space group (23). Table I shows the characteristic parameters of the silica network, that is, diameter of the mesopores and the wall thickness (WT).

X-ray diffraction pattern of MCM-41
Figure 4 shows the TEM images of the hexagonal arrangements of the porous system and the uniformity of the pores (24). The average pore diameter of the MCM-41 observed from TEM images is in good agreement with XRD patterns and N2 sorption measurements.

Transmission electron micrographs of a MCM-41 in the (100) direction and b MCM-41 in the (110) direction
Assessment of In Vitro Mineralization
Bioactive oxide materials (ceramics and glasses) form a bone-like apatite layer on their surfaces after being implanted in bone. The potential in vivo bioactivity of these materials may be inferred by their ability to mineralize apatite on their surface during immersion in SBF, as proposed by Kokubo and Takadama (in vitro Kokubo test) (25). SBF has inorganic ion concentrations close to that of human blood plasma and does not contain any cells or proteins. In such an environment, bone-like apatite is created by the chemical reactions of the biomaterial components with the SBF ions. The mineralization activity of oxide materials determines not only the chemical composition but also the surface properties, especially the porosity (the presence of pores larger than 2 nm) and hydrophilicity of the surface (e.g., the presence of reactive hydroxyl groups) (26). These indications lead to think that ordered mesoporous silica such as MCM-41, thanks to high surface area, appropriate size of pores, and the occurrence of silanol (Si–OH) reactive groups, is a promising candidate as bioactive bone tissue substitute.
To confirm the mineralization activity of a MCM-41 in the form of powder and granulate, the samples of silica were soaked in SBF and then the formation of apatite phase was detected by FTIR, XRD, and SEM-EDS techniques.
The FTIR spectra showed two new bands at about 550 and 600 cm−1 wavenumbers (the major absorption mode of the phosphate groups—the O–P–O bending mode), which indicates the formation of crystalline apatite (Fig. 5). Before incubation in the SBF, the FTIR spectra of silica showed a single, weak band observed at about 580 cm−1 (vibration of Si–O). However, following longer incubation of the samples in the SBF solution, a sharp doublet appeared at 564 and 602 cm−1, indicating the formation of crystalline hydroxyapatite. In addition, carbonate groups substituting the PO4 3− ions in the apatite structure can be detected in samples by the appearance of bands at 1410–1460 cm−1 (27).

FTIR spectra of a MCM-41 in the powder form and b MCM-41 in the granulate form after different days of mineralization studies in simulated body fluid
The induction period for the crystallinity of HA depends on the form of silica. It was observed that the incubation of powder in SBF solution results in fast growth of hydroxyapatite on its surface (5 days). In case of granules, the appearance of HA on their surface was noted after 3 weeks of the experiment. The differences in the HA growth on the samples surface are related to its formation mechanism. The OH– groups from silica powder form hydrogen bonds with the components of the SBF solution, especially with Ca2+, CO3 2−, and HPO4 2− ions, resulting in the growth of HA on its surface. In the case of granulate obtained using EC, part of −OH silica groups is probably blocked by the binder that could lead to weakened ability of HA formation.
Figure 6 shows the XRD patterns of two forms of carrier. After immersion in the SBF, the XRD analysis showed that all samples produced new peaks demonstrating the surface crystallization. The most intense peaks occur at 2θ 32°, 26° that are 2 1 1 and 0 0 2 diffractions, respectively of the apatite (according to the standard JCPDS cards (09-0432)) (28). As apparent from the diffraction pattern, the content of crystalline phase decreases for sample of granulate as shown by previous studies FTIR (Fig. 5).

X-ray diffraction (XRD) patterns of a MCM-41 in the powder form and b MCM-41 in the granulate form after different days of mineralization studies in simulated body fluid compared with hydroxyapatite reference sample
The formation of HA deposition was further proved by SEM/EDS analysis. Figure 7 showed that the needle-like flower-shaped crystallites—characteristic for apatite—were deposited on the surface of these materials after soaking in SBF for 1 month. EDS analysis suggested that the Ca/P ratio of the accumulated forms on sample surface was 1.63–1.67, which was close to that of the biological hydroxyapatite (1.67) (27).

Scanning electron microscope (SEM) images and corresponding EDS spectra of a MCM-41 in the powder form and b MCM-41 in the granulate form after 1 month of mineralization studies in simulated body fluid
Drug Release
Due to its porous properties and mineralization activity, the MCM-41 materials can fulfill several functions: they can be either an osteoconductive material or an implanting carrier of the drug.
In this study, we measured the extent of ChD and MT release from two forms (powder and granulate) over a period of 10 h. In addition, ChD release was continued over the next 2 days. Figure 8 shows the cumulative release profiles of ChD and MT in SBF solution. In general, the release of MT both from powder and granulate form was faster than ChD. While release behavior during the whole period was similar that is two-step release profile, an initial fast release and relatively slow subsequent release was observed. Simultaneously, the process of formulating drugs changed the rate and extent of drug release. For the powder (variant (I) in Fig. 8) and granulate (variant (II) in Fig. 8), the first one provided the greater drug release: 98% MT after 3 h and 21% ChD after 10 h, whereas the granulate form achieved drug release of 98% MT after 10 h and 14% ChD after 10 h. A further release of ChD proceeded in a linear but slow manner. The cumulative amount of ChD released after 72 h from the powder and granulate forms was 63 and 46%, respectively.

Mean cumulative release of a ChD and MT (wt%) from the powder (I) and granulate (II) during the first 600 min of the experiment and b ChD (wt%) from the powder (I) and granulate (II) during the following 2 days of the experiment
Based on the Peppas model (Eq. (1)), the release exponent, n, for the all drug-loaded carriers was between 0.42 and 0.53. These values are close to 0.5 and indicate a diffusion-controlled release.
To explain the differences in release profile of both substances from the dosage forms used, the following factors should be considered: (i) the properties of drug particles (polymorphic state, particle size, and drug solubility), (ii) interaction of drug molecules with carrier, and (iii) carrier form.
Both substances were present in the carrier in amorphous form, as evidenced by the lack of XRD diffraction peaks derived from the crystals of the substance. This is consistent with the literature data (29–31) suggesting that the dispersion of substance in the solid vehicle prevents its re-crystallization. The amorphous form favors the improvement of substance solubility and thus should accelerate the diffusion of the drug from the carrier. Medicinal substances were used variously in terms of the size of the molecule. MT is much smaller; therefore, it will be more easily removed from the pores of the matrix as compared to ChD. Another reason for the different release profiles of medicinal substances from carriers lies in a diverse strength of interaction between the drug and the carrier surface. MT as a compound with four acceptor sites for hydrogen and one donor site for proton binds to the surface of silica by hydrogen bonds. ChD is characterized by 12 hydrogen acceptor counts and six hydrogen donor counts; therefore, it is strongly bound to the silica surface through a hydrogen bonding. In addition, as a hydrochloride, it binds with a strong ionic bond to nitrogen cation–oxygen anion (Fig. 9). Given these factors, it seems that the most significant effect on the release rate is attributed to the particle size and the ability to interact with the carrier. For these reasons, huge ChD molecules, which are strongly bound to the carrier surface, are hardly released from the pores of the carrier compared to MT. A separate issue is the drug dosage form used. In general, the release of drugs occurs faster from silica in the form of powder than from the granulate. When powder is dispersed in water solution, it absorbs water easily due to its hydrophilic and adsorptive properties. It affects rapid penetration of pores and channels of the carrier by water, causing easier dissolution of medicinal substances and faster release. In the case of granulate with hydrophobic binder (EC), when granulate is dispersed in water solution, it contacts with water and forms gel layer with high viscosity. The resulting gel layer acts as a diffusion barrier to delay or slow down the drug release.

Scheme of the drugs-carrier interactions
CONCLUSION
In this work, we used MCM-41 silica with ordered structure to obtain a bone substitute to be able to regenerate bone tissue and also dual drug-loaded delivery system (ChD and MT). The synthesized materials were obtained in the form of powder, and subsequently, they were granulated using ethanolic solution of EC as a binder. The porous carriers (powder and granulate) were characterized to evaluate their porosity and in vitro apatite-forming activity, essential in the design of the drug formulation for a bone implant.
Our results showed that the carriers obtained were ordered nanoporous with excellent adsorption properties and a biphasic release pattern of tested drugs, namely, ChD and MT. Interactions of the medicinal substance with surface of the carrier and the form of carrier determines the drug release rate. Faster release was observed for small MT molecules characterized by weaker interactions with carrier (98% MT after 3 or 10 h from powder and granulate, respectively), whereas ChD binds more permanently to the carrier releasing itself slower and more monotonic (17 and 12% ChD after 10 h from powder and granulate, respectively in the initial stage) reaching 63 and 46% release from powder and granulate after 72 h, respectively.
Our results on the drug release confirmed our assumptions of an appropriate choice of treatment substances and prediction of drugs’ interactions with carrier. It also confirmed the choice of the carrier in which the anti-inflammatory substance will be first released, thereby protecting the implant against recoil or infection, followed by the release of anti-tumor substances in the treatment of bone cancer.
Our experiment on in vitro bioactivity showed that MCM-41 powder showed good mineralization. The crystalline layer of hydroxyapatite had grown on the powder surface within a few days. Our results also revealed that in spite of surface modification by EC binder, carriers in granular form retained their mineralization properties.
All of these results suggest that the nanoporous silica powder and granulate form are attractive materials for filling bone defects due to their ability to form apatite layer and actively bond to the surrounding tissue. In addition, they might serve as drug release systems with the simultaneous advantages of protection of the implant against infection/recoil and treatment of cancerous lesions.
REFERENCES
Hench LL. Bioceramics: from concept to clinic. J Am Ceram Soc. 1991;74:1487–510.
Arcos D, Vallet-Regi M. Sol-gel silica-based biomaterials and bone tissue regeneration. Acta Biomater. 2010;6:2874–88.
Vallet-Regi M, Ragel CV, Salinas AJ. Glasses with application. Eur J Inorg Chem. 2003;6:1029–42.
Yan X, Yu C, Zhou X, et al. Highly ordered mesoporous bioactive glasses with superior in vitro bone-forming bioactivities. Angew Chem Int Ed Engl. 2004;43:5980–4.
Vallet-Regi M, Balas F, Arcos D. Mesoporous materials for drug delivery. Angew Chem Int Ed. 2007;46:7548–58.
Vallet-Regi M, Ramila A, Del Real RP, et al. A new property of MCM-41: drug delivery system. Chem Mater. 2001;13(2):308–11.
Li Z, Su K, Cheng B, et al. Organically modified MCM-type material preparation and its usage in controlled amoxicillin delivery. J Colloid Interf Sci. 2010;342:607–13.
Vadia N, Rajput S. Study on formulation variables of methotrexate loaded mesoporous MCM-41 nanoparticles for dissolution enhancement. Eur J Pharm Sci. 2012;45:8–18.
Tang Q, Xu Y, Wu D, et al. A study of carboxylic-modified mesoporous silica in controlled delivery for drug famotidine. J Solid State Chem. 2006;179:1513–20.
Mellaerts R, Aerts CA, Van Humbeeck J, et al. Enhanced release of itraconazole from ordered mesoporous SBA-15 silica materials. Chem Commun. 2007;13:1375–7.
Xia W, Chang J. Well-ordered mesoporous bioactive glasses (MBG): a promising bioactive drug delivery system. J Control Release. 2006;110:522–30.
Doadrio AL, Sousa EMB, Doadrio JC, et al. Mesoporous SBA-15 HPLC evaluation for controlled gentamicin drug delivery. J Control Release. 2004;97:125–32.
Vialpando M, Aerts A, Persoons J, et al. Evaluation of ordered mesoporous silica as a carrier for poorly soluble drugs: influence of pressure on the structure and drug release. J Pharm Sci. 2011;100:3411–20.
Vialpando M, Backhuijs F, Martens JA, et al. Risk assessment of premature drug release during wet granulation of ordered mesoporous silica loaded with poorly soluble compounds itraconazole, fenofibrate, naproxen, and ibuprofen. Eur J Pharm Biopharm. 2012;81:190–8.
Czarnobaj K. The design of nanostructured metronidazole-loaded HPC/oxide xerogel composites: influence of the formulation parameters on in vitro characterization. AAPS Pharm Sci Tech. 2015;16:1160–8.
Prokopowicz M. Bioactive silica-based nanomaterials for doxorubicin delivery: evaluation of structural properties associated with release rate. Mater Sci Eng C. 2013;33:3942–50.
Kokubo T, Kushitani H, Sakka S, et al. Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W. J Biomed Mater Res Part A. 1990;24(6):721–6.
Grün M, Lauer I, Unger KK. The synthesis of micrometer- and submicrometer-size spheres of ordered mesoporous oxide MCM-41. Adv Mater. 1997;9(3):254–7.
Prokopowicz M, Czarnobaj K, Szewczyk A, et al. Preparation and in vitro characterization of bioactive mesoporous silica microparticles for drug delivery applications. Med Sci Eng C. 2016;60:7–18.
Peppas N. Analysis of Fickian and non-Fickian drug release from polymers. Pharm Acta Helv. 1985;60(4):110–5.
Al-Oweini R, El-Rassy H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and RSi(OR)3 precursors. J Mol Struct. 2009;919(1–3):140–5.
Fidalgo A, Ilharco L. Correlation between physical properties and structure of silica xerogels. J Non-Cryst Solids. 2004;347(1–3):128–33.
Ciesla U, Schüth F. Ordered mesoporous materials. Micropor Mesopor Mat. 1999;27:131–49.
Vallet-Regi M. Mesoporous silica nanoparticles: their projection in nanomedicine. ISRN Materials Science. 2012;2012:1–20.
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone activity? Biomaterials. 2006;27(16):2907–12.
Vallet-Regi M, Arcos D. Mesoporous materials for biomedical applications. In: Pannone PJ, editor. Trends in biomaterials research. New York: Nova Science Publishers, Inc.; 2007. p. 109–42.
Ito A, Onuma K. Growth of hydroxyapatite crystals. In: Byrappa K, Ohachi T, editors. Crystal growth technology. New York: William Andrew Inc.; 2003. p. 525–48.
JCPDS Data Cards, International Centre for Diffraction Data, Pennsylvania. 1996.
Jackson CL, McKenna GB. Vitrification and crystallization of organic liquids confined to nanoscale pores. Chem Mater. 1996;8:2128–37.
Sliwinska-Bartkowiak M, Dudziak G, Grass R. Freezing behavior in porous glasses and MCM-41. Colloids Surf A: Physicochem Engin Aspects. 2001;187-188:523–9.
Godec A, Maver U, Bele M, et al. Vitrification from solution in restricted space: formation and stabilization of amorphous nifedipine in a nanoporous silica xerogel carrier. Int J Pharm. 2007;343:131–40.
ACKNOWLEDGEMENTS
This project was supported by the National Centre for Science of the Polish State (“OPUS” project: 2012/07/B/NZ7/04385).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Czarnobaj, K., Prokopowicz, M. & Sawicki, W. Formulation and In Vitro Characterization of Bioactive Mesoporous Silica with Doxorubicin and Metronidazole Intended for Bone Treatment and Regeneration. AAPS PharmSciTech 18, 3163–3171 (2017). https://doi.org/10.1208/s12249-017-0804-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-017-0804-3