
Abstract
Ritlecitinib, an orally available Janus kinase 3 and tyrosine kinase inhibitor being developed for the treatment of alopecia areata (AA), is highly soluble across the physiological pH range at the therapeutic dose. As such, it is expected to dissolve rapidly in any in vitro dissolution conditions. However, in vitro dissolution data showed slower dissolution for 100-mg capsules, used for the clinical bioequivalence (BE) study, compared with proposed commercial 50-mg capsules. Hence, a biowaiver for the lower 50-mg strength using comparable multimedia dissolution based on the f2 similarity factor was not possible. The in vivo relevance of this observed in vitro dissolution profile was evaluated with a physiologically based pharmacokinetic (PBPK) model. This report describes the development, verification, and application of the ritlecitinib PBPK model to translate observed in vitro dissolution data to an in vivo PK profile for ritlecitinib capsule formulations. Virtual BE (VBE) trials were conducted using the Simcyp VBE module, including the model-predicted within-subject variability or intra-subject coefficient of variation (ICV). The results showed the predicted ICV was predicted to be smaller than observed clinical ICV, resulting in a more optimistic BE risk assessment. Additional VBE assessment was conducted by incorporating clinically observed ICV. The VBE trial results including clinically observed ICV demonstrated that proposed commercial 50-mg capsules vs clinical 100-mg capsules were bioequivalent, with > 90% probability of success. This study demonstrates a PBPK model–based biowaiver for a clinical BE study while introducing a novel method to integrate clinically observed ICV into VBE trials with PBPK models. Trial registration: NCT02309827, NCT02684760, NCT04004663, NCT04390776, NCT05040295, NCT05128058.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alopecia areata (AA) is an autoimmune disease with an underlying immuno-inflammatory pathogenesis characterized by non-scarring hair loss of the scalp, face, and/or body (1,2,3). AA can have a substantial impact on quality of life (4, 5), including psychosocial burdens such as anxiety or depression (6, 7). There is an unmet need for a consistently effective treatment as effective therapeutic options for AA are currently limited (8). Ritlecitinib is an orally bioavailable, covalent inhibitor of Janus kinase 3 and the hepatocellular carcinoma family kinases (9, 10). The efficacy and safety of ritlecitinib was demonstrated in a phase 2b/3 trial (9). The US Food and Drug Administration (FDA) and the Japanese Ministry of Health recently approved ritlecitinib (LITFULO®), a once-daily oral treatment, for individuals 12 years of age and older with severe AA. The approved recommended dose for ritlecitinib is 50 mg. Currently, ritlecitinib is the first and only treatment approved by the FDA for adolescents with severe AA.
Ritlecitinib is a tosylate salt with a dissociation constant (pKa) of 4.85. Ritlecitinib is highly soluble across the physiological pH range at the therapeutic dose of 50 mg (intrinsic solubility 6.7 mg/mL). In vitro permeability showed high apparent passive permeability in MDCK-LE cells (11). Based on the highly soluble and highly permeable nature of the molecule, the oral absorption of ritlecitinib is characterized by rapid absorption with a peak plasma concentration time of < 1 h (10). Tablet formulation was mainly used during the development of ritlecitinib. In vitro dissolution data showed that the tablet disintegration and dissolution was very rapid (Pfizer data on file). Clinical data showed that the pharmacokinetics (PK) of the ritlecitinib tablet is similar to that of an oral solution (10).
The proposed commercial dosage of ritlecitinib is 50-mg capsules. Bioequivalence (BE) of the capsule formulation to clinical tablets was demonstrated using 100-mg capsules (NCT04390776). The 100-mg capsule was the highest capsule strength developed to account for the possibility of using doses higher than 50 mg for commercialization. Both 50- and 100-mg capsules were manufactured using a common blend (i.e., the two strengths were quantitatively proportional). As 50-mg capsules were not used for clinical BE study, in vitro dissolution data were generated to demonstrate similarity between the 50- and 100-mg capsules. Due to the highly soluble characteristics of ritlecitinib, an immediate-release (IR) solid dosage form of ritlecitinib is expected to dissolve rapidly in any in vitro dissolution conditions. However, in vitro dissolution data showed slower dissolution with 100-mg capsules. The f2 similarity analysis showed that dissolution rates were dissimilar between 50- and 100-mg capsules. Even though the 50-mg capsules were faster than 100-mg capsules, the above results could be interpreted as an inadequate bridge between the strengths and may require a clinical BE study to bridge 50- and 100-mg capsules based on FDA and EMA guidance (12, 13).
Physiologically based PK (PBPK) modeling has been utilized to predict absorption, distribution, metabolism, and excretion of chemical substances of interest. During a relatively long history of PBPK modeling, the area that PBPK models have been most used is drug-drug interactions (14). In recent years, PBPK modeling has been used for biopharmaceutical purposes where the model is used to assess the impact of drug substance or product attributes on oral absorption. The term PBBM (physiologically based biopharmaceutics) modeling has been introduced to combine PBPK absorption modeling with biopredictive dissolution testing for use in biopharmaceutics (15, 16). This recent progress has led to many applications of PBPK model–based virtual BE trials (PBPK/VBE) in the place of clinical BE studies (17,18,19). Assessing the credibility of PBPK models based on the context of use is an important element of establishing mechanistic PBPK models (20). Developed PBPK models need to be verified using observed clinical data to demonstrate that the model can describe oral absorption of molecule mechanistically and is sensitive enough to detect modified PK with pharmaceutical or physiological changes. The PBPK model can also assess the population (inter-subject) variability (21).
Aside from the validity of the model, another important parameter when a PBPK model is utilized to assess BE risk is intra-subject coefficient of variation (ICV), as BE studies are generally performed as crossover studies (22) and ICV is the key determinant of the 90% confidence intervals and the number of subjects required for BE studies (23). Within-subject variability (WSV) can be estimated and incorporated in PBPK/VBE trials using changes in physiology, particularly related gastrointestinal (GI) physiology (16). ICV differs from WSV in that it includes other random sources of variability and is a more clinically relevant source of variability.
However, the PBPK model–estimated WSV is not derived with physiologically relevant ranges, and estimated WSV values have not been extensively validated with a wide variety of drug products, which could lead to poor estimates of clinically observed variability and will impact any PBPK model–based BE risk assessment. This article describes a novel approach to conducting VBE trials in which the clinically observed ICV was posteriori integrated into the PK parameters of a large population of virtual subjects generated with a validated PBPK model. A large number of VBE trials were conducted by randomly sampling the population of virtual subjects at the specified sample size to conduct VBE trials to assess the risk of BE failure. The described methodology represents a significant advance in improving the statistical robustness of PBPK-based risk assessments of the bioperformance of drug products.
Materials and Methods
In Vitro Dissolution Experiment
In vitro dissolution experiments were conducted in accordance with the requirements outlined in the regulatory guidance (13). The experiments were conducted using USP Apparatus II (paddle) at 50 rpm with 500 mL of test media. Three capsule formulations (30, 50, and 100 mg) and variant capsule formulations used for clinical studies were tested in three different pH media (pH 1.2, 4.5, and 6.8). The measurement was taken with 12 replicates and relative standard deviation was reported. The f2 values were calculated comparing 30- vs 100-mg capsules and 50- vs 100-mg capsules.
Modeling Software
A ritlecitinib PBPK model was developed and verified using Simcyp version 22 release 1 (Certara, Sheffield, UK). The simulation analysis to conduct VBE with clinically observed ICV was conducted using R® version 4.1.3 (The R Foundation for Statistical Computing, Vienna, Austria).
PBPK Model Development
Physiological properties and relevant inputs are summarized in Table I. All the simulations were conducted using the Sim-healthy volunteer population. Absorption of ritlecitinib was modeled using the ADAM (Advanced Dissolution, Absorption, and Metabolism) model. The permeation of ritlecitinib was modeled with the mechanistic passive regional permeability predictor (MechPeff) within Simcyp. Effective concentration at the epithelial surface was set as a free aqueous concentration (unbound only) and transcellular permeability did not include transcellular ion permeation, Ptrans,0. Based on the logP of ritlecitinib (1.55), Ptrans,0 was estimated as 106 × 10−6 cm/s and jejunum1 Peff as 8.38 × 10−4 cm/s within the MechPeff model. The formulation type selected was either an IR solution or an IR solid formulation depending on the dosage form used in each clinical study. Solution data was modeled with the “solution with precipitation” option. Simcyp default values were used for precipitation parameters, critical supersaturation ratio (CSR), precipitation rate constant (PRC), and secondary precipitation rate constant (sPRC), as ritlecitinib is not expected to precipitate. Tablet formulation was modeled with the diffusion layer model (DLM) with experimentally measured particle size information as input.

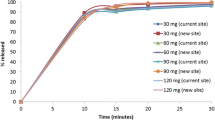
Capsule formulation dissolution profiles. a Variant capsule dissolution results in 500 mL 0.1 N hydrochloric acid (HCl) media, 50 rpm (left), and pH 6.8 media, 50 rpm (right). These profiles were used for Datasets 12, 13, and 14. b Capsule dissolution results in 500 mL 0.1 N HCl media, 50 rpm (left), and pH 6.8 media, 50 rpm (right). These profiles were used for Datasets 16, 18, 19, and 20. c Capsule dissolution results in 500 mL pH 4.5 media, 50 rpm. The 100-mg capsule data was used for Dataset 17
Although ritlecitinib is a high solubility drug (intrinsic solubility of 6.7 mg/mL and the highest therapeutic dose of 200 mg), in vitro dissolution data showed that capsule formulations exhibit a lag time and slower dissolution compared with the dissolution rate observed with tablet formulations. This is due to the slower disintegration of capsule formulations compared with tablet formulations, likely caused by the capsule shell material. To translate the slower disintegration of 100-mg capsule formulations to an in vivo oral absorption PK profile, the experimentally measured dissolution profile was entered as “dissolution profile” along with the experimentally determined relative standard deviation. The experimentally determined dissolution profile was entered as discrete for capsule formulations, and particle size data and solubility data were not input parameters for the capsule formulation simulation. For the tablet formulation modeling, D10, D50, and D90 values of the experimentally determined active pharmaceutical ingredient (API) particle size were entered and fitted to a log-normal distribution. The number of bins was set at 10. The Hintz-Johnson particle heff model was used to estimate the dissolution rate for tablet formulations with poly-dispersed particle size distribution.
Ritlecitinib total oral clearance was observed to be dose-dependent. The mechanism of this dose-dependent clearance is not clear. Therefore, clearance input was estimated from population PK analysis at each dose from the first-in-human single ascending dose study (24). A full PBPK distribution model using Method 1 (Poulin-Theil/Berezhkovskiy) was used for the volume of distribution setting. A tissue:plasma Kp scaler of 0.68 was applied to fit the clinically measured Vss of 1.0 L/kg, as without Kp scaler adjustment, the model did not predict the observed Vss.
PBPK Model Verification and Application
The established model was verified with multiple clinical studies (listed in Table II). The demographic input for model verification was the same age range and proportion of females used for each clinical study. A simulation was conducted with the same number of subjects in 10 trials. The predicted vs observed ratio between 0.8 and 1.25 is considered study-to-study variability and used as model verification acceptance criteria.
Sample Size Determination for VBE
The sample size was estimated using established ritlecitinib PBPK model–generated PK profiles of 5000 subjects for the 100-mg capsules, which are the reference formulation. PK concentrations were simulated at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 12, 16, and 24 h after the first dose with the same demographic information used for Simcyp VBE analysis. The sample size was determined prior to conducting any simulations for the 50-mg capsules. The non-compartmental analysis PK parameters for the 5000 PK profiles were calculated for the 100-mg capsules. PK parameters (area under the curve [AUC] and maximum concentration [Cmax]) were determined with the program written with R®. The sample size assessment was performed using the PK parameters for the reference 100-mg capsules only by duplicating the simulated PK parameters per subject as the test formulation. A difference between the duplicated dataset for the 100-mg capsules (test) and reference was implemented by multiplying the test dataset with factors of 0.9 and 0.95 to enable sample size assessment at assumed GMRs of 0.9 and 0.95. The virtual subjects from the test and reference datasets were randomly assigned to Sequence 1 (Period 1 = Treatment A; Period 2 = Treatment B) or Sequence 2 (Period 1 = Treatment B; Period 2 = Treatment A). The ICV values of 24% and 12% for Cmax and AUCinf were determined from a clinical study where 100-mg capsules were dosed in 12 subjects (NCT04004663). ICV was derived from the mean square error (MSE), which is obtained from a mixed effect model with treatment, sequence, and period as fixed effects and subject within the sequence as a random effect. The clinically observed ICVs for AUC and Cmax were simulated assuming a log-normal distribution and randomly assigned to each subject for Period 2. A total of 1000 VBE trials were simulated by randomly sampling the specified sample size (N) of subjects from the pool of 5000 subjects without replacement. The GMR and 90% CI of AUC and Cmax for each trial were calculated using linear mixed-effect analysis. Sample sizes (N) tested were 12, 16, 24, 36, 48, 56, and 60 for a GMR of 0.90, and 16, 24, and 36 for a GMR of 0.95. The probability of BE success at each sample size was calculated as the number of trials that met the BE criteria of 90% CI for GMR within 80–125%. The sample size for the VBE trial was fixed to an N value that met the prespecified criteria that > 90% of the simulated trials meet the BE criteria. A schematic for the conduct of this simulation is provided in Fig. 2. The sample size determined a priori using the described method above was also used for the Simcyp-based PBPK VBE.

Schematic for implementation of ICV to PBPK-based VBE. For sample size assessment, the above schematic was repeated for each evaluated value of sample size (N = 12, 16, etc) and probability of success of BE was calculated with results collated as shown in Fig. 5. For the final VBE, the assessment was conducted once for each PK parameter at the prespecified sample size
PBPK VBE Analysis
The bioequivalence of two capsule formulations (50 and 100 mg) was assessed using the Simcyp VBE module. Two separate compound files for the 100- and 50-mg capsules were generated using each dissolution profile. A population of 24 healthy adults with a male and female proportion of 0.5 and aged between 18 and 55 years old was generated from a virtual population library of healthy subjects. The VBE was conducted at the 100-mg dose in 10 trials with a crossover study design with two treatments, two periods, and two sequences (T1T2/T2T1). WSV input is summarized in Table III. The within-subject variability factors were selected to cover a range of GI physiological variabilities based on the reference by Bego et al (16).
VBE with Clinically Observed ICV
VBE between 100- and 50-mg capsules by including clinically observed ICV was conducted using the methodology described under sample size determination and Fig. 2 for VBE with the prespecified sample size. PBPK model–generated PK profiles of 5000 subjects for the 100-mg capsules (reference) and 2 × 50-mg capsules (test) were used for VBE assessment. The criteria for declaring BE were prespecified to be the same as those for sample size determination (i.e., probability of BE success of ≥ 90%). The R scripts are provided in the Supplement Material section.
Results
In Vitro Dissolution
The dissolution profiles for ritlecitinib capsule formulations, including variant formulations used in a clinical relative bioavailability study, are shown in Fig. 1. Two variant formulations were developed and tested in the clinic: (1) a 100-mg capsule with large particle size API and (2) an over-encapsulated reference 100-mg capsule. The particle size of the reference capsule was D50 of 37.5 μm, whereas a capsule with the large particle size API had D50 of 237 μm. Because ritlecitinib is highly soluble when 100 mg is dissolved in 500 mL of media, large particle size API did not slow down the dissolution rate compared with the reference 100-mg capsules. The dissolution rate of the over-encapsulated capsule (100 mg, size 1 in size 0 capsule shells) was slower in both pH 1.2 and 6.8 media (Fig. 1a).
The dissolution profile of 30-, 50-, and 100-mg capsules in 0.1 N hydrochloric acid (HCl) and pH 6.8 media and in pH 4.5 media are shown in Fig. 1b and c, respectively. The results showed that the dissolution profile of the 100-mg capsule had a longer lag time compared with 30- and 50-mg capsules. The f2 values from the comparison of 30- vs 100- and 50-mg capsules vs 100-mg capsules are summarized in Supplement Table S1, including the results of the f2 bootstrap. The f2 similarity analysis showed that the dissolution profiles between 30- and 100-mg capsules and between 50- and 100-mg capsules were dissimilar.
PBPK Model Verification
A single-dose oral administration of ritlecitinib in various formulations (solution, tablet, and capsule formulations) was used to verify the ritlecitinib absorption PBPK model. First, the Simcyp ritlecitinib PBPK model was verified using observed PK data obtained following single oral doses of 5 to 800 mg with a solution formulation in healthy male subjects (NCT02309827). The predicted to observed ratios were within ranges of 0.95 to 1.70 for AUCinf and 0.95 to 1.35 for Cmax (Dataset 1 to 7; Table II). Overall, using the oral clearance values obtained from the population PK analysis at various doses, the ritlecitinib PBPK model predicted the oral PK parameters and dose proportionality reasonably well (Fig. 3). Although the solution was modeled with “solution with precipitation,” the model predicted that there was no precipitation since ritlecitinib is a highly soluble drug.

Dose proportionality. Datasets 1, 2, 3, 4, 5, 6, and 7 from a single-dose-ascending study. Ritlecitinib was dosed as a solution at various doses under the fasted condition
The food effect of ritlecitinib was evaluated using tablet formulation at 50 mg (Datasets 9 and 10). Clinical data showed that a high-fat meal decreased Cmax by approximately 39% with no meaningful change of AUCinf (102.3%) when dosed in 50-mg tablets (NCT02684760). The model predicted a similar reduction in Cmax with food intake with tablet formulations (Tables II, IV; Datasets 9 and 10). The clinical data showed that the Cmax was reduced when 100-mg capsules were dosed under the fed condition without significant changes in AUCinf (Tables II and IV; Datasets 16 and 17). With the 100-mg capsule formulation, the predicted fed-to-fasted mean Cmax ratio was 56%, whereas the observed ratio was 67.83% (90% CI 54.08%, 85.08%; NCT04390776). Using pH 4.5 dissolution data as the stomach dissolution profile, the model predicted reduced Cmax with food intake with capsule formulation. Overall, these results suggested that the ritlecitinib PBPK model can predict tablet formulation performance mechanistically to predict observed food effect (Table IV). This result indicated that the model settings for permeation and GI physiological condition are appropriate to predict the PK performance of ritlecitinib with various conditions (fasted vs fed) and formulations (solution vs tablets).
For capsule formulation simulations, experimentally determined dissolution profiles in 0.1 N HCl media and pH 6.8 media were used to model capsule formulations for stomach and small intestinal dissolution profiles, respectively. The verification results demonstrated that the model was able to predict the observed ritlecitinib 100-mg capsules (Table II; Datasets 12 and 16), 30-mg capsules (Table II; Dataset 18), and 50-mg capsules (Table II; Datasets 19 and 20) under the fasted condition. Moreover, slower dissolution variant formulations were also modeled using experimentally determined dissolution data. Large particle size API did not significantly change the dissolution profiles in both 0.1 N HCl and pH 6.8 media. The model predicted no change in the PK, which was aligned with the observed clinical data (Table II; Dataset 13). Over-capsulated capsules slowed the dissolution profile (Fig. 1), and the model predicted a slight reduction in Cmax. Although clinical data showed no changes in Cmax, the predicted to observed ratios for Cmax were within the acceptance criteria of 0.8 to 1.25. Overall, the results confirmed that the model can predict ritlecitinib capsule performance using in vitro dissolution profiles (Fig. 4).

Simcyp predicted vs observed ritlecitinib PK profiles following single doses of capsule formulations. a One-hundred-milligram capsules (Dataset 12). b Thirty-milligram capsules (Dataset 18). c Fifty-milligram dose with a single 50-mg capsule (Dataset 19). d Two-hundred-milligram dose with four 50-mg capsules (Dataset 20). Prediction: green line = mean; gray lines = 5% and 95% percentiles
Sample Size Analysis
The probability of demonstrating BE at a given assumed GMR and sample size of N is shown in Fig. 5a. As Cmax has a larger ICV, the sample size was determined using the probability of VE success for Cmax. For Cmax for a GMR of 0.95, a sample size of 24 subjects achieved a probability of BE success of 90%. For a GMR of 0.90, a sample size of 48 would be needed to achieve a probability of BE success of 90%. For AUC for both the above conditions, the probability of BE success is > 90%. The distribution of the GMR at various sample sizes was assessed (Fig. 5b). Based on these results, a sample size of 24 was selected for the subsequent BE trial simulation for the 100-mg capsules vs two 50-mg capsules.

Sample size assessment. a Probability of success for AUC (left) and Cmax (right) with various sample sizes (N). b Geometric mean ratio (magenta) and upper (green) prediction interval (UPI) and lower (blue) prediction interval (LPI) for AUC (left) and Cmax (right)
PBPK Virtual BE Analysis
Virtual BE trials were conducted using the Simcyp VBE module with predicted WSV to compare the BE between 50- and 100-mg capsules. The trials were conducted with 24 subjects in 10 trials. The results demonstrated that 50-mg capsules were bioequivalent to 100-mg capsules, with a 100% probability of success (Fig. 6a). The average predicted intra-subject variability of 10 VBE trials was 7.4% for Cmax and 1.6% for AUCinf. This predicted estimate of intra-subject variability was much smaller than clinically observed ICV values of 24% and 12% for Cmax and AUCinf, respectively.

VBE output. a Simcyp virtual bioequivalence (VBE) output comparing 50- and 100-mg capsules dosed at 100 mg (2 × 50-mg capsules vs 1 × 100-mg capsule) under the fasted condition. The VBE analysis was conducted with 24 subjects in 10 trials. b VBE analysis with clinically observed ICV with R programming. The trials were conducted with 24 subjects in 1000 trials. The black solid line represents the point estimate. The red dotted line represents the lower confidence interval. The green dotted line represents the upper confidence interval
Virtual BE with Clinical ICV
To make the BE risk assessment more credible, clinically observed ICV was added to Simcyp-generated PK parameters. The results demonstrated that the probability of BE success for AUCinf and Cmax were 100% and 97.5%, respectively. As shown in Fig. 6b, the estimated GMRs for 100- and 50-mg capsules were 0.98 and 0.99 for AUCinf and Cmax, respectively. The comparison of two approaches (PBPK VBE with ICV vs. only PBPK-based VBE) for VBE trials indicated that using clinically observed ICV provides a more realistic BE assessment with all sources of clinical variability accounted.
Discussion
A ritlecitinib absorption PBPK model was implemented to predict the PK performance of ritlecitinib. The model was developed with available in vivo human oral clearance values for each dose determined with population PK analysis. The absorption part of the model was built only with parameters based on physiological principles and in vitro measured values. Model verification was performed by comparing the predicted and observed clinical data from various single-dose PK studies with various immediate-release oral dosage forms (solution, tablets, and capsules) and the effect of food on PK profiles. Capsule formulations were modeled with experimentally determined dissolution data to evaluate the impact of lag time and dissolution rate on ritlecitinib PK performance. Model verification results indicated that the model input parameters were of sufficient confidence to enable the prediction of capsule formulations in the fasted state using in vitro dissolution data for doses between 30 and 200 mg.
Of particular note, the clearance mechanism of ritlecitinib is complex (25). In vivo metabolism studies indicated that the primary metabolic pathways for the elimination of ritlecitinib were related to glutathione conjugation and cytochrome P-450 (CYP) oxidation (26). In vitro reaction phenotyping in recombinant human glutathione transferase enzyme indicated that multiple cytosolic and microsomal isoforms were involved in the metabolism of ritlecitinib. In vitro reaction phenotyping in hepatocytes indicated metabolism primarily by CYP3A4, followed by lesser contributions of CYP2C8, 1A2, and 2C9. In a clinical study, a marginal increase in the exposure of ritlecitinib was observed with itraconazole coadministration, indicating that CYP3A4 has a minor role in the metabolism of ritlecitinib. Collectively, multiple glutathione transferase and CYP metabolic enzymes are involved in the metabolism of ritlecitinib, with no single major enzyme pathway. While the mechanism is not clear, and is likely related to pathway saturation, total systemic clearance (CL) of ritlecitinib was observed to be dose-dependent. To avoid the complexity of CL mechanisms involved in ritlecitinib disposition, estimated CL values at each dose based on population PK analysis were incorporated into the current ritlecitinib PBPK model to evaluate the absorption of two capsule formulations.
Because ritlecitinib is a highly soluble and highly permeable drug, the rate of absorption was expected to be controlled by the GI transit time. Therefore, a PBPK VBE trial was first attempted by adding the variability in physiological parameters including gastric emptying time to WSV. However, this approach did not predict the clinically observed ICV. Simcyp predicted intra-subject variability of 7.4% for Cmax and 1.6% for AUCinf, whereas the clinically observed ICV was 24% and 12% for Cmax, and AUCinf, respectively. Instead of including additional parameters or readjusting the range of WSV input empirically to capture the observed variability, a novel approach was taken to incorporate the observed ICV into a PBPK model simulated PK profile to conduct VBE trials. One of the PBPK simulation tools, GastroPlus (Simulations Plus), provides the feature to enter ICV values as a user input (18). However, the tool does not provide or describe a method for sample size analysis based on known ICV nor does it provide criteria for interpretation or conclusions. In this study, a novel hybrid approach to conduct VBE by integrating the PBPK model generated PK with prior knowledge of clinically observed ICV was introduced along with criteria for interpretation and conclusions. The first critical step of this unique approach was to determine the sample size based on the observed ICV using the reference formulation alone. This approach approximates the design and conduct of clinical BE trials.
It is important to recognize the difference between WSV or intra-subject variability and ICV. WSV is the dose-to-dose variability within a subject and can specifically only be calculated from replicate design studies. Traditionally biopharmaceutic studies, especially estimation-type relative bioavailability studies, are run as crossover non-replicated designs where each formulation is only tested once. Analysis of crossover non-replicated study designs usually yields an estimate of random variability, which is referred to as ICV. The estimated ICV from crossover (non-replicated) study designs includes WSV in absorption, distribution, metabolism, and excretion (ADME) of the drug (physiologic variability in the subject), analytical variability, within-formulation variability, subject-by-formulation interaction, and unexplained random variability.
Although the ICV derived from crossover non-replicated design biopharmaceutic studies may not be able to identify the individual sources of variability, it represents a practical estimate of expected random variability within a subject for calculating sample sizes of BE trials, which are typically designed as two-sequence, two-period crossover studies. Simulation studies using mixed effect analysis have suggested that ICVs derived from non-replicated studies are a good estimate for WSV under certain conditions (27). Hence, when evaluating or assessing BE using PBPK models utilizing the traditional 2 × 2 crossover designs, it is important to account for random variability in the parameter estimates. This should be done even when the relative contribution of the individual sources of variability is not known, as it represents a more conservative approach and accounts for all the sources of variability in a composite manner. If the random variability observed in clinical trials is not implemented in PBPK models, it may lead to unrealistic expectations for equivalency between formulations, especially when the true GMR deviates beyond the range of 0.95 to 1.05.
VBE trials with observed ICV were used to evaluate the BE between 50- and 100-mg ritlecitinib capsules. In this study, the virtual simulations were conducted with 1000 trials by randomly sampling subjects (N) from a pool of 5000. These numbers were chosen to represent a large enough pool of study participants to sample from, to enable constructing a distribution of outcomes for objective assessment of the risk of BE failure. The analysis results demonstrated that, although in vitro dissolution data showed dissolution profiles were dissimilar, the BE assessment with ICV demonstrated that the 50-mg capsules were bioequivalent to the 100-mg capsules. A key assumption of the described methodology for implementing clinically observed ICVs to PBPK-based VBE assessments is that the test and reference formulations have similar ICVs. For IR formulations, this is a reasonable assumption as random sources of variability are unlikely to differ between 2 IR formulations especially when both formulations are rapidly disintegrating and dissolving. Nevertheless, the VBE conducted with this method by incorporating ICV is a pragmatic way of replicating in a virtual setting exactly how a clinical bioequivalence trial would be conducted. Through the power of simulation, instead of conducting the trial once as what would be done for a clinical bioequivalence study, we are conducting a large number of virtual BE trials with prespecified acceptance criteria of >90%, which provides an objective risk assessment and high confidence for declaring two products bioequivalent.
Conclusion
A verified PBPK model for ritlecitinib was developed and successfully applied to predict the oral absorption of solution, tablet, and capsule formulations including food effects. Although in vitro dissolution data showed that the dissolution profiles were dissimilar, the VBE assessment with ICV demonstrated that the 50-mg capsules were bioequivalent to the 100-mg capsules. Finally, this study demonstrated the value of using a novel approach to conduct VBE trials with greater statistical rigor.
Data Availability
Data are available upon reasonable request.
Change history
21 February 2024
A Correction to this paper has been published: https://doi.org/10.1208/s12248-024-00895-w
References
Villasante Fricke AC, Miteva M. Epidemiology and burden of alopecia areata: a systematic review. Clin Cosmet Investig Dermatol. 2015;8:397–403. https://doi.org/10.2147/ccid.S53985.
Ramírez-Marín HA, Tosti A. Evaluating the therapeutic potential of ritlecitinib for the treatment of alopecia areata. Drug Des Devel Ther. 2022;16:363–74. https://doi.org/10.2147/dddt.S334727.
Islam N, Leung PS, Huntley AC, Gershwin ME. The autoimmune basis of alopecia areata: a comprehensive review. Autoimmun Rev. 2015;14(2):81–9. https://doi.org/10.1016/j.autrev.2014.10.014.
Liu LY, King BA, Craiglow BG. Alopecia areata is associated with impaired health-related quality of life: a survey of affected adults and children and their families. J Am Acad Dermatol. 2018;79(3):556-8.e1. https://doi.org/10.1016/j.jaad.2018.01.048.
Aldhouse NVJ, Kitchen H, Knight S, Macey J, Nunes FP, Dutronc Y, et al. “‘You lose your hair, what’s the big deal?’ I was so embarrassed, I was so self-conscious, I was so depressed:” a qualitative interview study to understand the psychosocial burden of alopecia areata. J Patient Rep Outcomes. 2020;4(1):76. https://doi.org/10.1186/s41687-020-00240-7.
Colón EA, Popkin MK, Callies AL, Dessert NJ, Hordinsky MK. Lifetime prevalence of psychiatric disorders in patients with alopecia areata. Compr Psychiatry. 1991;32(3):245–51. https://doi.org/10.1016/0010-440x(91)90045-e.
Vallerand IA, Lewinson RT, Parsons LM, Hardin J, Haber RM, Lowerison MW, et al. Assessment of a bidirectional association between major depressive disorder and alopecia areata. JAMA Dermatol. 2019;155(4):475–9. https://doi.org/10.1001/jamadermatol.2018.4398.
Pratt CH, King LE, Messenger AG, Christiano AM, Sundberg JP. Alopecia areata. Nat Rev Dis Primers. 2017;3(1):17011. https://doi.org/10.1038/nrdp.2017.11.
King B, Zhang X, Harcha WG, Szepietowski JC, Shapiro J, Lynde C, et al. Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b–3 trial. The Lancet. 2023;401(10387):1518–29. https://doi.org/10.1016/S0140-6736(23)00222-2.
Eisman S, Sinclair R. Ritlecitinib: an investigational drug for the treatment of moderate to severe alopecia areata. Expert Opin Investig Drugs. 2021;30(12):1169–74. https://doi.org/10.1080/13543784.2021.2012149.
Thorarensen A, Dowty ME, Banker ME, Juba B, Jussif J, Lin T, et al. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) Allowing for the interrogation of JAK3 signaling in humans. J Med Chem. 2017;60(5):1971–93. https://doi.org/10.1021/acs.jmedchem.6b01694.
Guidance for industry. Bioavailability and bioequivalence studies submitted in NDAs or INDs - general considerations. Center for Drug Evaluation and Reseach (CDER) 2014.
Guideline of the Investigation of Bioequivalence. Committee for Medicinal Products for Human Use (EMA) 2010.
Yuvaneshwari K, Kollipara S, Ahmed T, Chachad S. Applications of PBPK/PBBM modeling in generic product development: an industry perspective. J Drug Deliv Sci Technol. 2022;69:103152. https://doi.org/10.1016/j.jddst.2022.103152.
Bermejo M, Hens B, Dickens J, Mudie D, Paixão P, Tsume Y, et al. A mechanistic physiologically-based biopharmaceutics modeling (PBBM) approach to assess the in vivo performance of an orally administered drug product: from IVIVC to IVIVP. Pharmaceutics. 2020;12(1):74.
Bego M, Patel N, Cristofoletti R, Rostami-Hodjegan A. Proof of concept in assignment of within-subject variability during virtual bioequivalence studies: propagation of intra-subject variation in gastrointestinal physiology using physiologically based pharmacokinetic modeling. AAPS J. 2022;24(1):21. https://doi.org/10.1208/s12248-021-00672-z.
Wu D, Sanghavi M, Kollipara S, Ahmed T, Saini AK, Heimbach T. Physiologically based pharmacokinetics modeling in biopharmaceutics: case studies for establishing the bioequivalence safe space for innovator and generic drugs. Pharm Res. 2023;40(2):337–57. https://doi.org/10.1007/s11095-022-03319-6.
Wu D, Li M. Current state and challenges of physiologically based biopharmaceutics modeling (PBBM) in oral drug product development. Pharm Res. 2023;40(2):321–36. https://doi.org/10.1007/s11095-022-03373-0.
Loisios-Konstantinidis I, Cristofoletti R, Fotaki N, Turner DB, Dressman J. Establishing virtual bioequivalence and clinically relevant specifications using in vitro biorelevant dissolution testing and physiologically-based population pharmacokinetic modeling. case example: naproxen. Eur J Pharm Sci. 2020;143:105170. https://doi.org/10.1016/j.ejps.2019.105170.
Kuemmel C, Yang Y, Zhang X, Florian J, Zhu H, Tegenge M, et al. Consideration of a credibility assessment framework in model-informed drug development: potential application to physiologically-based pharmacokinetic modeling and simulation. CPT Pharmacomet Syst Pharmacol. 2020;9(1):21–8. https://doi.org/10.1002/psp4.12479.
Wedagedera JR, Afuape A, Chirumamilla SK, Momiji H, Leary R, Dunlavey M, et al. Population PBPK modeling using parametric and nonparametric methods of the Simcyp Simulator, and Bayesian samplers. CPT Pharmacometrics Syst Pharmacol. 2022;11(6):755–65. https://doi.org/10.1002/psp4.12787.
Hauschke D, Steinijans WV, Diletti E, Schall R, Luus HG, Elze M, et al. Presentation of the intrasubject coefficient of variation for sample size planning in bioequivalence studies. Int J Clin Pharmacol Ther. 1994;32(7):376–8.
Wada S, Kagatani S, Nakagami H. Quantitative estimation of intra-subject variability in bioequivalence studies of generic solid oral dosage forms by multiple regression analysis. J Drug Del Sci Technol. 2021;64:102656. https://doi.org/10.1016/j.jddst.2021.102656.
Wojciechowski J, Purohit VS, Huh Y, Banfield C, Nicholas T. Evolution of ritlecitinib population pharmacokinetic models during clinical drug development. Clin Pharmacokinetics. 2023;62(1765):1779. https://doi.org/10.1007/s40262-023-01318-3.
Leung L, Yang X, Strelevitz TJ, Montgomery J, Brown MF, Zientek MA, et al. Clearance prediction of targeted covalent inhibitors by in vitro-in vivo extrapolation of hepatic and extrahepatic clearance mechanisms. Drug Metab Dispos. 2017;45(1):1–7. https://doi.org/10.1124/dmd.116.072983.
Liu J, Solan R, Wolk R, Plotka A, O’Gorman MT, Winton JA, et al. Evaluation of the effect of ritlecitinib on the pharmacokinetics of caffeine in healthy participants. Bri J Clin Pharmacol. 2023;89(7):2208–15. https://doi.org/10.1111/bcp.15695.
Kang WH, Lee JY, Chae JW, Lee KR, Baek IH, Kim MS, et al. Population pharmacokinetic method to predict within-subject variability using single-period clinical data. Pharmaceuticals (Basel). 2021;14(2):114. https://doi.org/10.3390/ph14020114.
Acknowledgements
The authors appreciate the help of Kieran Smith and Marilia Alvarenga De Oliveira for generating dissolution data. The authors also appreciate the many Pfizer colleagues for their input and discussion.
Funding
This study was sponsored by Pfizer Inc., New York, NY, USA.
Author information
Authors and Affiliations
Contributions
K. S., A. S., and V. P. wrote the manuscript. K. S., A. S., M. D., Y. H., and V. P. designed the research. K. S., A. S., M. D., Y. H., and V. P. conducted the research. K. S., A. S., and V. P. analyzed data. A. M. generated the dissolution data. M. W. formulated the ritlecitinib capsules. Y. H. and V. P. managed the clinical studies. All authors reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
All authors are employees of Pfizer Inc., New York, NY, USA, and may hold stock or stock options.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The PDF version has been corrected to replace the Figure 4 with the correct image. This error was introduced in production and not the fault of the authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saadeddin, A., Purohit, V., Huh, Y. et al. Virtual Bioequivalence Assessment of Ritlecitinib Capsules with Incorporation of Observed Clinical Variability Using a Physiologically Based Pharmacokinetic Model. AAPS J 26, 17 (2024). https://doi.org/10.1208/s12248-024-00888-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-024-00888-9