
Abstract
Qualifying critical reagents in ligand binding assays by parallel testing of current and candidate reagent lots is recommended by regulatory agencies and industry groups, but specific guidance on the format of reagent qualification experiments is limited. Equivalence testing is a statistically sound approach that is consistent with the objective of critical reagent qualification. We present power analysis for equivalence regions ranging from 1.25- to 1.5-fold multiples of the GM ratio (centered on 1) of current and candidate lots, over a range of assay variability from 5 to 30% coefficient of variation (CV). A 1.25-fold equivalence region can be tested using 6 to 12 plates per lot for assays with up to 15% CV but is not practical for more variable assays. For these assays, wider equivalence regions are justified so long as care is taken to avoid assay drift and the assay remains suitable for the intended use. The equivalence test method is illustrated using historical data from passing and failing reagent qualification experiments. Simulation analysis was performed to support the design of qualification experiments using 6, 12, or 18 plates per lot over a broad range of assay variability. A challenge in implementing the equivalence test approach is selecting an appropriate equivalence region. Equivalence regions providing 90% power using 12 plates/lot were consistent with 1.5σ bounds, which are recommended for equivalence testing of critical quality attributes of biosimilars.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Guidance documents from the International Congress on Harmonization (1, 2), regulatory agencies (3,4,5), and industry groups (6, 7) recommend qualifying critical reagents in ligand binding assays to ensure that the candidate reagent performs similarly to the original reagent in the assay. Specific guidance, however, on the format of the reagent qualification experiment and acceptance criteria is limited. If the reagent change is not expected to influence the assay, a single comparative accuracy and precision assessment may be sufficient for characterization, while additional validation experiments are recommended for major changes (2). Direct comparison of new and old lots in parallel is recommended (2, 6, 7). At minimum, one run with three control levels for minor changes in reagents or three runs with three or five control levels for major changes are recommended by industry groups (6, 7). These groups also concur with O’Hara et al. (8, 9) in recommending that a priori acceptance criteria be established.
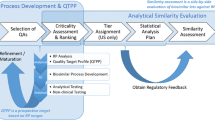
Our lab has developed, qualified, and validated multiple ligand binding immunoassays, including enzyme-linked immunosorbent assays (ELISAs) that measure anti-glycoprotein (GP) immunoglobulin G (IgG)-binding antibodies to Ebola virus (10, 11), Sudan virus (12), and Marburg virus (MARV) (12). These assays are used in multiple studies over an extended period of time. To ensure comparability of assay results between studies and over time, we use a standard method to qualify new lots of critical reagents (e.g., coating antigen, conjugate, plate lot, substrate) that is more rigorous than the minimum experiments recommended by industry groups. Our original qualification experiment used a “sister plate” design (described below) and acceptance criteria based on the mean percent difference in measured concentration for samples tested on plates that were identical except for the reagent being qualified. The experimental design was adapted from an earlier qualification design for an anthrax anti-protective antigen (PA) IgG ELISA with similar performance characteristics (13, 14) but a different plate layout with fewer test samples on each plate. This approach was used for the qualification of two candidate lots of recombinant glycoprotein (rGP) coating antigen in the anti-MARV GP IgG ELISA. These qualification experiments were used to motivate and illustrate a new reagent qualification strategy that uses equivalence test methodology to show that the candidate lot of rGP coating antigen is equivalent to the current lot when used in the assay.
Briefly, our original qualification strategy was as follows. Twelve ELISA plates were prepared as six pairs of “sister plates” consisting of one plate coated with the current lot of rGP and one plate coated with the candidate lot of rGP. Each pair of sister plates was otherwise identical, using the same reference standard, quality control, and test sample (TS) preparations. The sister plates were prepared by three independent test operators (two plates per operator) over multiple days. System suitability and sample suitability criteria were applied as usual. The percent difference for each TS was calculated using the mean results calculated over the six plates from each lot, and each TS was required to have a percent difference in means of less than or equal to 15%. If the acceptance criteria were met, the candidate lot was accepted with no further testing. If the acceptance criteria were not met, then per protocol the qualification was repeated two more independent times to confirm the failure. If the failure was not confirmed, then the passing results from the two independent runs were used to support qualification with acknowledgment of the initial failure.
Qualification experiments were performed for two lots of rGP coating antigen (MARV Ci67 Lot 29APR16 and MARV Angola Lot 13JAN17) for use in the anti-MARV GP IgG ELISA. Both qualifications compared a candidate lot of rGP coating antigen to the current lot of rGP (MARV Ci67 Lot 13DEC2013). Despite known differences between isolates, the MARV Angola rGP coating antigen was tested in the immunoassay to evaluate whether the assay could be modified to use the MARV Angola isolate, which had been used for vaccine development. In each case, the experiment was performed three times before the candidate lot was accepted (MARV Ci67 Lot 29APR16) or failed (MARV Angola Lot 13JAN17). Examination of the failed qualification of MARV Angola Lot 13JAN17 indicated that there was a strong correlation between the new and old coating antigens, but there was a shift in the reportable values for the failed lot that was not present in the passing lot. This outcome was anticipated. The need for replication before eventually accepting MARV Ci67 Lot 29APR16, however, was not expected, and the applicability of the qualification study design and acceptance criteria to the anti-MARV GP IgG ELISA were questioned following these qualification experiments. It appeared that the qualification acceptance criteria may have been too stringent given the number of test samples per plate for the anti-MARV GP IgG ELISA (ten TSs per plate with all ten required to pass) compared to the anti-PA IgG ELISA (four TSs per plate with all four required to pass) so that the first round of testing for MARV Ci67 Lot29APR16 failed. In addition, the original study design did not include enough test replicates to obtain sufficiently precise estimates to meet the 15% difference acceptance criterion for the anti-MARV GP IgG ELISA.
To address these issues, the critical reagent qualification experiment was redesigned to use equivalence testing as a more rigorous statistical approach that is consistent with the objective of the experiment, along with sample size and acceptance criteria that ensure the experiment is adequately powered. An advantage of the equivalence test approach is that both the mean and variability of the assay are accounted for in the analysis. Equivalence testing is based on two one-sided t-tests (TOST) (15,16,17,18) and is typically evaluated using a 90% confidence interval for the average difference or ratio between the reportable values for two test conditions, in this case, two lots of rGP coating antigen. The 90% confidence interval is compared to a predefined equivalence region and is expected to be contained wholly within the equivalence region in order for the lots to be considered sufficiently equal. If any portion of the 90% confidence interval falls outside the equivalence region, the lots are not considered to be equivalent. Historical data from the Ci67 vs Ci67 qualification experiments were used to demonstrate a model-based approach to estimating 90% confidence intervals for the TOST equivalence test method. The experimental design was developed using both traditional power analysis and simulation-based power analysis. Equivalence region–based power and sample size are acceptable in some contexts (19, 20) but do not necessarily ensure that the assay remains fit for purpose. Therefore, equivalence regions based on FDA guidance for equivalence testing of critical quality attributes of biosimilars (21,22,23,24) were computed and compared to those developed using simulation-based power analysis.
Materials and Methods
Traditional Power Analysis
A power analysis was conducted to determine appropriate sample sizes (number of tests per lot) for TOST equivalence tests assuming a nominal geometric mean (GM) ratio of one, which is consistent with perfect equivalence for a lognormal distribution, between current and candidate reagent lots. It was assumed that GM ratios of 0.95 to 1.05 between lots would be within an acceptable margin of equivalence. For simplicity, it was assumed that independent tests of a single test sample would be performed using the current and candidate lots. The number of independent tests per lot required to achieve 80% power was calculated for equivalence regions ranging from 1.25- to 1.5-fold multiples of the GM ratio (centered on one), over a range of assay variability from 5 to 30% coefficient of variation (CV). The equivalence regions and nominal ratio were selected to be compatible with the current 15% difference in means acceptance criterion while accommodating normal assay variability in the confidence interval. The upper bound of the equivalence region corresponds to the fold-change multiplier, and the lower bound is calculated as the reciprocal of the upper bound. The power analysis was generated using the SAS software version 9.4, POWER procedure (SAS Institute, Cary, NC).
Equivalence Test Method
Previously collected data from “sister plate” qualification experiments (described above) performed for two candidate lots of rGP coating antigen (MARV Ci67 Lot 29APR16 and MARV Angola Lot 13JAN17) using a qualified human anti-MARV GP IgG ELISA that has since been validated (12) were used to demonstrate the equivalence test approach for reagent qualification. Up to ten samples can be tested on each plate in the anti-MARV GP IgG ELISA. For each candidate lot, qualification experiments were performed using sets of ten test samples. Random samples of 6, 12, and 18 plates coated with the candidate reagent and corresponding samples of 6, 12, and 18 plates coated with the current reagent (MARV Ci67 Lot 13DEC2013) were drawn with replacement from the qualification experiments for each of the two candidate rGP coating antigens. The sister plate design used in the qualifications required that when one plate in a pair failed to meet the assay acceptance criteria, both were repeated. All plates that met the system suitability criteria were included in the sample set from which the random samples were drawn. The sampling of the plates ignored the sister plate design of the qualification in order to better represent the true distribution of the data. Linear mixed effects models were used to estimate the GM ratio between current and candidate reagent lots for each TS and the corresponding 90% confidence intervals. The models included the test sample, reagent lot, and their interaction as fixed effects and the plate number, test operator, and test date as random effects. Separate models were fit to each random sample of 6, 12, or 18 candidate and current reagent plates from each candidate qualification experiment. Model parameters were used to calculate 90% confidence intervals for the GM ratio (candidate/current) for each test sample. The 90% confidence intervals were compared to equivalence regions from the power analysis based on assay variability of 20% CV for illustration. The equivalence regions are conservative, as the variability is slightly less than the qualified anti-MARV GP ELISA (22.5% CV). The analysis was conducted using the SAS software version 9.4, MIXED procedure (SAS Institute, Cary, NC).
Simulation-Based Power Analysis for Equivalence Regions
To better account for multiple test samples and the sources of variability in the anti-MARV GP ELISA, a simulation-based power analysis was performed to supplement the traditional power analysis, given the more complex experimental design for the assay qualification. Simulation parameters included the number of plates per lot (6, 12, or 18 based on replicating our original qualification design up to three times), total assay variability (5 to 30% CV to cover a broad range of potential assays and intended uses), and number of independent test samples per plate (4, 5, or 10 based on typical plate layouts used in the laboratory). The total assay variability was apportioned with 70% applied to sources of intermediate precision (IP) and 30% to repeatability based on the variance components estimated in the anti-MARV GP IgG ELISA qualification (12). IP is defined here to include sources of variability within the lab, such as different test operators, test days, and plates, and repeatability is random variability that occurs in repeated testing under the same conditions. Additional simulations evaluated the sensitivity of the experiment to the sources of variability, assigning 60 or 90% of the total variability to IP, and included extreme cases of 1% and 99% of the variability attributed to IP. In each case, the remaining assay variability was allocated to repeatability. For each combination of the number of plate pairs, total variability and proportion attributed to IP, and the number of independent TSs, simulated samples representing the two lots were drawn from a normal distribution to represent a plate effect and from a lognormal distribution to represent the sample measurement, accounting for sample-to-sample variability. For each simulated experiment, a mixed effects model was fit and used to estimate the 90% CI for the ratio of geometric means between the two lots for each TS. This sampling was repeated 1000 times for each combination of interest. Using the 1000 simulated experiments, equivalence regions were established such that the 90% confidence intervals would fall within the regions for 90% of the 1000 simulated experiments to approximate 90% power. It was required that the 90% confidence interval must be wholly within the equivalence region for 75% of the TSs to meet the acceptance criteria. The increase to 90% power was to allow for a maximum of 10% of reagent qualifications to fail the equivalence test when the reagents are truly equivalent. The requirement that 75% of TSs must pass was motivated by the explorations of the rGP coating antigen qualifications, as it was plausible to have a TS fall outside of the expected behavior. The 75% cutoff was a practical choice for varying sample sizes by allowing a single TS to fail in the case of 4 or 5 TSs and up to two TSs to fail if 10 TSs are used. Simulation analysis was performed using R version 4.1.2 (25).
Biosimilar Method for Equivalence Regions
For biosimilar products, the FDA recommends equivalence testing to assess the difference in means between biosimilar and reference products for critical quality attributes identified as tier 1, the highest risk ranking (22,23,24). For the equivalence acceptance criteria for these tests, the FDA recommends using a regulatory constant (c) times the standard deviation of the reference product (σR), assuming a normal distribution. We adapted this approach to calculate equivalence regions for the GM ratio of candidate and current reagent lots based solely on assay capability, for assay variability of 5 to 30% CV and c = 1.5. The following formula was used to convert assay variability expressed as %CV to standard deviation.
Because a lognormal distribution was used to characterize assay precision in qualification experiments, a natural log transformation was used, and the resulting equivalence bounds were exponentiated to reflect the GM ratio of candidate and current reagent lots.
Results
Results of the traditional power analysis performed for a TOST equivalence test for the ratio of independent measurements between current and candidate lots for a single TS are shown in Table I. The number of tests per lot required to achieve 80% power was calculated for various equivalence region widths (1.25-fold to 1.50-fold) for assay variability of 5 to 30% CV.
Figure 1 illustrates the equivalence test approach for 6, 12, or 18 tests per lot, where the tests were randomly selected from the set of passing plates from qualification experiments for candidate lots MARV Ci67 Lot 29APR16 and MARV Angola Lot 13JAN17 of rGP coating antigen in the anti-MARV GP IgG ELISA. The sample sizes were selected based on the power analysis and also for convenience as they reflected one, two, or three replicates of our original qualification experiment. For each random sample, a mixed effects model was fit and used to estimate the 90% CI for the ratio of geometric means between the two lots for each TS. The 90% CIs were compared to equivalence regions of 1.5-fold for 6 tests, 1.3-fold for 12 tests, and 1.25-fold for 18 tests per lot based on assay variability of 20% CV (Table I; actual assay variability is 22.5% CV). The 90% confidence intervals for each TS must be contained wholly within the equivalence region in order for the lots to be considered equivalent. If any portion of the 90% confidence interval falls outside the equivalence region, the lots are not considered equivalent.

Ratio of the geometric means with 90% confidence intervals based on a random samples of 6 (left; 1.5 fold equivalence region), 12 (center; 1.3-fold equivalence region), or 18 (right; 1.25-fold equivalence region) plates per lot from the MARV Ci67 Lot 29APR16 qualification that passed (top) and MARV Angola Lot 13JAN17 qualification that failed (bottom)
Results of the simulation analysis are summarized in Table II. Upper bounds of the equivalence regions that approximate 90% power, corresponding to the fold-change multipliers, are displayed for each simulated scenario. Lower bounds of the equivalence regions can be calculated as the reciprocal of the upper bound. Equivalence regions calculated using the biosimilar method are presented in Table III.
Discussion
Power analysis was performed for a TOST equivalence test for the ratio of independent measurements between current and candidate lots for a single TS. The number of tests per lot required to achieve 80% power was calculated for various equivalence region widths (1.25-fold to 1.50-fold) for assay variability of 5 to 30% CV (Table I). The equivalence regions and nominal ratio were selected to accommodate normal assay variability that would be acceptable for immunoassays and consistent with the current 15% difference acceptance criterion. A 1.25-fold equivalence region width is well accepted by regulatory agencies in other contexts and was the narrowest region considered. For this equivalence region, sample sizes of up to 11 plates per lot provide 80% power for assay variability up to 15% CV. A qualification study of this size exceeds the guidance for critical reagent testing and is practical to implement. Thus, for assays with variability of up to 15% CV, equivalence testing can be implemented with a reasonable number of plates per lot and a narrow equivalence region. For immunoassays where variability greater than 15% CV is acceptable for the intended use, the sample sizes required to use a 1.25-fold equivalence region are larger and likely impractical. Therefore, wider equivalence regions may be appropriate for many immunoassays to allow for greater assay variability. When assay variability is 20 to 30% CV, equivalence regions of 1.3-fold to 1.5-fold can be tested with 6 to 12 plates per lot, consistent with the sample sizes for less variable assays using a narrower (1.25-fold) equivalence region, while still exceeding the published guidance for critical reagent testing as described above. For assays with greater than 30% CV, equivalence regions in the 1.25- to 1.5-fold range are not practical, but wider equivalence regions may be appropriate so long as care is taken to avoid introducing bias when changing a critical reagent, and the assay remains suitable for the intended use.
To illustrate the equivalence test approach, Fig. 1 shows the 90% confidence intervals for 6, 12, or 18 tests per lot, where the tests were randomly selected from the set of passing plates from qualification experiments for candidate lots MARV Ci67 Lot 29APR16 and MARV Angola Lot 13JAN17 of rGP coating antigen in the anti-MARV GP IgG ELISA. The sample sizes were selected based on the power analysis and also for convenience as they reflected one, two, or three replicates of our original qualification experiment. For each random sample, a mixed effects model was fit and used to estimate the 90% CI for the ratio of geometric means between the two lots for each TS. The 90% CIs were compared to equivalence regions of 1.5-fold for 6 tests, 1.3-fold for 12 tests, and 1.25-fold for 18 tests per lot based on assay variability of 20% CV (Table I; actual assay variability is 22.5% CV). The 90% confidence intervals for each TS must be contained wholly within the equivalence region in order for the lots to be considered equivalent. If any portion of the 90% confidence interval falls outside the equivalence region, the lots are not considered equivalent. As shown in Fig. 1, the 90% CIs for MARV Ci67 Lot 29APR16 (which previously passed) were contained wholly within the equivalence region for all TSs for the random sample of size 12 but only eight of ten TSs for random samples of size 6 and 18. In contrast, for MARV Angola Lot 13JAN17 (which previously failed), the 90% CI was contained wholly within the equivalence region for at most two TSs. Although limited, this exercise illustrates both the method and that the equivalence test approach can differentiate between good and bad reagent lots. The sample sizes used in this analysis provided 80% power for independent tests of a single TS, and the assumed assay variability was slightly lower (20% CV) than the assay variability for the anti-MARV GP IgG ELISA (22.5% CV). The power analysis did not account for multiple TSs nor the contribution of sources of IP to total assay variance. Therefore, the mixed results for MARV Ci67 Lot 29APR16 were not surprising and led us to further explore the effects of multiple test samples and assay variability on the study design.
A simulation analysis was performed to guide the experimental design for future assay qualifications. The effects of total assay variability and the ratio of IP to total assay variability, sample size, and number of TSs per plate on the size of the equivalence region can be supported for a given assay and experimental design. Our initial investigation showed that the original qualification experiment had low power due to the increased throughput of ten TSs per plate for the anti-MARV GP IgG ELISA (compared to earlier assays with just four TSs per plate) combined with the original acceptance criteria that required that all TSs pass. Therefore, the acceptance criteria for the simulation were relaxed to require that at least 75% of test samples pass, i.e., 3 of 4, 4 of 5, or 8 of 10 TSs must pass, as an informal adjustment for multiple test samples. The following assay and design parameters were considered in the simulation:
-
1)
Sample sizes of 6, 12, or 18 plates per sample
-
2)
4, 5, or 10 independent TSs per plate
-
3)
Acceptance criteria require that at least 75% of TSs must pass
-
4)
%CV ranging from 5 to 30%
-
5)
IP accounts for 1, 60, 70, 90, or 99% of total assay variability
Results of the simulation analysis are summarized in Table II. Upper bounds of the equivalence regions that approximate 90% power, corresponding to the fold-change multipliers, are displayed for each simulated scenario. Lower bounds of the equivalence regions can be calculated as the reciprocal of the upper bound. The simulation analysis showed a very strong concordance to the power analysis (Table I) that assumed independent tests of a single sample at 80% power. For assay variability of 10% CV and sample size of 6 plates per lot, the equivalence regions ranged from 1.2- to 1.24-fold. For assays with 15% CV and a sample size of 12 plates per lot, the equivalence regions ranged from 1.22- to 1.25-fold. In both cases, the combination of assay variability, sample size, and equivalence region was consistent with the power analysis (Table I). Similarly, for assays with 20% CV, the equivalence regions ranged from 1.44 to 1.53 for 6 plates per lot, 1.29 to 1.33 for 12 plates per lot, or 1.24 to 1.27 for 18 plates per lot, again consistent with the power analysis in Table I for similar equivalence regions and assay variability.
The informal adjustment to the acceptance criteria to require that 75% of TSs must pass was largely effective in reducing the influence of the number of TSs on the power of the study. In many cases, the equivalence regions were the same for 4, 5, or 10 TSs per plate or differed within a very narrow region. For example, at 20% assay variability with 70% attributed to sources of IP, consistent with the performance characteristics of the anti-MARV GP IgG ELISA, the simulated equivalence regions were 1.31- to 1.33-fold about the GM ratio of current and candidate reagents for layouts with 4, 5, or 10 TSs per plate. Simulated equivalence regions were more consistent relative to the number of TSs per plate for larger sample sizes, lower assay variability, or when large proportions of assay variability were attributed to sources of IP.
For assay variability up to 15% CV, varying the contribution of IP to total assay variability had little effect on the equivalence regions supported for the sample sizes considered. For assays with greater total variability, the equivalence regions were slightly narrower when variability was primarily random but generally consistent over the range of IP considered. The effect of IP on the equivalence regions decreased as the number of samples increased.
The experimental design was developed using both traditional power analysis and simulation-based power analysis. However, equivalence regions based on power and sample size do not necessarily ensure that the assay remains fit for purpose. Therefore, the FDA guidance for equivalence testing of critical quality attributes of biosimilars was adapted for reagent qualification. These equivalence regions are based on assay capability, expressed as assay variability of 5 to 30% CV, and used a regulatory constant of c = 1.5 as recommended by the FDA for similarity assessment (22,23,24). The upper bounds of these regions (1.5σ) were exponentiated for comparison to equivalence regions for the GM ratio of candidate to current reagent lots. Equivalence regions calculated using the biosimilar method are consistent with simulated equivalence regions using 12 plates per lot that provide 90% power over a range of assumptions (Table III).
The anti-MARV GP IgG ELISA is a qualified assay with 22.2% CV total assay variability. Using the biosimilar method for this assay, a 1.39-fold equivalence region would ensure that the assay performs equivalently with the candidate reagent. For a validated assay, the validation acceptance criteria for precision could be used to derive a standard deviation that is fit for purpose (σFFP) and the biosimilar method applied to calculate an equivalence region that would ensure the assay remains fit for purpose.
Conclusions
The original qualification experimental design, while larger than those recommended by industry groups for routine lot changes, did not have sufficient power for the anti-MARV GP IgG ELISA in the context of a critical reagent change, given the assay variability and the number of test samples per plate. The equivalence test method was introduced as a more rigorous statistical approach that is consistent with the experimental objective and takes both the mean and variability of the data into account. The experimental design was developed using both traditional power analysis and simulation-based power analysis. Based on the power analysis, a 1.25-fold equivalence region can be tested using 6 to 12 plates per lot for assays with up to 15% CV but is not practical for more variable assays. Equivalence regions based on power and sample size are acceptable in some contexts but do not necessarily ensure that the assay remains fit for purpose. Therefore, equivalence regions based on FDA guidance for equivalence testing of critical quality attributes of biosimilars were developed and compared to those developed using simulation-based power analysis. Equivalence regions calculated using the biosimilar method were consistent with simulated equivalence regions using 12 plates per lot that provide 90% power over a range of assumptions. Therefore, a design using 12 plates per lot has become standard in our laboratory for qualifying critical reagents. While this experiment is substantially larger than the minimum testing recommendations of industry groups, a larger qualification experiment provides greater confidence in new reagents, which is important in our lab as assay results are compared between studies and over time. A larger qualification experiment may not be practical in every case. Individual laboratories should assess the criticality of reagent lot changes in the context of their experiments to determine whether this approach is warranted for reagents that have the potential to affect the assay reportable values.
Data Availability
The data presented in this study are available upon reasonable request from the corresponding author.
References
International Congress on Harmonization (ICH) Tripartite Guideline. Validation of analytical procedures: text and methodology Q2(R1). 2005. Available from https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf. Accessed 21 June 2023.
ICH Harmonized Guideline. Bioanalytical method validation and study sample analysis M10. 2022. Available from https://database.ich.org/sites/default/files/M10_Guideline_Step4_2022_0524.pdf. Accessed 21 June 2023.
Food and Drug Administration (FDA) Guidance Document. Bioanalytical method validation guidance for industry. 2018. Available from https://www.fda.gov/media/70858/download. Accessed 21 June 2023.
European Medicines Agency. Guidelines for bioanalytical method validation – scientific guideline. 2011. Available from https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf. Accessed 21 June 2023.
Ministry of Health, Labour and Welfare. Guideline on bioanalytical method (ligand binding assay) validation in pharmaceutical development. 2014. Available from http://www.nihs.go.jp/drug/BMV/260530_LBA-GL_E.pdf. Accessed 21 June 2023.
King LE, Farley E, Imazato M, Keefe J, Khan M, Ma M, et al Ligand binding assay critical reagents and their stability: recommendations and best practices from the global bioanalysis consortium harmonization team. AAPS J. 2014;16(3):504–15. https://doi.org/10.1208/s12248-014-9583-x.
Pihl S, van der Strate BW, Golob M, Ryding J, Vermet L, Jaitner B, et al EBF recommendation on practical management of critical reagents for antidrug antibody ligand-binding assays. Bioanalysis. 2019;11(19):1787–98. https://doi.org/10.4155/bio-2019-0248. Erratum in: Bioanalysis. 2020;12(5):349.
O’Hara DM, Theobald V, Egan AC, Usansky J, Krishna M, TerWee J, et al Ligand binding assays in the 21st century laboratory: recommendations for characterization and supply of critical reagents. AAPS J. 2012;14(2):316–28. https://doi.org/10.1208/s12248-012-9334-9.
O’Hara DM, Theobald V. Life cycle management of critical ligand-binding reagents. Bioanalysis. 2013;5(21):2679–96. https://doi.org/10.4155/bio.13.241.
Rudge TL Jr, Sankovich KA, Niemuth NA, Anderson MS, Badorrek CS, Skomrock ND, et al Development, qualification, and validation of the filovirus animal nonclinical group anti-Ebola virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay for human serum samples. PLoS One. 2019;14(4):e0215457. https://doi.org/10.1371/journal.pone.0215457.
Niemuth NA, Rudge TL Jr, Sankovich KA, Anderson MS, Skomrock ND, Badorrek CS, et al Method feasibility for cross-species testing, qualification, and validation of the filovirus animal nonclinical group anti-Ebola virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay for non-human primate serum samples. PLoS One. 2020;15(10):e0241016. https://doi.org/10.1371/journal.pone.0241016.
Rudge TL Jr, Machesky NJ, Sankovich KA, Lemmon EE, Badorrek CS, Overman R, et al Assays for the evaluation of the immune response to Marburg and Ebola Sudan vaccination-filovirus animal nonclinical group anti-Marburg virus glycoprotein immunoglobulin G enzyme-linked immunosorbent assay and a pseudovirion neutralization assay. Vaccines (Basel). 2022;10(8):1211. https://doi.org/10.3390/vaccines10081211.
Quinn CP, Semenova VA, Elie CM, Romero-Steiner S, Greene C, Li H, et al Specific, sensitive, and quantitative enzyme-linked immunosorbent assay for human immunoglobulin G antibodies to anthrax toxin protective antigen. Emerg Infect Dis. 2002;8(10):1103–10. https://doi.org/10.3201/eid0810.020380.
Semenova VA, Schiffer J, Steward-Clark E, Soroka S, Schmidt DS, Brawner MM, et al Validation and long term performance characteristics of a quantitative enzyme linked immunosorbent assay (ELISA) for human anti-PA IgG. J Immunol Methods. 2012;376(1–2):97–107. https://doi.org/10.1016/j.jim.2011.12.002.
Schuirmann DJ. On hypothesis testing to determine if the mean of a normal distribution is contained in a known interval. Biometrics. 1981;37:617.
Anderson S, Hauck WW. A new procedure for testing equivalence in comparative bioavailability and other clinical trials. Commun Stat Theory Methods. 1983;12(23):2663–92. https://doi.org/10.1080/03610928308828634.
Hauck WW, Anderson S. A new statistical procedure for testing equivalence in two-group comparative bioavailability trials. J Pharmacokinet Biopharm. 1984;12(1):83–91. https://doi.org/10.1007/BF01063612.
Rocke DM. On testing for bioequivalence. Biometrics. 1984;40:225–30.
Kelter R. Bayesian Hodges-Lehmann tests for statistical equivalence in the two-sample setting: power analysis, type I error rates and equivalence boundary selection in biomedical research. BMC Med Res Methodol. 2021;21:171. https://doi.org/10.1186/s12874-021-01341-7.
Lakens D. Equivalence tests: a practical primer for t tests, correlations, and meta-analyses. Soc Psychol Personal Sci. 2017;8(4):355–62. https://doi.org/10.1177/1948550617697177.
FDA Guidance Document. Scientific considerations in demonstrating biosimilarity to a reference product guidance for industry. 2015. Available from https://www.fda.gov/media/82647/download. Accessed 21 June 2023.
Wang T, Chow SC. On the establishment of equivalence acceptance criterion in analytical similarity assessment. J Biopharm Stat. 2017;27(2):206–12. https://doi.org/10.1080/10543406.2016.1265539.
Chow SC, Song F, Bai H. Analytical similarity assessment in biosimilar studies. AAPS J. 2016;18(3):670–7. https://doi.org/10.1208/s12248-016-9882-5.
Tsong Y, Dong X, Shen M. Development of statistical methods for analytical similarity assessment. J Biopharm Stat. 2017;27(2):197–205. https://doi.org/10.1080/10543406.2016.1272606.
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. 2021. URL https://www.R-project.org/. Accessed 21 June 2023.
Acknowledgements
We would like to acknowledge Nick Skomrock and Alisha Rajbhandari for simulation modeling and Larry Wolfraim, Janet Lathey, Nancy Ulbrandt, Aparna Kohlekar, Chris Badorrek, and Greg Rutkowski for review of the manuscript.
Funding
This research was funded by the National Institute of Allergy and Infectious Diseases, contract number HHSN272201200003I, task order number HHSN27200009.
Author information
Authors and Affiliations
Contributions
Conceptualization: Nancy A. Niemuth, Cheryl A. Triplett, and Michael S. Anderson.
Data acquisition: Karen A. Sankovich and Thomas L. Rudge, Jr.
Writing—original draft: Nancy A. Niemuth.
Writing—review and editing: Nancy A. Niemuth, Cheryl A. Triplett, Michael S. Anderson, Karen A. Sankovich, and Thomas L. Rudge, Jr.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Nancy A. Niemuth is retired from Battelle.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Niemuth, N.A., Triplett, C.A., Anderson, M.S. et al. A Case Study for Critical Reagent Qualification for Ligand Binding Assays Using Equivalence Test Methodology. AAPS J 25, 89 (2023). https://doi.org/10.1208/s12248-023-00857-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-023-00857-8