
Abstract
The genome content of adeno-associated virus (AAV) vectors is critical to the safety and potency of AAV-based gene therapy products. Empty capsids are considered a product-related impurity and a critical quality attribute (CQA) of the drug product, thus requiring characterization throughout the production process to demonstrate they are controlled to acceptable levels in the final drug product. Anion exchange chromatography has been used to achieve separation between empty and full capsids, but requires method development and gradient optimization for different serotypes and formulations. Here, we describe an alternative approach to quantitation that does not rely on achieving separation between empty and full capsids, but instead uses the well-established relationship between absorbance at UV A260/A280 and relation to DNA/protein content, in combination with anion-exchange chromatography to allow one to calculate the relative proportion of empty and full capsids in AAV samples from a single peak. We call this approach ACUVRA: Anion-exchange Chromatography UV-Ratio Analysis, and show the applicability of the method through a case study with recombinant AAV2 (rAAV2) process intermediates and drug substance. Method qualification and GMP validation in a quality control (QC) laboratory results show that ACUVRA is a fit-for-purpose method for process development support and characterization, while also being a QC-friendly option for GMP release testing at all stages of clinical development.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recombinant adeno-associated virus (rAAV) vectors have become the leading platform for in vivo delivery of gene therapies for a diverse array of human diseases (1). Advancements in manufacturing processes using transient triple transfection of plasmid DNA into adherent or suspension-adapted mammalian cell culture has enabled major improvements in scaling up production to be suitable for clinical and commercial batches (1). The gene of interest is packaged through the natural biological mechanism in a viral protein capsid shell consisting of ~ 60 subunits of viral protein VP1, VP2, and VP3 in a ratio of ~ 1:1:10 (2). However, AAV production is an imperfect process, creating empty capsids that are difficult to separate and similarly challenging to quantify (3). Historically, density gradient centrifugation purification was the most effective technique to separate empty capsids, and is still commonly used for small-scale research production, but is challenging to scale up for larger clinical batch supply. This bottleneck has been increasingly supplanted by more scalable removal methods involving a chain of affinity and ion-exchange chromatography (IEC) steps, with empty capsid removal rarely complete. Quantitation of these empty capsids is necessary for both process characterization and dosing determination, due to the possibility of empty capsids limiting efficacy and producing adverse immunogenic effects (4, 5).
Genome size may play a role in packaging efficiency and ease of empty capsid separation. In designing a therapeutic viral vector, it is necessary to consider the length of the therapeutic component. Typical genome length for AAV packaging is ~ 4.7 kb, but variability in packaging efficiency can lead to overpacked or partially filled particles, where the gene of interest is not packaged at exactly one copy per particle as intended (6). The length of the packaged genome or its absence in the final AAV capsid particle contribute to differences in isoelectric point (pI)—the key attribute leveraged to remove empty capsids downstream using ion-exchange chromatography.
Multiple methods have been used in attempt to separate and quantify empty capsids. Analytical ultracentrifugation (AUC) is able to accurately measure empty, partial, and full capsid species, as well as aggregates and other impurities (7). Currently, AUC is the only well-established technique that can reliably resolve partials from full and empty capsids. However, it has several limitations such as requiring large sample volumes making it problematic when samples are limited, as is the case with low yield processes and small batches that are common in pre-clinical and early clinical gene therapy manufacturing. In addition, the SedFit data processing software is not 21 CFR Part 11 compliant, further complicating the validation of AUC for routine GMP testing (8). Charge detection mass spectrometry (CDMS) is still in its infancy and carries the weight of validation challenges in GMP and QC environments (9, 10). Transmission electron microscopy with negative staining (TEM) is also commonly used to identify and count empty and full capsids. However, its complex instrumentation requires significant analyst expertise and quantitative count results can be susceptible to analyst bias (11), making it less suitable for QC routine use. Another common method combining qPCR to measure genome titer with ELISA capsid titer has been used to compare empty-to-full capsid ratio. However, these results rely on the precision of both methods, resulting in an estimation of the ratio that has been shown to be inaccurate (12,13,14). Imaged capillary isoelectric focusing (icIEF) has been applied to the analysis of empty and full AAV, although the accuracy of the quantitation and reproducibility of the profile has not yet been well established, and peaks cannot be collected for orthogonal confirmation of identity (15). More recently, SEC-UV-RI-MALS has been used for AAV characterization and reporting of multiple attributes from a single analysis, including the full and empty capsid content (16). While this technique is well established for analysis and characterization of biomolecules and polymers, the complexity of the instrumentation and data analysis software require substantial training and user expertise, which poses additional burdens on QC for validation and implementation for routine use.
Alternatively, cuvette-based UV measurements at 260 and 280 nm have shown to be a viable method to accurately determine the amount of empty capsids in a given sample (17). However, the method requires precise calculation of both the genome and capsid extinction coefficients. This calculation is theoretical in nature and does not take into consideration anomalies such as capsid post-translational modifications (18). Interference from DNA, protein, chromatin, other process contaminants, and excipients such as histidine can bias results, requiring highly purified samples. Moreover, this method requires denaturation of the AAV capsid, preventing sample recovery for any further characterization by orthogonal techniques. A more recent example used SEC-UV and the ratio of 230 and 260 nm to establish a linear relationship for empty and full quantitation (19). However, SEC is isocratic and there is an upper limit on sample injection volume, which limits the range of sample concentrations that can be analyzed while minimizing band broadening. In addition, SEC has limited ability to separate out impurities with similar molecular weight or hydrodynamic radius, which could make this approach unsuitable for analysis of process intermediates.
Anion-exchange chromatography is now increasingly used at the process development scale as a method for the removal of empty capsids (1, 19, 20). More recently, it has been used as an analytical tool as well, usually coupled with UV, or fluorescence detection for improved sensitivity (21). All these methods use peak area percentages as the basis for quantitation of empty and full capsid percentage (21,22,23). Theoretically, full capsids containing DNA have a more negative surface potential and therefore bind more tightly to a positively charged column in a high-pH, low-salt environment. This causes empty capsids to elute first in a shallow salt gradient. However, the difference in pI between empty and full capsids is small (~ 0.4 pI) and complete separation is difficult to achieve, often requiring extensive column screening and gradient optimization (21, 23). Such method development efforts take time and require the availability of representative material in sufficient quantities, which is often not feasible in early stages of product development. In addition, the separation conditions are optimized for one specific serotype of AAV and will likely require further optimization for other serotypes (23). The assumption that full capsids will always be more negatively charged than empty capsids may be an oversimplification, as it does not take into consideration capsid permeability, the presence of counterions in the capsid interior (24, 25), or post-translational modifications (PTMs), such as phosphorylation, glycosylation, and deamidation that may alter the pI and affect column binding (26). The type and extent of post-translational modifications (PTMs) has been found to vary between different batches, even from the same production process (27). Differences in formulation at various stages in the process can further complicate this issue. In prior published methods, consistent separation of empty and full capsids with adequate resolution is essential for a quantitation strategy based on peak area percentages. For products in early stages of process development, changes to the upstream process can lead to altered post-translational modification profiles which can impact the capsid pI value, and thereby may require re-development of the ion-exchange gradient to achieve separation.
The method we present here shifts the goal of anion-exchange chromatography away from complete separation of empty and full capsids. Instead, the intention is to contend with the main limitations surrounding the UV absorbance ratio measurement method (17), namely, the interference from host cell DNA and proteins, or residuals in process intermediates. In this method, which we refer to as ACUVRA: Anion-exchange Chromatography UV-Ratio Analysis, we examine the ratio of absorbance at 260 and 280 nm from the integrated area of the capsid peaks and construct a standard curve from a range of %Empty standards characterized by an established orthogonal technique, in this case TEM, eliminating the need to calculate precise extinction coefficients for DNA and capsid. The finalized method was designed specifically to be “QC-friendly” (28): simple sample preparation, widely available instrumentation, and performing all the calculations within the 21 CFR part 11 compliant chromatography data system (CDS), thus preserving data integrity.
The method has additional utility for analytical testing to support process development. Our alternative approach to quantitation allows for reduced time required for HPLC gradient development and is more resilient to changes in charge heterogeneity of the product that can arise during process development. We show that the final method is suitable for calculating the % Empty capsid in rAAV2 process intermediates, while also being successfully validated for release testing of drug substance in a GMP Quality Control setting.
Materials and Methods
rAAV2 Samples
The recombinant AAV (rAAV) used in this study is derived from wild-type AAV2 containing a single-stranded DNA genome of approximately 4.2 kb. rAAV2 samples were produced by Biogen. Briefly, cell culture harvest material was clarified and processed by tangential-flow filtration (TFF), iodixanol density gradient ultracentrifugation, and affinity column chromatography. rAAV2%Empty calibration standards were prepared from materials that were further purified using anion-exchange chromatography to generate enriched Full and Empty samples.
Capsid Titer
AAV2 capsid titer was performed using a PROGEN AAV2 Titration ELISA kit (PROGEN Biotechnik GmbH). The microtiter plate-based assay utilizes an A20 antibody to identify the conformational epitope in assembled capsid proteins based on the sandwich ELISA method. In brief, serial dilutions are performed in assay buffer to bring samples into appropriate concentration range. Samples are incubated with the biotin-conjugated A20 antibody at 37°C, washed with assay buffer, and then incubated with the streptavidin peroxidase conjugate at 37°C. Samples are then incubated with tetramethylbenzidine substrate for 15 min at room temperature, causing a color reaction that is proportional to bound AAV particles. The reaction is stopped using sulfuric acid, and absorbance is read at 450 nm.
Calibration Standards
Calibration standards were prepared from in-house manufactured rAAV2 materials. The Full capsid enriched material was prepared from cell culture harvest subject to a tangential flow filtration (TFF) step, followed by purification on a preparative-scale monolith anion exchange column and collecting the Full peak. The Empty capsid enriched material was prepared by collecting the Empty peak from purification of the starting material via preparative-scale anion exchange chromatography. The Empty and Full collected fractions were determined by negative stain TEM (TEM) to be approximately 100% and 5% Empty, respectively. Four calibration standards were created by co-mixing proportions of the Empty and Full standard materials to achieve the following approximate %Empty designations: 51%, 30%, 17%, and 5%. Each co-mixture was analyzed by TEM (duplicate grids prepared and imaged on two different days) to assign the %Empty value, then sub-aliquoted at a volume sufficient to use one aliquot per assay. Aliquots of each calibrant were stored at –80°C until immediately prior to use.
Anion-Exchange Chromatography
The anion-exchange method used in this study was developed on a Waters Alliance e2695 HPLC system equipped with either a PDA detector (2996/2998; Waters) or a dual-wavelength UV detector (2489; Waters); data were acquired at 260 and 280 nm. A Protein-Pak Hi Res Q anion-exchange column (5 µm, 4.6 × 100 mm) (Waters Corporation, Milford, MA, USA) and a salt gradient was used to perform the separation.
The mobile phases were (A) 20 mM Bis–Tris propane (BTP), 1 mM MgCl2, pH 9.5; and (B) 20 mM BTP, 1 mM MgCl2, 1 M NaCl, pH 9.5, delivered at a flow rate of 0.3 mL/min.
Finalized run conditions for analysis of rAAV2 samples started at initial conditions of 100 mM NaCl (10% B), then a linear gradient from 100 to 300 mM NaCl (10–30% B) over 10 min, followed by a 1-M NaCl (100% B) wash for 6 min. The column was equilibrated at initial conditions for 17 min between each injection.
Sample Preparation
Drug substance (DS) samples and calibration standards were prepared by diluting with mobile phase A to achieve a final salt concentration of no more than 100 mM, to allow sufficient binding to the column. Sample injections contained approximately 8.0E + 10 total capsids, with varying injection volumes up to 100 µL.
Process intermediate samples were buffer exchanged using Amicon Ultra 30 K MWCO centrifugal filter units (MilliporeSigma, Burlington, MA). Filter units were prewetted with DS formulation buffer prior to buffer exchange. One buffer exchange step constituted the addition of 400 µL formulation buffer followed by centrifugation at 14,000 × g for 10 min. Final retentate volumes of ~ 30 µL were recovered.
Data Processing
Calibration standards were run alongside testing samples, and A260/A280 ratios attained were correlated with %Empty values determined from TEM to construct a calibration curve using a quadratic fit. All data processing for routine use was performed with Empower 3 software (Waters Corporation, Milford, MA). The capsid peaks from the 260-nm channel are considered the main component, while the peak in the 280-nm channel is defined as an internal standard. A mean smoothing factor of 21 was applied to both data channels prior to integration of peaks. The smoothing factor was chosen to improve consistency of automatic integration and did not significantly change the calculated ratio (data not shown).
TEM
rAAV2 samples were applied to copper electron microscopy grids (Electron Microscopy Sciences, Hatfield, PA), rinsed with distilled water, negatively stained with 0.75% (w/v) uranyl formate, and dried by vacuum. The grids were imaged using a Philips CM10 transmission electron microscope. VAS software (Vironova, Stockholm, Sweden) was used for image processing. The software uses “support vector machines,” a supervised machine-learning method in which the software is trained by a manually classified set of images using predefined features such as shape, size, and intensity profile to determine particle counts, empty, and full particles. Empty particles take up the stain and appear dark in the center, making them distinguishable from full particles. To ensure representative counts, a minimum of ten images were taken at a magnification between 21,000 × and 52,000 × , with a target particle density of 100 particles per image.
Results and Discussion
Anion-Exchange Chromatography UV-Ratio Analysis
Anion-exchange chromatography utilizes the difference in surface charge, resulting in species separation. Theoretically, empty capsids lacking the encapsulated DNA have a less negative charge than full capsids, causing them to bind less tightly to the anion-exchange column and elute first in shallow salt gradient conditions. There are many examples in the literature of IEC separations developed and optimized to exploit this net charge difference to achieve resolution between full and empty capsids for a range of serotypes (21, 22). However, each of these published methods is only applicable to the serotype it was developed for, and often required extensive method development and gradient optimization to achieve adequate resolution between empty and full capsids. Moreover, none of the reported applications have shown that the final optimized separation conditions were robust to product charge heterogeneity changes that may arise from batch-to-batch process variability and other product-related related variants that are often encountered in gene therapy early phase process development and scale-up.
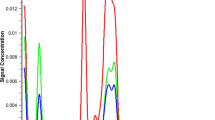
We took a novel approach, where the goal was to develop a method that could become a platform, which would enable shortened method development timelines without sacrificing the method performance that is necessary for successful deployment and validation in a GMP QC setting. We use a shallow salt gradient to first elute DNA and other process-related impurities, followed by AAV capsids. Some empty/full separation may occur, but is not the objective and does not pose challenges to data interpretation, as integration of all capsid-related peaks is used to determine the A260/A280 ratio (Fig. 1).

Overlays of 260 nm (solid) and 280 nm (dashed) ACUVRA chromatograms for a buffer blank, b rAAV2 5% Empty capsid calibration standard, and c rAAV2 51% Empty capsid calibration standard. Peaks in (a) labeled with * are buffer-related or system peaks observed in all standards and samples. Integration window for the capsid peak is denoted in inset in (b) and (c). Representative TEM images taken at 39,000 × magnification for d 5% Empty calibration standard and e 51% Empty calibration standard. A representative empty capsid (red circle) and full capsid (green circle) are shown in (d)
Calibration Model for Accurate Determination of %Empty Capsids
The A260/A280 ratio has previously been used for estimating vector genome titer in purified samples (17). It is noted that the correlation between this ratio and %Empty capsids in a sample is not linear due to changing DNA and protein contribution to UV absorbance as the %Empty capsids changes. The ACUVRA calibration curve (Fig. 2) was created by creating mixtures from mostly full and mostly empty rAAV2 material, as determined by TEM. While AUC is considered the “gold standard,” the large volumes required precluded its use in this instance. The small sample volumes required for TEM allowed for analysis of multiple replicates on multiple days, thus reducing the uncertainty in the final reported %Empty results. In this study, we evaluated two calibration models: a quadratic model, and a more complex alternative model, derived from physical principles (see Supplementary Information), which we call the “r3” model. As demonstrated, both quadratic and r3 models performed similarly within the range of the calibration curve. The decision to use the quadratic model (Fig. 2) for the final method was based on it being the simplest to implement for routine QC testing with the ability to perform all calculations within the validated Chromatography Data System (CDS) software. However, the r3 model does have advantages in certain situations, which is discussed further in the Supplementary Information.

Calibration curve with quadratic fit. Calibration standards were prepared by co-mixing of rAAV2 materials to final levels of 5%, 17%, 31%, and 51% Empty capsids, as determined from TEM analysis. The A260/A280 response is the ratio of the integrated peak areas for the rAAV2 capsid peaks in the 260- and 280-nm channels. The mean of n = 2 measurements is shown with each error bar constructed from the min and max of the data
Proteins have an absorbance maximum at 280 nm, but DNA also contributes to absorbance at this wavelength. This relationship is complicated by the effect of changing the empty/full sample composition, making the conversion of the A260/A280 ratio to a %Empty capsid result less straightforward. Recently, SEC-UV-RI-MALS has been applied for analysis of AAV, combining the signals from each detector to calculate the Empty and Full content, along with capsid and genome titer (16). This approach also uses the A260/A280 ratio, but avoids the need for external calibration standards by using the RI signal to calculate the sample mass in absolute terms. However, this calculation relies on several assumptions: dn/dc of protein, dn/dc of DNA, extinction coefficient of protein, extinction coefficient of DNA, 1:1:10 ratio of VP proteins, and that AAV particles are isotropic scatterers. While any one of these assumptions is derived from established theory, the compound error of this many assumptions could be sizable. The approach we take here makes no assumptions except that TEM can measure %Empty accurately. An additional drawback of SEC is its being an isocratic separation, and there is an upper limit on the volume of sample that can be injected while maintaining good peak shape (29, 30). IEC has advantages when analyzing low concentration samples, as injection volume does not impact the efficiency of the separation, and the sample is effectively concentrated at the top of the column prior to the gradient elution. Unfortunately, the increasing salt gradient poses a challenge for coupling with RI detection, thus precluding UV-RI-MALS quantitation approach with this mode of chromatography.
Our %Empty standard calibration curve approach eliminates the necessity for assumptions surrounding extinction coefficient calculations or for the resolution of empty and full capsid peaks to characterize the %Empty capsids in a sample. This also helps control for any instrument-to-instrument differences that could impact the absorbance response, such as UV detector lamp age and flow-cell dimensions.
It should be noted that the A260/A280 ratio range which we measured was small, with the 51% Empty calibrant measuring ~ 1.2 and 5% Empty calibrant measuring ~ 1.45. This narrow range requires that assay precision be given due consideration, as small changes in these values can have significant impact on results. Therefore, we took steps to uniformly integrate all capsid peaks in the chromatograms at each wavelength and found that application of a smoothing factor greatly improved the consistency of the baseline integrations. Precision was further controlled by performing n = 2 injections of the calibrants and samples and setting sample acceptance criteria around the relative difference of the A260/A280 ratio between replicates. Calibrants were not created for sample with more than 51% Empty capsids because this is outside of the expected range for this rAAV2’s process intermediates, drug substance, and drug product samples. However, a wider range could be advantageous, especially in research and academic settings, to allow for analysis of a broader range of samples. As noted in previous studies, samples with compositions of predominantly empty capsids have been modeled to show that precision of the absorbance ratio decreases as the percentage of empty capsids increases (17). In our work, we generated four calibration levels. While additional levels may improve the fitting of the calibration model, we demonstrate that four levels are sufficient to achieve acceptable accuracy and precision for quantitation of %Empty capsids in the range relevant for our rAAV2 samples.
Analysis of rAAV2 Process Intermediates and Drug Substance
Two rAAV2 process intermediates and a drug substance sample were analyzed using the above-described ACUVRA method. Process intermediate 1 and process intermediate 2 represent different fractions recovered following the iodixanol density gradient ultracentrifugation step, which is the first purification step in the downstream process. The presence of iodixanol in the process intermediates caused significant interference in detection of the capsid peaks at both 260 and 280 nm, as shown in Fig. 3. However, we found that buffer-exchanging six times was effective in removing this interference and enabled successful detection and quantitation of the capsid peaks in these process intermediate samples. Drug substance samples with salt concentrations > 100 mM NaCl required only an initial dilution in mobile phase A buffer prior to analysis to enable binding to the anion-exchange resin.

a 260-nm and b 280-nm chromatograms representative of rAAV2 process intermediates from the post-iodixanol density gradient ultracentrifugation step showing overlaid profiles before (red) and after (blue) performing six buffer exchanges into drug substance formulation buffer. Enhanced view of capsid peak at ~ 7 min is shown in inset panels, showing effective removal of iodixanol by buffer exchanging to enable detection and integration of the capsid peak at both 260 and 280 nm. Buffer-related peaks are denoted with *
Representative chromatograms and TEM images for rAAV2 process intermediate 1, process intermediate 2, and drug substance are shown in Fig. 4. A comparison of the %Empty capsid results by ACUVRA to orthogonal TEM analysis (Table I) demonstrates the good agreement between the two methods and confirms that the calibration curve quantitation approach is suitable for both process intermediates and purified drug substance. While the accuracy of 143% for process intermediate 1 is outside the typical 70–130% acceptable range per ICH Q2(R1) Validation of Analytical Procedures guidance (31), the relative accuracy requirements for our purposes are less stringent for samples from early process steps than for drug substance. The ACUVRA results for process intermediates are suitable for characterizing trends across the multiple downstream process steps.

Overlays of 260 nm (solid) and 280 nm (dashed) ACUVRA chromatograms for rAAV2: a process intermediate 1, b process intermediate 2, and c drug substance. Blue-shaded region on the chromatograms indicates the integration window for the capsid peaks, and buffer-related peaks are labeled with *. Representative TEM images for each sample are shown to the right of each chromatogram, with a representative empty (red circle) and full (green circle) capsid labeled in each image. TEM images in panels (a) and (b) were acquired at 21,000 × magnification, while (c) was acquired at 39,000 × magnification
Method Qualification and Validation for rAAV2
The goal of this work was to develop a QC-friendly method to measure %Empty capsids in the rAAV2 drug substance and drug product for batch release and stability studies. Qualification experiments were conducted in accordance with ICH Q2(R1) to assess Repeatability, Intermediate Precision, Accuracy, and Range. Method performance attributes are summarized in Table II, and show repeatability of 7.9% RSD, intermediate precision of 7.4% RSD, and accuracy (expressed as recovery to TEM results) of 112.9%. The range of the method is defined by the low and high level calibration standards, which in this instance is 5–51% Empty capsids. The qualification results were used to set the acceptance criteria for method validation in a quality control laboratory. Method validation results (Table II) show consistent performance between the development and QC laboratories, meeting all validation acceptance criteria, thus demonstrating this as a viable QC-friendly approach for %Empty capsid determination.
Additional Considerations
It should be mentioned that the established ACUVRA approach is not able to distinguish between partially filled and empty capsids. This is also a limitation of IEC methods that do separate the empty from full capsids, as well as the SEC-UV-RI-MALS (16) and SEC-UV (19) approaches. Currently, AUC is the only broadly available technique that has the resolving power to separate the partially filled from empty and full capsids (7, 23), although more recently icIEF (15) and mass photometry (32) have been shown to resolve partially filled capsids in certain circumstances, while CDMS has been used to characterize AAV genome packaging (10). None of these techniques, including ACUVRA, can identify if the capsid is packed with DNA from host cells or other exogenous debris; however, this quality and safety concern can be addressed with specifically designed residual impurity assays. From a Chemistry, Manufacturing, and Controls (CMC) and patient-safety perspective, we classify as product-related impurities any capsids that do not contain the complete target genome. In our method, we report “%Empty capsids” and treat this as an impurity method, but one could also report results as “%Full capsids” and consider it as an expression of product purity.
Conclusions
In this paper, we showed the suitability of an anion-exchange HPLC assay using A260/A280 absorbance ratio (ACUVRA) for the determination of %Empty capsids in rAAV2 samples. The standard curve approach provides a straightforward way to translate the A260/A280 ratio into a %Empty capsid result, using equipment that is common in most QC laboratories and does not require specialized training or additional data processing software. Method performance assessed during qualification and QC validation studies show ACUVRA to be an accurate and precise method for determining %Empty capsid content in drug substance, while also being suitable for analyzing process intermediates, overcoming matrix interference that was a key limitation of previous non-chromatographic methods relying on the UV-ratio for quantitation (17). The ACUVRA chromatography gradient, coupled with sample-appropriate %Empty capsid calibration standards, could potentially be adapted for other serotypes without extensive gradient development to optimize chromatographic separation of empty and full capsids. In addition, there are advantages to being able to use the same method for monitoring %Empty capsids throughout the process, from characterization of process intermediates all the way through to drug substance and drug product release and stability testing, enabling an end-to-end analytical control strategy for this important product-related impurity.
References
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18(5):358–78.
Wörner TP, Bennett A, Habka S, Snijder J, Friese O, Powers T, et al. Adeno-associated virus capsid assembly is divergent and stochastic. Nat Commun. 2021;12(1):1642.
Clément N, Grieger JC. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol Ther - Methods Clin Dev. 2016;3:16002.
Basner-Tschakarjan E, Mingozzi F. Cell-mediated immunity to AAV vectors, evolving concepts and potential solutions. Front Immunol. 2014;5:350. https://doi.org/10.3389/fimmu.2014.00350.
Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B, et al. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: implications for gene therapy using AAV vectors. Hum Gene Ther. 2006;17(4):440–7.
Bennett A, Mietzsch M, Agbandje-McKenna M. Understanding capsid assembly and genome packaging for adeno-associated viruses. Future Virol. 2017;12(6):283–97.
Burnham B, Nass S, Kong E, Mattingly M, Woodcock D, Song A, et al. Analytical ultracentrifugation as an approach to characterize recombinant adeno-associated viral vectors. Hum Gene Ther Methods. 2015;26(6):228–42.
Savelyev A, Gorbet GE, Henrickson A, Demeler B. Moving analytical ultracentrifugation software to a good manufacturing practices (GMP) environment. PLOS Comput Biol. 2020;16(6):e1007942.
Contino NC, Jarrold MF. Charge detection mass spectrometry for single ions with a limit of detection of 30 charges. Int J Mass Spectrom. 2013;345–347:153–9.
Wörner TP, Snijder J, Friese O, Powers T, Heck AJR. Assessment of genome packaging in AAVs using Orbitrap-based charge-detection mass spectrometry. Mol Ther - Methods Clin Dev. 2022;24:40–7.
Subramanian S, Maurer AC, Bator CM, Makhov AM, Conway JF, Turner KB, et al. Filling adeno-associated virus capsids: estimating success by cryo-electron microscopy. Hum Gene Ther. 2019;30(12):1449–60.
Dobnik D, Kogovšek P, Jakomin T, Košir N, Tušek Žnidarič M, Leskovec M, et al. Accurate quantification and characterization of adeno-associated viral vectors. Front Microbiol. 2019;10:1570. https://doi.org/10.3389/fmicb.2019.01570
Fagone P, Wright JF, Nathwani AC, Nienhuis AW, Davidoff AM, Gray JT. Systemic errors in quantitative polymerase chain reaction titration of self-complementary adeno-associated viral vectors and improved alternative methods. Hum Gene Ther Methods. 2012;23(1):1–7.
Kuck D, Kern A, Kleinschmidt JA. Development of AAV serotype-specific ELISAs using novel monoclonal antibodies. J Virol Methods. 2007;140(1–2):17–24.
Li T, Gao T, Chen H, Demianova Z, Wang F, Malik M, et al. Determination of full, partial and empty capsid ratios for adeno-associated virus (AAV) analysis. SCIEX Application Note. https://sciex.com/content/dam/SCIEX/pdf/tech-notes/all/AAV-Full-Partial-Empty.pdf. Accessed 17 Feb 2021.
McIntosh NL, Berguig GY, Karim OA, Cortesio CL, De Angelis R, Khan AA, et al. Comprehensive characterization and quantification of adeno associated vectors by size exclusion chromatography and multi angle light scattering. Sci Rep. 2021;11(1):3012.
Sommer JM, Smith PH, Parthasarathy S, Isaacs J, Vijay S, Kieran J, et al. Quantification of adeno-associated virus particles and empty capsids by optical density measurement. Mol Ther. 2003;7(1):122–8.
Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182(2):319–26.
Meng H, Sorrentino M, Woodcock D, O’Riordan CR, Dhawan V, Verhagen MF, et al. Size exclusion chromatography with dual wavelength detection as a sensitive and accurate method for determining the empty and full capsids of recombinant adeno-associated viral vectors. Hum Gene Ther. 2021;33(3–4):202–12.
El Andari J, Grimm D. Production, processing, and characterization of synthetic AAV gene therapy vectors. Biotechnol J. 2021;16(1):2000025.
Wang C, Mulagapati SHR, Chen Z, Du J, Zhao X, Xi G, et al. Developing an anion exchange chromatography assay for determining empty and full capsid contents in AAV6.2. Mol Ther - Methods Clin Dev. 2019;15:257–63.
Qu G, Bahr-Davidson J, Prado J, Tai A, Cataniag F, McDonnell J, et al. Separation of adeno-associated virus type 2 empty particles from genome containing vectors by anion-exchange column chromatography. J Virol Methods. 2007;140(1–2):183–92.
Fu X, Chen WC, Argento C, Clarner P, Bhatt V, Dickerson R, et al. Analytical strategies for quantification of adeno-associated virus empty capsids to support process development. Hum Gene Ther Methods. 2019;30(4):144–52.
Heffron J, Mayer BK. Improved virus isoelectric point estimation by exclusion of known and predicted genome-binding regions. Appl Environ Microbiol. 2020;86(23):e01674-e1720.
Heffron J, Mayer BK, Semrau JD. Virus isoelectric point estimation: theories and methods. Appl Environ Microbiol. 2021;87(3):e02319-e2320.
Mary B, Maurya S, Arumugam S, Kumar V, Jayandharan GR. Post-translational modifications in capsid proteins of recombinant adeno-associated virus (AAV) 1-rh10 serotypes. FEBS J. 2019;286(24):4964–81.
Thadani NN, Yang J, Moyo B, Lee CM, Chen MY, Bao G, et al. Site-specific post-translational surface modification of adeno-associated virus vectors using leucine zippers. ACS Synth Biol. 2020;9(3):461–7.
Xin Bu, John A. Castoro. Method robustness considerations for successful product commercialization. 2015. Available from: http://www.pharmoutsourcing.com/Featured-Articles/174550-Method-Robustness-Considerations-for-Successful-Product-Commercialization/. Accessed 16 Feb 2021.
Hong P, Koza S, Bouvier ESP. A review size-exclusion chromatography for the analysis of protein biotherapeutics and their aggregates. J Liq Chromatogr Relat Technol. 2012;35(20):2923–50.
Ricker RD, Sandoval LA. Fast, reproducible size-exclusion chromatography of biological macromolecules. J Chromatogr A. 1996;743(1):43–50.
ICH-Guidelines Q2(R1), Validation of analytical procedures: text and methodology. [Internet]. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2006. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf. Accessed 17 Feb 2021.
Wu D, Hwang P, Li T, Piszczek G. Rapid characterization of adeno-associated virus (AAV) gene therapy vectors by mass photometry. Gene Ther. 2022;20:1–7.
Acknowledgements
The authors thank Dong Xu and George Bou-Assaf for valuable scientific discussions and advice on experimental design and data analysis.
Funding
All work was funded by Biogen Inc.
Author information
Authors and Affiliations
Contributions
RF, DT, BB, NL, MS, and CB contributed to the experimental design and data analysis. RF, DT, BB, NL, MS, and CB did the experimental work. RF, DT, BB, and NL wrote the manuscript. RF, SB, ZS, and BY revised the manuscript. All authors edited the final manuscript version.
Corresponding author
Ethics declarations
Conflict of Interest
All authors are employees of Biogen Inc. and Biogen shareholders.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Frenkel, R., Tribby, D., Boumajny, B. et al. ACUVRA: Anion-Exchange Chromatography UV-Ratio Analysis—A QC-Friendly Method for Monitoring Adeno-Associated Virus Empty Capsid Content To Support Process Development and GMP Release Testing. AAPS J 25, 3 (2023). https://doi.org/10.1208/s12248-022-00768-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-022-00768-0