
Abstract
In recent years, global sensitivity analysis (GSA) has gained interest in physiologically based pharmacokinetics (PBPK) modelling and simulation from pharmaceutical industry, regulatory authorities, and academia. With the case study of an in-house PBPK model for inhaled compounds in rats, the aim of this work is to show how GSA can contribute in PBPK model development and daily use. We identified two types of GSA that differ in the aims and, thus, in the parameter variability: inter-compound and intra-compound GSA. The inter-compound GSA aims to understand which are the parameters that mostly influence the variability of the metrics of interest in the whole space of the drugs’ properties, and thus, it is useful during the model development. On the other hand, the intra-compound GSA aims to highlight how much the uncertainty associated with the parameters of a given drug impacts the uncertainty in the model prediction and so, it is useful during routine PBPK use. In this work, inter-compound GSA highlighted that dissolution- and formulation-related parameters were mostly important for the prediction of the fraction absorbed, while the permeability is the most important parameter for lung AUC and MRT. Intra-compound GSA highlighted that, for all the considered compounds, the permeability was one of the most important parameters for lung AUC, MRT and plasma MRT, while the extraction ratio and the dose for the plasma AUC. GSA is a crucial instrument for the quality assessment of model-based inference; for this reason, we suggest its use during both PBPK model development and use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Due to the opportunity of directly targeting the biophase of interest, the inhalation route has been considered a convenient way of drug administration for local treatment of lung-specific diseases, such as asthma and chronic obstructive pulmonary disease (COPD). This route allows the administration of drugs at lower dosages, minimizing potential side effects driven by high systemic exposures. Topically active compounds for lung diseases have normally an adequate, and generally sustained, lung residence time (1,2,3). However, efforts have to be placed in the optimization of drug lung disposition looking for an optimal lung retention. In fact, an increased residence time in the airways could potentially translate into the risk of drug removal from the lung due to mucociliary clearance or into the risk of unsafe drug accumulation in pulmonary tissues. For this reason, it is necessary to maintain an appropriate balance between lung retention and absorption by the modulation of the interplay of some key properties, such as solubility, permeability and lung tissue binding (1,3).
Nowadays, administration by inhalation to rodents is still an important step in preclinical development of new drugs designed for the inhalation route (1). A mechanistic model able to predict pharmacokinetic quantities of interest after inhalation of new compounds in preclinical species could be extremely beneficial during drug discovery (4), for example for prioritizing compounds before animal experiments or for preclinical to clinical translation.
For these reasons, in a previous work we developed a physiologically based pharmacokinetic (PBPK) model for inhaled drugs (5). This model was used to predict the compounds lung disposition in preclinical species (e.g. rodents), starting from physiological and in vitro parameters, such as mucociliary clearance rate, drug solubility and permeability.
In recent years sensitivity analysis, including global sensitivity analysis (GSA), has gained interest in PBPK modelling and simulation from both industry and academia (6,7,8,9,10,11). In a recent work, sensitivity, uncertainty and variability analyses were highlighted as tools able to improve the confidence in the context of inhalation PBPK modelling (12). Moreover, regulatory agencies highlighted that a sensitivity analysis should be performed during the process of development and refinement of PBPK models (13,14). Sensitivity analysis could be defined as “The study of how uncertainty in the output of a model (numerical or otherwise) can be apportioned to different sources of uncertainty in the model input” (15). GSA differs with respect to other types of sensitivity analysis, such as the local one, as it performs a multivariate variation of all the considered input parameters taken in their whole range of variation (16).
Like all the mechanistic models, physiological models have both uncertainty and variability in the model (input) parameters. Uncertainty refers to an incomplete understanding of the system, lack of data or error in the measurement/estimation of certain parameters. Variability instead refers to the inherent heterogeneity of the system properties or parameters, for example among subjects, experiments, or drugs. It is possible to reduce the uncertainty by performing better experiments and collect more data. Instead, it is impossible to reduce the variability (given a certain context); however, it could be better characterized (https://www.epa.gov/expobox/uncertainty-and-variability). Given the uncertainty (or variability) in the model parameters, it follows that also the predicted model outputs are uncertain (or variable) too (16). In our case, for example, the rat weight could be seen as a variable input parameter, while the values of the active and passive permeability across lung tissues as uncertain parameters. Pulmonary and plasma AUC are examples of pharmacokinetic output metrics of interest, whose uncertainties are driven by those of the model parameters.
Sensitivity analysis helps to get insights into the model behaviour as a function of the parameter variation (16). By performing this type of analysis, it is in fact possible to assess how much each parameter, with its variation, impacts the variation of the model outputs (16). Consequently, sensitivity analysis could be a valuable instrument to help in understanding if the model behaves as expected or what the parameters are that need to be more precisely known to allow reliable model predictions.
In this context, the aim of our work was to understand how GSA can be integrated in the process of PBPK model development and use. This was done within the case study of the in-house PBPK model for inhaled compounds in rats. Among all GSA methods, we choose the variance-based method because of its ability to detect interaction effects and to handle nonlinear and nonmonotonic relationships between the parameters and the model output (16,17,18).
In this work, we performed two types of GSA that differ in the aim and thus in the considered drug-specific parameter variability range: inter-compound and intra-compound GSA. Inter-compound GSA resembles the analysis done in Melillo et al. (6) for intestinal absorption models. Each of the drug-related model parameters was considered variable in a range given by the minimum and the maximum value of the considered set of compounds. Thus, inter-compound GSA mainly focalize on the “between-drug” parameter variability and it is useful to understand the model behaviour in the subspace of all the considered compounds. The aim of this analysis is to understand what the key parameters are that mostly explain the differences in the model predictions among drugs. On the other hand, intra-compound GSA is focused on the parameter uncertainties related to a specific compound. This analysis has the purpose of finding the most important parameters that, with their uncertainty, mostly cause the model output uncertainty. In this work, intra-compound GSA was performed on three representative compounds belonging to the Chiesi Farmaceutici portfolio (namely A, B and C). For each compound, drug-related parameters were considered to vary in a range representing the parameter uncertainty.
MATERIALS AND METHODS
Variance-Based GSA
Let us consider the generic model
with Y a scalar model output (e.g. plasma drug AUC), f the input-output relationship and X the vector of the k uncorrelated input parameters. GSA methods consider each Xi, i = 1…k, as a random variable, with associated a probability distribution (16,17). Thus, Y is a random variable too and can be obtained through model evaluation after sampling from the joint probability distribution of X. For each Xi, variance-based GSA derives two sensitivity indices from the decomposition of the variance (V) of Y, the so-called main (or first order) effect (Si) and total effect (ST,i), in Eqs. (2) and (3), respectively (16,17).
E is the expected value and X~i is the vector containing all the parameters, except Xi. Both Si and ST, i are always included in [0, 1]. Si is related with the part of V(Y) explained by the variation of Xi taken alone, and ST, i is the sum of Si with the interaction effects of Xi with all the other inputs. Interaction effects can arise when more than one parameter varies at the same time. The relationship Si ≤ ST, i is always valid. The higher Si and ST, i are, the more the variation of Xi explains V(Y) and so, the more Xi is considered important. Instead, ST, i = 0 means that Xi is not influent on V(Y). The difference ST, i − Si gives information about the extent of interaction effect involving the ith parameter (16,17).
Pulmonary Absorption Model
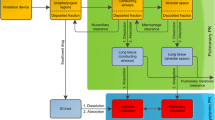
The considered physiologically based model was originally presented in Grandoni et al. (5) and was inspired by the work of Boger et al. (19). The model is composed of three parts describing the pulmonary absorption, the intestinal absorption and the systemic disposition. The pulmonary absorption model was built to take into account the principal PK processes occurring when a drug is inhaled: deposition, mucociliary clearance, dissolution, absorption in lung tissue and in blood circulation (20). In the model, lungs were divided into two parts, the central and the peripheral regions. The central region corresponds approximatively to the tracheobronchial region, while the peripheral region to the alveolar region. Both the regions were further divided in four compartments: the undissolved drug, the dissolved drug, the extravascular and vascular lung tissue. The central region was considered perfused by the systemic circulation, while the peripheral region by the pulmonary circulation. The model structure is shown in Fig. 1.

Physiologically based model structure. The model is composed of three parts: pulmonary absorption model, intestinal absorption model and systemic PBPK. Red and blue arrows represent arterial and venous blood flows, respectively. Black dotted arrows represent the clearance processes
Drugs were considered intra-tracheally administered to rats. Only a fraction of the drug amount administered to the animal actually reaches lungs (Finh), whereas the rest it is supposed to be deposited in the oropharyngeal region and gets swallowed (Fswa). Of the fraction delivered to lungs, a part is deposed in the central region (FC) and a part reaches the peripheral region (FP). FC was calculated from the mass median aerodynamic diameter (MMAD) and the geometric standard deviation (GSD) of the drug particles with the software MPPD2.11 (21,22), as explained in the supplementary materials, section 2. Once deposited, in both central and peripheral regions, the drug dissolves in the physiological fluids and then is supposed to be passively absorbed in the lung tissues. Here, the drug can diffuse to the vascular compartment or back to the dissolved drug compartment. A monodirectional transport from the tissue to the dissolved drug compartment was included to account for the possible action of efflux transporters, such as the P-glycoproteins. The mucociliary clearance mechanism has been considered acting only on the undissolved drug compartment of the central region, since this mechanism should be negligible in the alveoli (20).
To describe the systemic drug disposition and the intestinal absorption, the pulmonary absorption model was coupled with the whole body PBPK model presented in Grandoni et al. (23), as shown in Fig. 1. All the model equations are reported in the supplementary materials, section 1. Physiological lung-related parameters are reported in Table SII. The ranges of variability of physiological parameters are reported in Table I. All the remaining physiological parameter values (e.g. organ volumes and blood flows) of the whole body PBPK model are reported in the supplementary materials, section 4.
Inter-compound and Intra-compound GSA
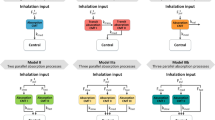
Inter-compound and intra-compound are two ways of performing GSA that differ in the aim and, thus, in the considered drug-specific parameter variability range. Figure 2 didactically shows the difference.

Difference in the parameter space used for inter-compound and intra-compound GSA. Inter-compound GSA (green area) considers the “between-drug” variability, while intra-compound GSA (red area) considers the uncertainty associated to the parameters in a specific compound. As shown in the figure, generally variability is wider than uncertainty
We performed the inter-compound GSA on the physiological pulmonary absorption model decoupled from the distribution PBPK and the intestinal absorption models. This was done to characterize the absorption process and to simplify the model and the results understanding. To decouple the model, blood inflow and outflow values of both peripheral and central lung vascular compartments were set equal to zero. Moreover, the fluxes due to passive permeability from the vascular to the extravascular compartments were set equal to zero too, in both the lung regions. Thus, lung vascular compartments behave like traps (integrators). The outputs considered in this analysis are the fraction absorbed (fa) and the AUC and MRT of the drug concentration in the whole lung. Whole lung concentration was obtained as the sum of the solid and dissolved amounts in the epithelial lining fluids with the amounts in the tracheobronchial and alveolar extravascular compartments, divided by the total volume. fa was obtained as fa = 1 − fCL, where fCL is the fraction of the drug eliminated by the mucociliary clearance at the steady state.
In the inter-compound GSA, we considered each of the drug-related parameters varying in a range given by the minimum and the maximum values observed in a set of nine compounds belonging to the Chiesi Farmaceutici portfolio (Table II). The parameter distributions were considered uniform in these ranges. The PBPK model performances on these nine compounds are shown in the supplementary materials, section 5.
To have more homogeneous behaviour, inter-compound GSA was performed separately for highly and poorly soluble compounds. The criterion used to classify compounds in the two groups was inspired to the one adopted for orally administered compounds (24,25,26). A dose number for inhaled compounds was defined as in Eq. (4).
Drug dose was fixed to 10 μg (i.e. the typical selected dose in the considered experiments), Velf is the lung epithelial lining fluid (ELF) volume (sum of the central and peripheral ELF volumes) and \( {C}_{\mathrm{S},\mathrm{p}{\mathrm{H}}_{\mathrm{elf}}} \) is the drug solubility measured in simulated lung fluid at pH 6.9 (27,28). A compound was classified as highly soluble if D0, inh < 1 or poorly soluble if D0, inh ≥ 1. During GSA, we first extracted the values of D0, inh and then we computed the \( {C}_{\mathrm{S},\mathrm{p}{\mathrm{H}}_{\mathrm{elf}}} \) using Eq. (4) (6).
The intra-compound GSA was performed on the whole body PBPK model for three representative compounds, characterized by different properties. The considered outputs are the drug whole lung and plasma concentration AUC and MRT. Here, whole lung concentration was calculated as the sum of the drug amount in all the pulmonary absorption model compartments, divided by the lung total volume. All the drug-specific model parameters were considered uniformly distributed between the ranges reported in Table III, except for the dose that was considered normally distributed with a CV equal to 15% (29). When no experimental data supporting the uncertainty range definition were available, ranges reflecting the perceived parameter uncertainties were used.
To account for the population variability of rat weight, all the volumes and blood flows were multiplied for (wsubj/wmean) and (wsubj/wmean)0.75, respectively, where wsubj is the extracted rat weight and wmean is the mean rat weight equal to 250 g, as used in (23).
All the analyses were performed by using MATLAB R2019a (30) on a 64-bit computer configured with Intel® Core™ i7-7700 @ 3.60 GHz x8 processors, running Ubuntu 16.04 LTS. The differential equations were solved by using the ‘ode15s’ MATLAB solver, for a time span ranging from 0 to 400 h. An ad hoc MATALB code to implement variance-based GSA was developed. To perform the GSA in both inter- and intra-compound cases, we used 20,000 samples (n) extracted from a unit hypercube. Then, samples were brought back to each parameter distribution by using simple linear transformations for the uniformly distributed parameters and the inverse cumulative density function for the other parameters. n number of samples corresponds to n(k + 2) number of model evaluation needed to compute the sensitivity indices, with k the number of parameters (17). Uncertainty on the calculation of the sensitivity indices was estimated by using 10,000 bootstrap samples (31).
RESULTS
Inter-compound GSA
Here, the results of the inter-compound GSA on the pulmonary absorption model decoupled from the whole-body PBPK, for both highly and poorly soluble compounds, are reported. References to figures reported in the supplementary materials are indicated with an “S”, followed by an Arabic number. In Fig. S7, the distributions of the selected model outputs are shown. In Fig. 3 the sensitivity indices for fa and for the logarithms of drug concentration AUC and MRT in lungs are shown. We choose the log scale for the AUC and MRT to avoid problems in the computation of the variance-based sensitivity index estimation due to the skewness of AUC and MRT distributions in natural scale (32).

Inter-compound GSA results for the output of interests: a fraction absorbed for highly soluble compounds; b logarithm of whole lung AUC for highly soluble compounds; c logarithm of whole lung MRT for highly soluble compounds; d fraction absorbed for poorly soluble compounds; e logarithm of whole lung AUC for poorly soluble compounds; f logarithm of whole lung MRT for poorly soluble compounds. Concerning the parameters: mw is the compound molecular weight; MMAD is the mass median aerodynamic diameter; GSD is the geometric standard deviation; rho the drug true density; D0_inh is the dose number; F_inh the inhaled fraction; fu_t is the fraction unbound in lung tissue; fu_elf is the fraction unbound in the epithelial lining fluid; P_p is the passive permeability; P_a is the active permeability; weight is the rat body weigh; k_MC is the mucociliary clearance; alpha is the scaling factor for the peripheral lung passive permeability
For highly soluble compounds, the parameters that mostly explain the fa variability are D0, inh and MMAD. Concerning D0, inh, it is probably important because it controls the solubility, and so the dissolution rate. The higher the dissolution rate is, the faster the drug is removed from the solid compartment in the central region. That is in fact the region in which the drug could be eliminated via mucociliary clearance. However, the fa variability is quite low, as shown in Figure S7. So, D0, inh and MMAD are the most important parameters, but the variation of the model output is in any case limited.
For poorly soluble compounds, both MMAD and D0, inh, even if with a minor contribution of GSD and the active permeability (Pa), still impact the fa variability. Concerning D0, inh, the reasons of its importance are probably the same of highly soluble compounds. MMAD could be important in determining fa variability mainly for two reasons: first of all, the MMAD value could impact the dissolution rate when the solubility is low; second, it determines FC and, mainly for poorly soluble compounds, fa could be sensitive to the repartition between central and peripheral regions. In fact, in peripheral region the mucociliary clearance does not occur. The main difference with respect to the high solubility case is that fa variability is quite high. Thus, MMAD and D0, inh are responsible for a great variation of the model output.
Concerning lung AUC, the parameters that mostly explain its variability for highly soluble compounds are fu, t (fraction unbound in the lung tissue), Pp (passive permeability) and Pa. This probably happens because fu, t and the permeabilities are parameters that determine the drug retention into the lungs and thus they control the AUC. For poorly soluble compounds, in addition to parameters that control the drug retention into the lungs, D0, inh and MMAD are also important. This happens because, as explained before, they could impact fa.
Concerning lung MRT, the parameters that mostly explain the variability for highly soluble compounds are still fu, t and both passive and active permeabilities. The reasons of their importance are probably similar to the one for the AUC: these are the parameters responsible to the drug retention in lungs. For poorly soluble compounds, the most important parameters are the same of highly soluble compounds, with the addition of D0, inh and MMAD.
Intra-compound GSA
Here the results of the intra-compound GSA for three representative compounds belonging to the Chiesi Farmaceutici portfolio, namely A, B and C, are reported. With respect to all the other compounds, compound A is characterized by a lower solubility, a higher permeability and a low fu, t. Compound B has a higher solubility, a lower permeability and a higher fu, t. Finally, compound C has an intermediate solubility and permeability and a low fu, t. The parameter values and associated uncertainty or variability are reported in Table III.
Compound A
The distribution of drug plasma and whole lung AUC and MRT and the sensitivity indices relative to compound A are reported in Figure S8 and Fig. 4, respectively.

Intra-compound GSA results for a whole lung AUC, b whole lung MRT, c plasma AUC and d plasma MRT for compound A. Concerning the parameters: dose is the drug dose; weight is the rat body weight; k_MC is the mucociliary clearance; logP_ow is the logarithm of the octanol-water partition coefficient; pKa is the drug pKa; P_p is the passive permeability; P_a is the active permeability; Caco2AB is the permeability calculated using the CACO2 cell layer; BP is the blood to plasma ratio; Er is the extraction ratio; S is the drug solubility; MMAD is the mass median aerodynamic diameter; GSD is the geometric standard deviation; F_inh the inhaled fraction; fu_elf is the fraction unbound in the epithelial lining fluid; fu_t is the fraction unbound in lung tissue; fu_p is the fraction unbound in plasma; GFR is the glomerular filtration rate; rho is the drug true density; alpha is the scaling factor for the peripheral lung passive permeability
Whole lung AUC variance is mainly explained by the Pp uncertainty, together with a minor contribution of the dose, fu, t, rat weight and kMC (mucociliary clearance) variabilities. Even if the drug has a low solubility, it seems that the highest impact on the AUC is attributed to the passive permeability. This probably happens because compound A has very low fraction unbound in tissue; thus, even if the permeability of the free compound is relatively high, the overall drug flux across the cell membrane results to be the rate limiting step. In addition to that, it should be noted that the uncertainty associated to the passive permeability is higher with respect to those associated to other parameters. The passive permeability is still the most important parameter when whole lung MRT is considered. The reasons seem to be similar to those discussed for the AUC. Plasma AUC variability is mainly explained by the extraction ratio and, to a minor extent, by the dose, MMAD and GSD variabilities. These results highlight that the elimination process plays a major role in determining the plasma AUC variability. Unless nonlinear processes are involved, the importance of the dose and the extraction ratio on the plasma AUC is completely expected from a PK point of view. Concerning the plasma MRT, the most important parameter is the passive permeability. This probably happens because the drug is slowly absorbed from the lungs into the systemic circulation.
Compound B
The distribution of drug plasma and whole lung AUC and MRT and the sensitivity indices for relative to compound B are reported in Figure S9 and Fig. 5, respectively.

Intra-compound GSA results for a whole lung AUC, b whole lung MRT, c plasma AUC and d plasma MRT for compound B. For the parameters’ acronyms, please refer to Fig. 4 label
The parameter that mostly explains compound B whole lung AUC and MRT variation is Pp. This probably happens because compound B has a lower permeability and higher solubility with respect to the other compounds of interest; thus, the absorption rate is probably permeability limited. Moreover, as explained for compound A, with respect to all the other parameters, the permeability has associated a greater uncertainty; thus, it is more likely that they have a relevant impact in explaining the AUC variation.
Concerning the plasma AUC, the most important parameters are the dose and the extraction ratio, followed by rat weight and the blood to plasma ratio (BP). As for compound A, the elimination process is more important than the distribution or absorption processes in determining the AUC variation. The most important parameter in explaining the MRT variance is the passive permeability. As for compound A, this probably happens because the drug is slowly absorbed from the lungs into the systemic circulation.
Compound C
The distribution of plasma and whole lung AUC and MRT and the sensitivity indices relative to compound C are reported in Figure S10 and Fig. 6, respectively.

Intra-compound GSA results for a whole lung AUC, b whole lung MRT, c plasma AUC and d plasma MRT for compound C. For the parameters’ acronyms, please refer to Fig. 4 label
For both whole lung AUC and MRT, the parameter that with its variation mainly explains their variability is Pp. The reasons are probably similar to those reported for compounds A and B. Moreover, compound C has a higher solubility, but lower permeability than compound A and, together with a low fraction unbound, this could explain the slightly higher importance of the passive permeability with respect to compound A.
Concerning the plasma AUC, the parameters that mainly explain its variation are the dose and the extraction ratio, followed by the passive permeability, rat weight and BP. These results highlight that probably, for this drug, the elimination process has a greater role in determining the AUC variability with respect to the distribution or absorption. Concerning the plasma MRT, the situation resembles the one of compound A.
DISCUSSION
In this manuscript, we showed how GSA techniques were used to assess model behaviours and support the development of a mechanistic model describing pulmonary absorption for inhaled compounds. We identified two ways of performing GSA that differ in the aims and, thus, in the considered parameter variability: inter-compounds and intra-compound. Both the approaches helped in understanding different model aspects.
The inter-compound GSA was performed for the absorption model decoupled from the distribution PBPK. This analysis can be performed in more “homogeneous” sub-spaces of the whole parameter space, as we have done distinguishing highly soluble from poorly soluble compounds. Looking at the model output distributions gives the possibility of assessing the extent of the inter-compound variability of the metrics of interest. Then, GSA helps in understanding what the parameters are that mostly determine the observed variation of the output predictions between different compounds. For example, from our analysis it was possible to understand that the between-compound differences in the lung AUC for the low solubility compounds are mainly driven by the variation of both solubility- and permeability-related parameters and that strong interaction effects are present. In particular, the latter means that the effect of solubility and permeability on the lung AUC is not additive. This information may be particularly useful if the inter-compound GSA results are used to inform the optimization of the compounds’ physicochemical properties. As a consequence of performing inter-compound GSA, we gained insights on the model behaviour and, consequently, we increased the knowledge of the model. For these characteristics, inter-compound GSA is particularly useful for the error detection. In fact, in case of discrepancies between the expected and the actual model behaviours, GSA gives useful information that helps in identifying the reasons and, thus, possible errors in the model assumptions or implementation. When the inter-compound GSA was applied for the first time to our in-house PBPK model (that was still in the development phase), we immediately noticed that the model did not behave as expected. Guided by the GSA results, we found and corrected the implementation error. The implementation of complex PBPK models often consists of thousands of lines of code and hundreds of parameters (this is particularly true for commercial PBPK software). A ‘little bug’ in the code may have a huge impact on the model behaviour. For its ability to assess the model behaviour, to help in the error detection and, as reported by Iooss and Saltelli, for the general GSA capability of helping in identifying sensitive assumptions (33), the inter-compound GSA is particularly useful during the process of model development. Theoretically, if the model structure and the physiological and inter-compound parameter variabilities are correctly identified and fixed, this analysis can be performed just once (e.g. when the model is firstly presented or at the PBPK platform release).
The intra-compound GSA was instead performed for three representative compounds on the whole body PBPK model. The parameter variation was defined to represent the uncertainties associated to their values for a specific compound. With this analysis it is possible to know how much the model output variation is apportioned to the uncertainty of the parameters. When doing this analysis, it is useful to look at the output distribution, to determine if it is narrow enough to be considered acceptable. If not, GSA helps in selecting which parameters should be known with less degree of uncertainty in order to give a more accurate prediction. For example, we believe that the uncertainty associated to compound A lung MRT is too high. So, if one is interested in using this model for lung MRT predictions (e.g. for different dosages or species), from the GSA results we know that a better characterization of the passive permeability is needed in order to reduce uncertainties of the considered metrics. This situation probably does not happen for compound A lung AUC, given that we believe that the uncertainty associated with the prediction of this metric can be considered acceptable. Differently from inter-compound GSA, intra-compound GSA should be performed each time the model (or the PBPK platform) is used for a specific drug.
One challenge that we have faced in performing GSA was that, in certain situations, determining the uncertainty or variability ranges was not an easy task, in particular for the intra-compound GSA. In fact, due to the lack of available data, it was sometimes difficult to appropriately quantify the parameters’ degree of uncertainty. In these cases, expert opinion can be used to fill this gap.
In our work, we presented the applications of inter-compound and intra-compound GSA in a preclinical setting involving animal models. However, the use of these analyses is not limited to this context. The results of the inter-compound GSA highlight how a simultaneous variation of the compounds’ physicochemical properties impacts the model outputs. This information could be particularly precious in guiding compound optimization in early discovery phases. Moreover, similarly to what was shown here, the inter-compound GSA can be used in clinics to understand how the model behaves varying together both drug- and subject-specific parameters. Moreover, a comparison of the clinical inter-compound GSA results with the ones from the preclinical setting could give insights on how physiological changes across the species impact the model results. Intra-compound GSA can be useful outside of the preclinical setting as well. For its characteristics, this analysis is particularly suitable when the model is used to predict unseen scenarios, such as for the first in human translation and for predicting the PK in different populations (e.g. paediatrics).
CONCLUSION
In this work, we showed how GSA can be used within PBPK modelling and simulation. Performing GSA during both the model development and routine use increases the knowledge of the model; it helps in finding errors and in identifying the parameters that must be known with higher confidence, if one is interested in reducing the model prediction uncertainties. GSA is a crucial instrument for the quality assessment of model-based inference; for this reason, we suggest its use during both PBPK model development and use.
References
Forbes B, Asgharian B, Dailey LA, Ferguson D, Gerde P, Gumbleton M, et al. Challenges in inhaled product development and opportunities for open innovation. Adv Drug Deliv Rev. 2011;63(1):69–87.
Labiris NR, Dolovich MB. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol. 2003;56(6):588–99.
Strong P, Ito K, Murray J, Rapeport G. Current approaches to the discovery of novel inhaled medicines. Drug Discov Today. 2018;23(10):1705–17.
Bäckman P, Arora S, Couet W, Forbes B, de Kruijf W, Paudel A. Advances in experimental and mechanistic computational models to understand pulmonary exposure to inhaled drugs. Eur J Pharm Sci. 2018;113:41–52.
Grandoni S, Cesari N, Melillo N, Brogin G, Puccini P, Magni P. Development and evaluation of a PBPK model to study the pharmacokinetics of inhaled drugs in rats. In: PAGE 28, Abstr 9047 [Internet]. Stockholm, Sweden.; 2019. Available from: www.page-meeting.org/?abstract=9047
Melillo N, Aarons L, Magni P, Darwich AS. Variance based global sensitivity analysis of physiologically based pharmacokinetic absorption models for BCS I–IV drugs. J Pharmacokinet Pharmacodyn. 2019;46(1):27–42.
Melillo N, Darwich AS, Magni P, Rostami-Hodjegan A. Accounting for inter-correlation between enzyme abundance: a simulation study to assess implications on global sensitivity analysis within physiologically-based pharmacokinetics. J Pharmacokinet Pharmacodyn. 2019;46(2):137–54.
Zhang X-Y, Trame M, Lesko L, Schmidt S. Sobol sensitivity analysis: a tool to guide the development and evaluation of systems pharmacology models. CPT Pharmacometrics Syst Pharmacol. 2015;4(2):69–79.
McNally K, Cotton R, Loizou GD. A workflow for global sensitivity analysis of PBPK models. Front Pharmacol. 2011;2:31.
Daga PR, Bolger MB, Haworth IS, Clark RD, Martin EJ. Physiologically based pharmacokinetic modeling in lead optimization. 2. Rational bioavailability design by global sensitivity analysis to identify properties affecting bioavailability. Mol Pharm. 2018;15(3):831–9.
Yau E, Olivares-Morales A, Gertz M, Parrott N, Darwich AS, Aarons L, et al. Global sensitivity analysis of the Rodgers and Rowland model for prediction of tissue: plasma partitioning coefficients: assessment of the key physiological and physicochemical factors that determine small-molecule tissue distribution. AAPS J. 2020;22(2):41.
Kolli AR, Kuczaj AK, Martin F, Hayes AW, Peitsch MC, Hoeng J. Bridging inhaled aerosol dosimetry to physiologically based pharmacokinetic modeling for toxicological assessment: nicotine delivery systems and beyond. Crit Rev Toxicol. 2019;49(9):725–41.
CHMP (EMA). Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation - draft. London, UK: Committee for Medicinal Products for Human Use (CHMP), European Medicines Agency (EMA); 2016 Jul. Report No.: EMA/CHMP/458101/2016.
CDER (FDA). Physiologically based pharmacokinetic analyses - format and content: guidance for industry, draft. Rockville, MD: Center for Drug Evaluation and Research (CDER), U.S. Food and Drug Administration (FDA); 2016
Saltelli A, Tarantola S, Campolongo F, Ratto M. Sensitivity analysis in practice: a guide to assessing scientific models [internet]: John Wiley & Sons, Ltd; 2004. Available from:. https://doi.org/10.1002/0470870958.
Saltelli A, Ratto M, Andres T, Campolongo F, Cariboni J, Gatelli D, et al. Global sensitivity analysis. The Primer [Internet]. John Wiley & Sons, Ltd; 2008. Available from: https://doi.org/10.1002/9780470725184
Saltelli A. Making best use of model evaluations to compute sensitivity indices. Comput Phys Commun. 2002 May 15;145(2):280–97.
Sobol IM. Sensitivity estimates for nonlinear mathematical models. Math Model Comput Exp. 1993;1(4):407–14.
Boger E, Evans N, Chappell M, Lundqvist A, Ewing P, Wigenborg A, et al. Systems pharmacology approach for prediction of pulmonary and systemic pharmacokinetics and receptor occupancy of inhaled drugs. CPT Pharmacometrics Syst Pharmacol. 2016;5(4):201–10.
Borghardt JM, Weber B, Staab A, Kloft C. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 2015;17(4):853–70.
Anjilvel S, Asgharian B. A multiple-path model of particle deposition in the rat lung. Fundam Appl Toxicol. 1995;28(1):41–50.
National Institute for Public Health and the Environment (RIVM). Multiple path particle dosimetry model (MPPD v 1.0): a model for human and rat airway particle dosimetry. Bilthoven, The Netherlands. Bilthoven, The Netherlands.: RIVA Report 650010030.; 2002.
Grandoni S, Cesari N, Brogin G, Puccini P, Magni P. Building in-house PBPK modelling tools for oral drug administration from literature information. ADMET and DMPK. 2019;7(1):4–21.
Dahan A, Miller JM, Amidon GL. Prediction of solubility and permeability class membership: provisional BCS classification of the world’s top oral drugs. AAPS J. 2009;11(4):740–6.
Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20.
Hastedt JE, Bäckman P, Clark AR, Doub W, Hickey A, Hochhaus G, et al. Scope and relevance of a pulmonary biopharmaceutical classification system AAPS/FDA/USP Workshop March 16-17th, 2015 in Baltimore, MD. AAPS Open. 2016;2(1):1.
Hassoun M, Royall PG, Parry M, Harvey RD, Forbes B. Design and development of a biorelevant simulated human lung fluid. J Drug Deliv Sci Technol. 2018;47:485–91.
Stefani M, Cesari N, Corsaletti R, Fioni A, Saccani F, Volta R, et al. Development of an in vitro solubility test as a tool for predicting lung retention of poorly water soluble compounds. In: The Aerosol Society [Internet]. Edinburgh; 2018 [cited 2019 Aug 6]. Available from: https://aerosol-soc.com/abstracts/development-of-an-in-vitro-solubility-test-as-a-tool-for-predicting-lung-retention-of-poorly-water-soluble-compounds/
Fioni A, Selg E, Cenacchi V, Acevedo F, Brogin G, Gerde P, et al. Investigation of lung pharmacokinetic of the novel PDE4 inhibitor CHF6001 in preclinical models: evaluation of the PreciseInhale technology. J Aerosol Med Pulm Drug Deliv. 2017;31(1):61–70.
MATLAB R2019a. Natick: The MahWorks, Inc.; 2019.
Archer GEB, Saltelli A, Sobol IM. Sensitivity measures, ANOVA-like techniques and the use of bootstrap. J Stat Comput Simul. 1997;58(2):99–120.
Iman RL, Hora SC. A robust measure of uncertainty importance for use in fault tree system analysis. Risk Anal. 1990;10(3):401–6.
Iooss B, Saltelli A. Introduction to sensitivity analysis. In: Ghanem R, Higdon D, Owhadi H, editors. Handbook of uncertainty quantification [internet]. Cham: Springer International Publishing; 2016 [cited 2020 Jul 6]. p. 1–20. Available from: https://doi.org/10.1007/978-3-319-11259-6_31-1.
Hofmann W, Asgharian B. The effect of lung structure on mucociliary clearance and particle retention in human and rat lungs. Toxicol Sci. 2003;73(2):448–56.
Magdalena A-G, Brands MW, Zappe DH, Hall John E. Hypertension in obese Zucker rats. Hypertension. 1996;28(6):1047–54.
Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, et al. Large porous particles for pulmonary drug delivery. Science. 1997;276(5320):1868–72.
Gehr P, Annexe A. Anatomy and morphology of the respiratory tract. Ann ICRP. 1994;24(1–3):121–66.
Mercer RR, Russell ML, Roggli VL, Crapo JD. Cell number and distribution in human and rat airways. Am J Respir Cell Mol Biol. 1994;10(6):613–24.
Nie Y-C, Wu H, Li P-B, Luo Y-L, Long K, Xie L-M, et al. Anti-inflammatory effects of naringin in chronic pulmonary neutrophilic inflammation in cigarette smoke-exposed rats. J Med Food. 2012;15(10):894–900.
Parent RA. Treatise on pulmonary toxicology: comparative biology of the normal lung (Vol 1): CRC Press; 1992. p. 884.
Pinkerton KE, Gehr P, Castañeda A, Crapo JD. Architecture and cellular composition of the air-blood tissue barrier. In: Comparative Biology of the Normal Lung: Second Edition [Internet]. Elsevier Inc.; 2015 [cited 2019 Jul 29]. p. 105–17. Available from: https://ucdavis.pure.elsevier.com/en/publications/architecture-and-cellular-composition-of-the-air-blood-tissue-bar
Plopper CG, Hill LH, Mariassy AT. Ultrastructure of the nonciliated bronchiolar epithelial (Clara) cell of mammalian lung. III. A study of man with comparison of 15 mammalian species. Exp Lung Res. 1980;1(2):171–80.
Reynolds SD, Pinkerton KE, Mariassy AT. Epithelial cells of trachea and bronchi. In: Comparative biology of the normal lung: second edition [Internet]. Elsevier Inc.; 2015 [cited 2019 Jul 29]. p. 61–81. Available from: https://ucdavis.pure.elsevier.com/en/publications/epithelial-cells-of-trachea-and-bronchi
Acknowledgments
The study was sponsored by Chiesi Farmaceutici S.p.A.
Funding
Open access funding provided by Università degli Studi di Pavia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 694 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Melillo, N., Grandoni, S., Cesari, N. et al. Inter-compound and Intra-compound Global Sensitivity Analysis of a Physiological Model for Pulmonary Absorption of Inhaled Compounds. AAPS J 22, 116 (2020). https://doi.org/10.1208/s12248-020-00499-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-020-00499-0