
Abstract
A super-saturated self-nanoemulsifying drug delivery system (super-SNEDDS), containing the poorly water-soluble drug halofantrine (Hf) at 150% of equilibrium solubility (S eq), was compared in vitro and in vivo with a conventional SNEDDS (75% of S eq) with respect to bioavailability and digestibility. Further, the effect of digestion on oral absorption of Hf from SNEDDS and super-SNEDDS was assessed by incorporation of the lipase inhibitor tetrahydrolipstatin (orlistat) into the SNEDDS. The SNEDDS contained soybean oil/Maisine 34-I (1:1), Kolliphor RH40, and ethanol at a ratio of 55:35:10, w/w percent. For the dynamic in vitro lipolysis, the precipitation of Hf at 60 min was significantly larger for the super-SNEDDS (66.8 ± 16.4%) than for the SNEDDS (18.5 ± 9.2%). The inhibition of the in vitro digestion by orlistat (1% (w/w)) lowered drug precipitation significantly for both the super-SNEDDS (36.8 ± 1.7%) and the SNEDDS (3.9 ± 0.7%). In the in vivo studies, the super-SNEDDS concept proved valid in a rat model with a significantly larger C max for the super-SNEDDS (964 ± 167 ng/mL) than for the SNEDDS (506 ± 112 ng/mL). The bioavailability of Hf dosed in super-SNEDDS (32.9 ± 3.6%) and SNEDDS (22.5 ± 6.3%) did not change significantly with co-administration of orlistat (45.5 ± 7.3% and 21.9 ± 6.5%, respectively). However, the pharmacokinetic parameters changed; the t max of the super-SNEDDS (1.3 ± 0.1 h) and SNEDDS (2.8 ± 1.2 h) were significantly lower when dosed with orlistat (6.0 ± 1.3 and 6.3 ± 1.2 h, respectively). These findings suggest that the role of lipid digestion for the absorption of drugs from SNEDDS may be less important than previously thought.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The physico-chemical properties of highly lipophilic compounds often result in poor and variable bioavailability (1,2). This calls for enabling drug delivery systems in order to decrease variability in absorption and to increase bioavailability. One strategy to solve this problem is to utilize lipid- and surfactant-based drug delivery systems, such as self-nanoemulsifying drug delivery systems (SNEDDS). SNEDDS preconcentrates are water-free isotropic systems consisting of oil, surfactant, co-solvent, and drug, which upon mild agitation in aqueous environments (such as in the gastrointestinal tract) form a nanoemulsion. The delivery of lipophilic drugs in SNEDDS often results in an increased oral absorption and hence an improved bioavailability (3).
Traditionally, drugs are loaded into SNEDDS at concentrations below their equilibrium solubility (S eq) (4). The potentially resulting low drug load, however, can be an obstacle, and a reason not to develop a SNEDDS for a given drug, for example if the required dose cannot be obtained in one single capsule. In contrast, the recently introduced super-saturated SNEDDS (super-SNEDDS) contain the drug at concentrations above S eq and thereby both the dose and the thermodynamic activity of these delivery systems are increased. Recent studies in dogs and minipigs have shown that the bioavailability of a drug in super-SNEDDS is equal to or better than that in conventional SNEDDS when the same dose is given. Thus, the number of capsules to be ingested to obtain the desired dose can be decreased (5–7). However, due to the lower cost and the ease of handling of rats compared to dogs and minipigs, studies in rats are often preferred.
Among the many variables that are thought to be affecting bioavailability of a drug dosed in SNEDDS and super-SNEDDS, the most prominent are the physico-chemical properties of the drug, the type and composition of the lipid vehicle used, and the emulsification tendency and digestibility of the SNEDDS (3,8,9). Lipid digestion and the physico-chemical behavior of the digestion products have been extensively studied since the early 1960s (10). Digestion is a dynamic process, and studies have shown that various colloidal phases with different morphologies are formed during lipid digestion. Specifically, the digestion of SNEDDS is thought to play an important role for the absorption of the drugs due to the altered solubilizing capacity of the various and transient colloidal structures (11,12). For example, Fatouros and coworkers subjected a SNEDDS composed of long-chain triacylglycerides to in vitro lipolysis and the formed structures were analyzed by small-angle X-ray scattering (SAXS) (13). Initially, oil droplets coexist with micelles, but after 60 min of lipolysis (corresponding to 90% digestion of the triacylglycerides), predominantly mixed micelles were observed along with few unilamellar vesicles. The traditional theory on drug absorption from SNEDDS thus revolves around the solubilization of the drug in the formed colloidal structures and the formation of mixed micelles, promoting increased absorption. The general belief is that the mixed micelles containing the drug will diffuse to the unstirred water layer at the epithelial membrane and, due to the pH gradient found there, they disintegrate. The digestion products, the mixed micelles are composed of, will be absorbed and the bile salts re-circulated. The drug will be released from the mixed micelles upon their disintegration and hence be absorbed as free drug molecules.
One strategy to evaluate the role of digestion on drug absorption is the use of tetrahydrolipstatin (orlistat). Orlistat is a potent inhibitor of different lipases such as pancreatic lipase, gastric lipase, and carboxyl ester lipase, but not phospholipase A2 (14). In vitro studies with orlistat have shown that when it interacts with porcine pancreatic lipase, the interaction is irreversible; however, the same may not be true for all lipases (14). A more recent study with human pancreatic lipase has shown that the inhibitory effect exerted by orlistat may in fact be reversible (15). Orlistat has previously been shown to inhibit rat pancreatic lipase effectively and can therefore be used as a tool to inhibit lipolysis (digestion) of nanoemulsion droplets in vivo in rats (16). Orlistat is lipophilic and forms a stable monolayer at the oil-water interface; the availability of orlistat at the interface is of great importance to the inhibition of lipases, whereas orlistat in the core of an emulsion droplet will act merely as a reservoir (17). Hence, orlistat is expected to be present at the oil-water interface of the emulsion droplets, where it forms a complex with the lipase at its active site and blocks the activity of the lipase (18). In a previous study applying orlistat as a tool, de Smidt et al. found that the digestibility of medium-chain triacylglycerides and surfactant-based formulations did not significantly influence the absorption of penclomidine in rats. However, when using crude medium-chain triglyceride as a vehicle, the absorption was to a higher degree dependent on digestion (9).
The aims of the present study were twofold: to evaluate if the super-SNEDDS concept is also valid in rats and to elucidate the effect of digestion on drug absorption from SNEDDS using the lipase inhibitor orlistat as a tool.
MATERIALS AND METHODS
Materials
Halofantrine hydrochloride was purchased from APAC Pharmaceutical LLC (Hangzhou, China), and the halofantrine base was subsequently prepared from halofantrine hydrochloride as previously described (19). Lipoid E 80 was kindly donated by Lipoid (Ludwigshafen, Germany). Soybean oil (long-chain (LC) glycerides), glycerol, ethylenediaminetetraacetic acid tripotassium salt dihydrate (EDTA), 4-bromophenyl-boronic acid (BBBA), bile extract, tris-(hydroxymethyl)aminomethane (tris), maleic acid, calcium chloride, sodium hydroxide, and porcine pancreatic lipase were obtained from Sigma-Aldrich (St. Louis, MO, USA). Maisine 35-1 (a mixture of LC mono-, di-, and triglycerides) was kindly donated by Gattefossé (St. Priest, France), and Kolliphor RH 40 was donated by BASF (Ludwigshafen, Germany). Euthanyl® (sodium pentobarbital 240 mg/mL) was supplied by Bimeda-MTC (Cambridge, Ontario, Canada). Purified water was obtained from a Millipore Milli-Q Ultra Pure water purification system (Billerica, MA, USA). tert-Butyl methyl ether (TBME), UPLC-grade acetonitrile, and glacial acetic acid were all from Fisher Scientific (Waltham, MA, USA). All other chemicals were of analytical grade.
Preparation of Formulations
Preconcentrates were prepared as previously described (20). The SNEDDS were composed of soybean oil (27.5% (w/w)), Maisine 35-1 (27.5% (w/w)), Kolliphor RH40 (35% (w/w)), and absolute ethanol (10% (w/w)). Briefly, the LC glycerides (soybean oil) and molten Maisine 35-1 (heated to 50°C) were mixed. Kolliphor RH 40 was heated to 50°C and added to the lipid mixture. Ethanol was added and the mixture was left until homogenous.
The drug was weighed into the preconcentrates followed by sonication for 5 min for the SNEDDS and 30 min for the super-SNEDDS. The super-SNEDDS were then subjected to heating at 60°C for 5 h and then left at 37°C overnight to equilibrate, and a clear solution was obtained. The super-SNEDDS stayed physically stable and Hf stayed chemically stable for at least 6 months (5). The drug was added at 75% equilibrium solubility (S eq) and 150% S eq for the SNEDDS and super-SNEDDS, respectively. The equilibrium solubility was previously determined by Thomas et al. (5). Orlistat was dissolved in ethanol and added to the formulations, giving a final concentration of 1% (w/w). Immediately before the in vivo experiments, the SNEDDS were emulsified using Milli-Q water at a 25% (w/w) lipid/water ratio.
The intravenous o/w emulsion contained 0.1% (w/w) Hf, 20% (w/w) soybean oil, 2% (w/w) lecithin, 2.5% (w/w) glycerol, and 75.4% (w/w) purified water. The emulsion was prepared by dissolving Hf and lecithin in soybean oil under gentle heating (50°C). Glycerol was added to the water, and the solution was heated to 50°C before the two phases were combined. The mixture was homogenized for 3 min. To reduce the droplet size of the resulting emulsion further, the emulsion was homogenized on ice for 5 min using an ultrasonicator with a microtip at power output 5 (Sonics Vibra-Cell, Sonics and Materials, Newtown, USA). The final emulsion was filtered through a sterile 0.45-μm filter.
In Vitro Lipolysis
Dynamic in vitro lipolysis was carried out as previously described (20–22). Briefly, the SNEDDS was weighed into a vessel containing 25 mL fasted state intestinal lipolysis medium (2.5 mM bovine bile salt, 0.26 mM phospholipid, 2 mM Tris, 2 mM maleic acid, and 50 mM sodium chloride). After equilibration for 3 min, the pH was adjusted to 6.5 by an automated pH-stat (Metrohm Titrino 744, Tiamo version 1.3, Switzerland). The in vitro lipolysis was initiated by the addition of 5 mL freshly prepared pancreatic lipase (pH 6.5, 37°C). The pancreatic lipase was prepared by weighing the lipase into a polypropylene tube and adding water. The mixture was centrifuged (7 min, 4000 rpm, 37°C) and the pH of the supernatant was adjusted to 6.5, resulting in a total lipase activity of 550 u/mL. The rate of lipolysis was controlled by addition of calcium in the form of CaCl2 (0.6 M; 0.045 mmol/min) throughout the lipolysis (60 min). The liberated fatty acids generated during the lipolysis were continuously titrated with NaOH (0.4 M) to maintain the pH at 6.5. After 60 min, the lipolysis was terminated and a back titration was performed at pH 9 to determine the exact amount of liberated free fatty acids.
One milliliter of digestion medium was withdrawn at time zero and after 60 min of lipolysis. The lipase activity was quenched with 5 μL 4-BBBA (1 M in methanol) followed by ultracentrifugation (50,000 rpm, 50 min at 37°C).
Quantitative Analysis of Samples from Lipolysis
Samples were analyzed for Hf content after 60 min of lipolysis in the aqueous and pellet phases after ultracentrifugation and dilution, using an isocratic high-performance liquid chromatography (HPLC) method. The system consisted of a Dionex ASI-100 Automated sample injector, P680 HPLC pump, and a PDA-100 photo diode array detector (Thermo Fisher Scientific, Waltham, MA, USA). The column used was a Waters x-bridge C8 column (Waters, Milford, MA, USA). The mobile phases consisted of acetonitrile and purified water containing 0.2% SDS and glacial acetic acid in the ratio 80:20 (v:v). A constant flow of 0.8 mL/min was employed with an injection volume of 10 μL. The chromatograms were analyzed using Thermo Scientific Dionex Chromeleon 7 Chromatography Data System software (Thermo Fisher Scientific, Waltham, MA, USA).
In Vivo Study
The animal protocols used in this study were approved by the University of British Columbia’s Animal Care Committee and conform to the Canadian Council on Animal Care guidelines. Male Sprague Dawley (SD) rats (270–310 g) with jugular vein cannulation were obtained from Charles River Laboratories (Wilmington, MA, USA). The animals were kept on standard feed and had free access to water during the experiment. Before initiation of the experiment, the rats were fasted for 10–15 h. The rats received 6.7 mg/kg Hf and 140 or 70 mg/kg of lipid from the SNEDDS and super-SNEDDS, respectively, by oral gavage. Two hundred twenty-five microliters of blood samples was drawn from the jugular vein at 0 and 30 min and 1, 1.5, 2, 4, 6, 8, 12, 24, and 30 h after administration and collected into Eppendorf tubes containing EDTA. The withdrawn blood was replaced by an equal volume of normal saline containing 100 units heparin to avoid hypovolemia and to flush the jugular vein catheter. The plasma was subsequently harvested by centrifugation (10 min, 5000 rpm) and stored at −80°C until analysis.
The group receiving intravenous treatment received a dose of 1.7 mg/kg Hf in the jugular vein. Blood samples were additionally collected at 0.5, 2, 10, and 20 min. The animals were euthanized 30 h after dosing. The doses chosen were based on existing literature (23,24).
Quantitative Analysis of Plasma Samples
Quantification of Hf in the plasma samples obtained from the in vivo study was performed using a Waters Acquity UPLC system equipped with a binary solvent delivery system and a PDA detector (Waters, Milford, MA, USA). The analysis was based on a previously validated method described by Humberstone et al. (25). Briefly, 100 μL of plasma was aliquoted to a polypropylene tube and the plasma proteins were precipitated with 1 mL of acetonitrile and the samples vortexed for 2 min. Four milliliters of TBME was added and the samples were again vortexed for 2 min. The samples were then centrifuged for 5 min (4000 rpm, 4°C) and the supernatant transferred to a polypropylene tube containing 100 μL of 5 mM HCl in acetonitrile. The content was then evaporated to dryness under a stream of nitrogen at 45°C for 90 min (TurboVap LV, Zymark, MA, USA) and the residue reconstituted in acetonitrile. Twenty-five microliters of the reconstituted residue was injected onto a Waters x-bridge C8 column (Waters, Milford, MA, USA) maintained at 30°C. A constant flow rate of 1 mL/min and a mobile phase consisting of a mixture of acetonitrile and water containing 0.2% SDS and 0.2% glacial acetic acid in the ratio 85:15 were employed, and the data was collected at 257 nm. Standard curves were prepared daily for the quantification of Hf in the plasma samples. The chromatograms were analyzed using Empower 3 Chromatography Data Software (Waters, Milford, MA, USA).
Pharmacokinetic Analysis
The pharmacokinetic parameters were analyzed using WinNonlin Professional Version 6.3 (Pharsight Corporation, Mountain View, CA, USA). The area under the curve (AUC) was determined using the linear trapezoidal model from t = 0 until the last plasma sample was taken at t = 30 h. The maximum plasma concentration of Hf (C max), the time where the maximum concentration occurred (t max), and the time required to reduce the plasma concentration to half of the C max (t ½) were determined from the individual curves. The data was normalized to dose. The absolute bioavailability F a was calculated as follows:
Statistical Analysis
The data sets are expressed as mean ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism 6.03 (GraphPad Software, San Diego, CA, USA). For the in vitro data, ANOVA followed by Tukey’s posttest was used (α = 0.05), and for the in vivo data, ANOVA followed by either Šídák or Dunnet’s posttest was used to analyze statistical differences between groups (α = 0.05).
RESULTS AND DISCUSSION
The aims of the present study were to evaluate the absorption of Hf from SNEDDS and super-SNEDDS in rats and to elucidate the effect of digestion on absorption of Hf using the lipase inhibitor orlistat. Hf as the free base was employed as a model drug.
Dynamic In Vitro Lipolysis
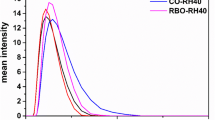
The titration curves from the in vitro lipolysis of the SNEDDS and super-SNEDDS with and without orlistat are shown in Table I and Fig. 1. When orlistat is present, the activity of pancreatic lipase is almost completely inhibited. The difference between the titration curves for the SNEDDS and the super-SNEDDS without orlistat can be ascribed to the amount of lipid present in the vessel. Since the same dose of drug was added in all the lipolysis experiments, the lipid content was 50% for the super-SNEDDS compared to the SNEDDS (the SNEDDS have a drug load of 75% of the S eq, whereas the super-SNEDDS are super-saturated and contain 150% of the S eq).

Amount of free fatty acids (FFA) generated from hydrolysis of the lipids in the SNEDDS (grey circles), the super-SNEDDS (black circles), the SNEDDS with orlistat (grey diamonds), and the super-SNEDDS with orlistat (black diamonds) during dynamic in vitro lipolysis. The data represents mean ± SD, n = 3
The relative amount of Hf found in either the pellet or the aqueous phase after in vitro lipolysis at 0 and 60 min is depicted in Fig. 2. When comparing the SNEDDS and the super-SNEDDS, the amount precipitated from the super-SNEDDS after 60 min of lipolysis is significantly higher than that of the SNEDDS (P < 0.1). Before initiation of lipolysis, the solubilization capacity can be ascribed to the dispersion of the undigested formulation in the presence of the bile salts and phospholipids. During lipolysis, the continuous digestion of the SNEDDS and super-SNEDDS and the formation of digestion products (free fatty acids and monoglycerides) result in a decrease in the solubilization capacity and precipitation of Hf. Since the lipid to drug ratio is higher for the SNEDDS than for the super-SNEDDS, the solubilizing capacity of the SNEDDS is higher. Upon hydrolysis, the higher amounts of lipids will generate more digestion products and this will facilitate drug solubilization due to the colloidal structures that are formed. From Fig. 2, it is clear that addition of orlistat to the SNEDDS results in less Hf precipitated at 60 min (P < 0.1). The same trend can be seen for the super-SNEDDS with and without orlistat (P < 0.1). The amount of precipitation for the super-SNEDDS with orlistat is higher than that for the SNEDDS with orlistat even though there is no digestion. In agreement with a previous study, the Hf precipitating during in vitro lipolysis was amorphous. Interestingly, the drug precipitates in the presence of orlistat were also found to be amorphous by X-ray powder diffraction (XRPD) (data not shown) (7). Precipitation in the presence of orlistat can be ascribed to the physical instability of the super-saturated system where the drug precipitates over time because of the energy induced by the stirring.

Relative amount of Hf found in the pellet (black) and the aqueous phase (grey) at time zero and after 60 min of in vitro lipolysis for the SNEDDS and super-SNEDDS, with and without orlistat. The data has been normalized and represents the mean ± SEM, n = 3. *indicates significant differences between the data points obtained after 60 min
In Vivo Study
The mean plasma concentration profiles of Hf administered orally to rats are presented in Fig. 3, and the pharmacokinetic parameters are provided in Table II. All SNEDDS were dosed at 6.7 mg Hf/kg body weight of the rats resulting in 140 mg lipid/kg for the SNEDDS and 70 mg lipid/kg for the super-SNEDDS.

Plasma concentration of Hf following oral administration as a SNEDDS and b super-SNEDDS to SD rats. Treatment included SNEDDS (black circles), SNEDDS with orlistat (black squares), super-SNEDDS (black circles), and super-SNEDDS with orlistat (black squares). The data represents mean ± SEM, n = 6
Super-SNEDDS Effect
Administration of Hf in super-SNEDDS results in a significantly larger C max compared to the SNEDDS (P < 0.05). There were no differences in the time to reach the maximum concentration (t max) and the half-life (t ½) between the SNEDDS and the super-SNEDDS. Although the differences in AUC between the SNEDDS and super-SNEDDS have not reached significance, there is a tendency of a higher AUC for the super-SNEDDS and hence also a higher relative bioavailability of the drug than for the SNEDDS. The same differences can be observed for the SNEDDS and super-SNEDDS when administered with orlistat.
The exact mechanism behind the increased absorption of Hf from SNEDDS and super-SNEDDS is still unknown. Traditionally, it is thought that the amount of lipids present in the gastrointestinal fluids is positively correlated to the absorption due to increased solubilization capacity. However, although super-SNEDDS in the current study only provide half the amount of lipids with respect to the drug concentration compared to the SNEDDS, the overall performance measured from the PK parameters is still similar or even improved. Thus, the solubilizing capacity of the digested lipids does not seem to be the only factor important for the absorption of HF. Based on the XRPD in vitro examination, the solid-state form of the drug precipitate during lipolysis is amorphous. It can thus be speculated that the increased precipitation of the drug in an amorphous form from the super-SNEDDS is positively correlated to increased absorption, due to promotion of local supersaturation close to the intestinal lumen upon re-dissolution of the amorphous precipitate. This is in agreement with a previous in vivo study on dogs (5).
The GI physiology of dogs and that of rats differ greatly, but the obtained data indicates that the rat is also a suitable model for evaluation of the performance of super-SNEDDS.
Orlistat Effect
Considering the effect of orlistat, as seen in Fig. 3 and Table II, t max is significantly longer for both the SNEDDS (6.3 ± 1.2 h) and the super-SNEDDS (6.0 ± 1.0 h) with orlistat compared to the SNEDDS (2.8 ± 1.2 h) and super-SNEDDS (1.3 ± 0.1 h) without orlistat. Orlistat reduces the C max for both the SNEDDS and super-SNEDDS; however, this reduction does not reach significant levels (P = 0.13).
Thus, orlistat changes the pharmacokinetic parameters when added to both SNEDDS and super-SNEDDS, but the total bioavailability does not change significantly (P > 0.05). The super-SNEDDS containing orlistat have a larger AUC than the other groups, but due to the relatively large variability, there is no statistical difference.
In a previous study applying orlistat as a tool, de Smidt et al. found that the digestibility of medium-chain triacylglycerides and surfactant-based formulations did not significantly influence the absorption of penclomidine in rats, which is in agreement with the data presented here. However, when using medium-chain triglyceride solution as a vehicle, the absorption was to a higher degree dependent on digestion (9). It is well known that medium-chain lipids to a lesser degree are dependent on digestion compared to LC lipids (26); therefore, it is difficult to compare these data directly with the those of the current study, in which LC lipids were used. Extended t max can in some cases be ascribed to gastro-retentivity; however, since orlistat previously has been reported to accelerate gastric emptying, the increased t max can therefore be attributed to the effect of digestion on absorption from the SNEDDS (27).
Comparison of In Vivo and In Vitro Data
In vitro lipolysis is often used to assess lipid formulations such as SNEDDS (28). Traditionally, it has been assumed that precipitation during in vitro lipolysis will lead to decreased in vivo absorption. On these terms, the current in vitro data cannot fully explain the obtained in vivo data; the increased precipitation in the case of the super-SNEDDS would lead to the expectation that the SNEDDS would perform better in vivo than the super-SNEDDS, which is not the case. Even though in vitro lipolysis can be useful for predicting the in vivo behavior, the model does not take absorption into consideration and also the extent of in vivo precipitation is, to the authors’ knowledge, still unknown. This is in accordance with a previous study by Thomas et al., also studying super-SNEDDS with Hf, but in a dog model. In this case, the findings were explained by the solid-state properties of precipitated Hf, which precipitates in an amorphous form (5). From the current study, it was shown that amorphous precipitation of Hf is most likely not due to a complex formation with digestion products (since digestion is completely blocked in the presence of orlistat). The amorphous nature of the precipitate is thus either an inherent property of the drug or due to complex formation with other components being present in the digestion medium. In this context, it is worth mentioning that Humberstone et al. suggested the possibility of the formation of a Hf-taurocholate complex (29).
It is evident from the effect of orlistat on the pharmacokinetic parameters (t max, C max, and t ½) that absorption of Hf from SNEDDS is affected by digestion, whereas the bioavailability is not changed significantly. The traditional mechanism proposed for absorption of poorly water-soluble drugs from SNEDDS involves the formation of different colloidal structures and mixed micelles, which will facilitate transport of the drug to the epithelial membrane (30). The formation of these structures is highly dependent on the action of the digestive lipases. In the current study, the in vitro data shows almost complete blockage of the digestion by addition of 1% (w/w) orlistat to the SNEDDS, which suggest that this also can be the case in vivo. It is therefore possible that colloidal structures are not generated when orlistat is present, and therefore, some other mechanisms of absorption will be important. It can be speculated that the absorption is merely facilitated by the nanoemulsion droplets, serving as a reservoir containing the drug in a predissolved form. Partitioning of Hf from the nanoemulsion droplets to the existing bile salt micelles will, in this case, enable absorption.
However, it is also possible that the in vivo lipase activity is not fully inhibited by the amount of orlistat dosed; orlistat is inhibiting the pancreatic lipase at a stoichiometry of around 1:1 (31). In the rat, pancreatic lipase is continuously secreted (32). It is therefore also possible that the orlistat inhibition is overcome by the continuous secretion of pancreatic lipase. This will lead to a reduced and/or delayed digestion of the SNEDDS, which can also result in the observed changes of the pharmacokinetic parameters, e.g., the delayed t max. In this case, orlistat will not lead to changes in the absorption mechanism but merely in the pharmacokinetics.
CONCLUSION
The present study has demonstrated that the super-SNEDDS principle is also effective in rats; a significantly higher C max of Hf was found when dosed in a super-SNEDDS compared to a conventional SNEDDS with the same dose of drug.
The pharmacokinetic parameters change when co-dosing SNEDDS and super-SNEDDS with orlistat; the C max decreases and the absorption phase seems to be extended, especially for the super-SNEDDS. However, these changes do not influence the overall bioavailability. Thus, orlistat can possibly be used as a tool to change pharmacokinetics of drugs dosed in SNEDDS. It can be speculated that the increased absorption of poorly soluble drugs, often seen from SNEDDS, is less than previously thought due to digestion of the lipids. It is possible that the partitioning directly from the nanoemulsion droplet to the bile salt micelles facilitates the absorption; however, further studies are needed in order to understand the mechanism of absorption from SNEDDS.
References
Stegemann S, Leveiller F, Franchi D, de Jong H, Lindén H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31(5):249–61.
Amidon G, Lennernäs H, Shah V, Crison J. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–20. English.
Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997;25(1):103–28.
Christensen JØ, Schultz K, Mollgaard B, Kristensen HG, Mullertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23(3):287–96.
Thomas N, Holm R, Müllertz A, Rades T. In vitro and in vivo performance of novel supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS). J Control Release. 2012;160(1):25–32.
Thomas N, Holm R, Garmer M, Karlsson J, Müllertz A, Rades T. Supersaturated self-nanoemulsifying drug delivery systems (super-SNEDDS) enhance the bioavailability of the poorly water-soluble drug simvastatin in dogs. AAPS J. 2013;15(1):219–27. English.
Thomas N, Richter K, Pedersen T, Holm R, Müllertz A, Rades T. In vitro lipolysis data does not adequately predict the in vivo performance of lipid-based drug delivery systems containing fenofibrate. AAPS J. 2014;16(3):539–49. English.
Bloedow DC, Hayton WL. Effects of lipids on bioavailability of sulfisoxazole acetyl, dicumarol, and griseofulvin in rats. J Pharm Sci. 1976;65(3):328–34.
de Smidt PC, Campanero MA, Trocóniz IF. Intestinal absorption of penclomedine from lipid vehicles in the conscious rat: contribution of emulsification versus digestibility. Int J Pharm. 2004;270(1–2):109–18.
Hofmann AF, Borgström B. The intraluminal phase of fat digestion in man: the lipid content of the micellar and oil phases of intestinal content obtained during fat digestion and absorption. J Clin Investig. 1964;43(2):247–57.
Yeap YY, Trevaskis NL, Quach T, Tso P, Charman WN, Porter CJH. Intestinal bile secretion promotes drug absorption from lipid colloidal phases via induction of supersaturation. Mol Pharm. 2013;10(5):1874–89.
Yano K, Masaoka Y, Kataoka M, Sakuma S, Yamashita S. Mechanisms of membrane transport of poorly soluble drugs: role of micelles in oral absorption processes. J Pharm Sci. 2010;99(3):1336–45.
Fatouros D, Deen GR, Arleth L, Bergenstahl B, Nielsen F, Pedersen J, et al. Structural development of self nano emulsifying drug delivery systems (SNEDDS) during in vitro lipid digestion monitored by small-angle X-ray scattering. Pharm Res. 2007;24(10):1844–53. English.
Weibel EK, Hadvary P, Hochuli E, Kupfer E, Lengsfeld H. Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. I. Producing organism, fermentation, isolation and biological activity. J Antibiot (Tokyo). 1987;40(8):1081–5.
Tiss A, Lengsfeld H, Carrière F, Verger R. Inhibition of human pancreatic lipase by tetrahydrolipstatin: further kinetic studies showing its reversibility. J Mol Catal B Enzym. 2009;58(1–4):41–7.
Fernandez E, Borgström B. Effects of tetrahydrolipstatin, a lipase inhibitor, on absorption of fat from the intestine of the rat. Biochim Biophys Acta. 1989;1001(3):249–55.
Tiss A, Lengsfeld H, Hadváry P, Cagna A, Verger R. Transfer of orlistat through oil–water interfaces. Chem Phys Lipids. 2002;119(1–2):41–9.
Tiss A, Ransac S, Lengsfeld H, Hadvàry P, Cagna A, Verger R. Surface behaviour of bile salts and tetrahydrolipstatin at air/water and oil/water interfaces. Chem Phys Lipids. 2001;111(1):73–85.
Porter CJH, Charman SA, Charman WN. Lymphatic transport of halofantrine in the triple-cannulated anesthetized rat model: effect of lipid vehicle dispersion. J Pharm Sci. 1996;85(4):351–6.
Thomas N, Müllertz A, Graf A, Rades T. Influence of lipid composition and drug load on the in vitro performance of self-nanoemulsifying drug delivery systems. J Pharm Sci. 2012;101(5):1721–31.
Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model: I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–22.
Zangenberg NH, Müllertz A, Gjelstrup Kristensen H, Hovgaard L. A dynamic in vitro lipolysis model: II: evaluation of the model. Eur J Pharm Sci. 2001;14(3):237–44.
Holm R, Porter CH, Müllertz A, Kristensen H, Charman W. Structured triglyceride vehicles for oral delivery of halofantrine: examination of intestinal lymphatic transport and bioavailability in conscious rats. Pharm Res. 2002;19(9):1354–61. English.
Karpf D, Holm R, Kristensen H, Müllertz A. Influence of the type of surfactant and the degree of dispersion on the lymphatic transport of halofantrine in conscious rats. Pharm Res. 2004;21(8):1413–8. English.
Humberstone AJ, Currie GJ, Porter CJH, Scanlon MJ, Charman WN. A simplified liquid chromatography assay for the quantitation of halofantrine and desbutylhalofantrine in plasma and identification of a degradation product of desbutylhalofantrine formed under alkaline conditions. J Pharm Biomed Anal. 1995;13(3):265–72.
Chow BPC, Shaffer EA, Parsons HG. Absorption of triglycerides in the absence of lipase. Can J Physiol Pharmacol. 1990;68(4):519–23.
Borovicka J, Schwizer W, Guttmann G, Hartmann D, Kosinski M, Wastiel C, et al. Role of lipase in the regulation of postprandial gastric acid secretion and emptying of fat in humans: a study with orlistat, a highly specific lipase inhibitor. Gut. 2000;46(6):774–81.
Dahan A, Hoffman A. Use of a dynamic in vitro lipolysis model to rationalize oral formulation development for poor water soluble drugs: correlation with in vivo data and the relationship to intra-enterocyte processes in rats. Pharm Res. 2006;23(9):2165–74. English.
Humberstone AJ, Porter CJH, Charman WN. A physicochemical basis for the effect of food on the absolute oral bioavailability of halofantrine. J Pharm Sci. 1996;85(5):525–9.
MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25(1):33–46.
Hadvary P, Lengsfeld H, Wolfer H. Inhibition of pancreatic lipase in vitro by the covalent inhibitor tetrahydrolipstatin. Biochem J. 1988;256:357–61.
Erlanson-Albertsson C, Larsson A, Duan R. Secretion of pancreatic lipase and colipase from rat pancreas. Pancreas. 1987;2(5):531–5.
Acknowledgments
The authors would like to thank Yang Hwan Yun for help with the in vitro lipolysis data. Funding for this project was provided by the Canadian Institute of Health Research to KMW. A special thanks to University of Copenhagen, Denmark, and University of British Columbia, Canada, for providing a platform for collaboration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The animal protocols used in this study were approved by the University of British Columbia’s Animal Care Committee and conform to the Canadian Council on Animal Care guidelines.
Rights and permissions
About this article

Cite this article
Michaelsen, M.H., Wasan, K.M., Sivak, O. et al. The Effect of Digestion and Drug Load on Halofantrine Absorption from Self-nanoemulsifying Drug Delivery System (SNEDDS). AAPS J 18, 180–186 (2016). https://doi.org/10.1208/s12248-015-9832-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9832-7