
Abstract
One of the major challenges in the development of central nervous system (CNS)-targeted drugs is predicting CNS exposure in human from preclinical data. In this study, we present a methodology to investigate brain disposition in rats using a physiologically based modeling approach aiming at improving the prediction of human brain exposure. We specifically focused on quantifying regional diffusion and fluid flow processes within the brain. Acetaminophen was used as a test compound as it is not subjected to active transport processes. Microdialysis probes were implanted in striatum, for sampling brain extracellular fluid (ECF) concentrations, and in lateral ventricle (LV) and cisterna magna (CM), for sampling cerebrospinal fluid (CSF) concentrations. Serial blood samples were taken in parallel. These data, in addition to physiological parameters from literature, were used to develop a physiologically based model to describe the regional brain pharmacokinetics of acetaminophen. The concentration–time profiles of brain ECF, CSFLV, and CSFCM indicate a rapid equilibrium with plasma. However, brain ECF concentrations are on average fourfold higher than CSF concentrations, with average brain-to-plasma AUC0 − 240 ratios of 121%, 28%, and 35% for brain ECF, CSFLV, and CSFCM, respectively. It is concluded that for acetaminophen, a model compound for passive transport into, within, and out of the brain, differences exist between the brain ECF and the CSF pharmacokinetics. The physiologically based pharmacokinetic modeling approach is important, as it allowed the prediction of human brain ECF exposure on the basis of human CSF concentrations.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Central nervous system (CNS) disorders are currently estimated to affect hundreds of millions of people worldwide (1). While established treatments are currently available for most of these disorders, significant unmet medical needs still remain, as currently available drugs are treating symptoms rather than curing the disease (2). Therefore, novel treatments or drugs with a different mechanism of action are needed. However, the failure rate of development of new CNS drugs is very high. The actual problem lies in the inability to predict human (wanted and unwanted) CNS drug effects.
It is known that unbound plasma concentrations may not necessarily represent the unbound brain concentrations available for target interaction, due to distributional mechanisms (3). In recent years, much attention has been given to blood–brain barrier (BBB) permeability, as this is assumed to be the main determinant of CNS exposure (4–9). However, even though BBB permeability is a very important determinant, it is not the only relevant process. The brain is a dynamic multi-compartmental system, in which all processes of drug entry, within brain diffusion, metabolism, binding, and elimination determine actual CNS target site concentrations (3). To ultimately be able to predict CNS drug effects in humans, a more mechanistic understanding is needed of the individual contributions of the processes involved in brain target site distribution and ultimately drug effects.
The use of cerebrospinal fluid (CSF) as a surrogate for unbound brain target site concentrations has been discussed previously by De Lange et al. (10), Shen et al. (11), Lin et al. (12), and Westerhout et al. (3). In short, it has been concluded that a generally applicable relationship between CSF concentrations and brain extracellular fluid (ECF) concentrations does not exist due to qualitative and quantitative differences in processes that govern the pharmacokinetics (PK) at these sites. However, CSF concentrations are often considered the best available surrogate for brain target site concentrations in humans (3,10–12).
In rats, the most common method for collecting a CSF sample is by a single puncture or the implantation of a cannula in either the lateral ventricle or the cisterna magna (10,13–16). However, taking a CSF sample significantly affects the CSF volume, of which the impact is not yet known (17). The intracerebral microdialysis technique can be used for monitoring CSF concentrations, with minimal disturbance of the normal CSF physiology, and the application of multiple microdialysis probes to single animals allows for a direct comparison between unbound concentrations in brain ECF, CSFLV, and CSFCM.
In this study, we investigated the passive blood–brain transport processes that govern the relationship between the PK at different sites in the brain for acetaminophen, as a paradigm compound for passive transport (10,18). Concentration–time profiles of acetaminophen were obtained by microdialysis at different sites in the brain (striatum, lateral ventricle, and cisterna magna), and by serial parallel blood sampling, plasma kinetics were obtained.
Materials and Methods
Chemicals and Solutions
Acetaminophen and saline were obtained from the Leiden University Medical Center Pharmacy (Leiden, The Netherlands). Sodium chloride, potassium chloride, calcium chloride, magnesium chloride, ethylenediaminetetraacetic acid, perchloric acid, sodium acetate, and l-(+)-ascorbic acid were obtained from J.T. Baker (Deventer, The Netherlands). Disodium hydrogen phosphate dihydrate and sodium dihydrogen phosphate monohydrate were obtained from Merck (Amsterdam, The Netherlands). 3,4-Dihydroxybenzylamine hydrobromide (DHBA), l-cysteine, and 1-octanesulfonic acid were obtained from Sigma (Zwijndrecht, The Netherlands). Methanol and acetic acid were obtained from Biosolve B.V. (Valkenswaard, The Netherlands). Microdialysis perfusion fluid was prepared as previously described (19), containing 140.3 mM sodium, 2.7 mM potassium, 1.2 mM calcium, 1.0 mM magnesium, and 147.7 mM chloride.
Animals
The study protocol was approved by the Animal Ethics Committee of Leiden University (UDEC no. 07068) and all animal procedures were performed in accordance with Dutch laws on animal experimentation. A total of 36 male Wistar WU rats (225–275 g, Charles River, Maastricht, The Netherlands) were randomly divided into two groups: the first group (n = 12) was used for the determination of the in vivo microdialysis probe recovery and the second group (n = 24) was used for brain disposition experiments.
After arrival, all animals were housed in groups for 5–7 days (Animal Facilities, Gorlaeus Laboratories, Leiden, The Netherlands), under standard environmental conditions (ambient temperature 21°C; humidity 60%; 12/12 h light/dark cycle, background noise, and daily handling), with ad libitum access to food (laboratory chow, Hope Farms, Woerden, The Netherlands) and acidified water. Between surgery and experiments, the animals were kept individually in three Makrolon type cages for 7 days to recover from the surgical procedures.
Surgery
All surgical procedures were performed under isoflurane (2%) anesthesia, while maintaining the body temperature at 37°C by an electric heating pad (CMA/150, CMA/Microdialysis AB, Stockholm, Sweden). First, cannulas were implanted in the left femoral artery and vein for blood sampling and drug administration, respectively. The cannulas were disinfected with 0.1% benzalkonium chloride for at least 24 h prior to the implantation. The arterial cannula consisted of 4 cm of non-sterile polyethylene tubing [inner diameter (ID) 0.28 mm and outer diameter (OD) 0.61 mm, Portex Fine Bore polythene tubing, Smiths Medical International Ltd, Kent, England] connected to 16 cm of non-sterile polyethylene tubing (ID 0.58 mm and OD 0.96 mm, Portex Fine Bore polythene tubing, Smiths Medical International Ltd, Kent, England). The thinner part of the cannula was inserted 4 cm into the artery. The venous cannula consisted of 19 cm non-sterile polyethylene tubing (ID 0.58 mm and OD 0.96 mm) with a small silicon ring round the tubing at 3 cm from the tip. The cannula was inserted 3 cm into the vein. Both cannulas were subcutaneously led to the back of the head and fixated in the neck with a rubber ring.
Subsequently, after placing the rat in a stereotaxic frame (David Kopf Instruments, Tujunga, USA), the animals were chronically instrumented with two CMA/12 microdialysis guides (CMA/Microdialysis AB, Stockholm, Sweden) in different combinations of striatum (ST), for sampling in brain ECF, and lateral ventricle (LV) and/or cisterna magna (CM) for sampling in CSF (ST + LV, ST + CM, or LV + CM; each group with n = 8). For the ST, the position of the microdialysis guide is: 1.0 mm anterior, 3.0 mm lateral, 3.4 mm ventral, relative to bregma. For the LV, the position of the microdialysis guide is: 0.9 mm posterior, 1.6 mm lateral, 2.9 mm ventral, relative to the bregma. For the CM, the position of the microdialysis guide is: 1.93 mm posterior, 3.15 mm lateral, 8.1 mm ventral, at an angle of 25° from the dorsoventral axis (towards anterior) and 18° lateral from the anteroposterior axis relative to lambda. The microdialysis guides were secured to the skull with three anchor screws and dental cement.
After the surgery, the animals received 0.03 ml Temgesic ® intramuscular (Schering-Plough, Amstelveen, The Netherlands) and 0.3 ml Ampicillan® (Alfasan B.V., Woerden, The Netherlands) subcutaneously. One day prior to the experiment, the microdialysis dummies were replaced by the microdialysis probes (CMA/12, polycarbonate, molecular weight cutoff 20 kDa, CMA/Microdialysis AB, Stockholm, Sweden, with a semipermeable membrane length of 4 mm for ST and 1 mm for LV and CM).
Experimental Setup
All experiments started between 9:00 and 10:00 a.m. to minimize the influences of circadian rhythms. First, microdialysis vials were weighed prior to the experiment to be able to validate the probe perfusion rate (a maximal deviation of 5% was allowed for the sample to be included in the data). All microdialysis vials were then placed in a cooled fraction collector (Univentor 820 Microsampler, AgnTho's AB, Lidingö, Sweden) to collect the microdialysate samples. Microdialysis perfusion fluid was prepared as described above. The microdialysis probes were connected to FEP tubing (CMA/Microdialysis AB, Stockholm, Sweden) with tubing adapters (CMA/Microdialysis AB, Stockholm, Sweden). The microdialysis probes were then continuously flushed with microdialysis perfusion fluid (2 μl/min, Bee-Hive, Bioanalytical Systems Inc., West Lafayette, USA), being equilibrated for 60 min before acetaminophen administration.
The in vivo microdialysis probe recovery of acetaminophen was determined on the basis of reverse dialysis (20). The concept of the reverse dialysis method assumes directional independence of the solute through the dialysis membrane and that recovery is independent of the perfused concentration (21), as was true for acetaminophen under in vitro conditions. In short, the microdialysis probes in striatum, lateral ventricle, and cisterna magna were perfused with different concentrations of acetaminophen (50, 200, and 1,000 ng/ml) in perfusion fluid. The in vivo recovery for each microdialysis probe location is defined as the ratio of the concentration difference between the dialysate (C dial) from striatum, lateral ventricle, or cisterna magna and perfusion fluid (C in) over the concentration in the perfusion fluid (Eq. 1) (22).
For the brain disposition experiments, the rats received an intravenous infusion of 15 mg/kg acetaminophen dissolved in saline (200 μl/min/kg for a period of 10 min) with an automated pump (Pump 22 Multiple Syringe Pump, Harvard Apparatus, Holliston, USA). Start and duration of infusion were corrected for internal volume of the tubing so that infusion started at t = 0 min; 10-min interval samples were collected between t = −1 h and t = 2 h, followed by 20-min interval samples until t = 4 h. After weighing the microdialysis vials, they were stored at −80°C before analysis.
For the determination of acetaminophen plasma concentrations, 100-μl blood samples were obtained, in parallel to the microdialysate samples, from the arterial cannula at t = −5 (blank), 2, 7, 10, 15, 30, 60, 120, 180, and 240 min. For the determination of plasma protein binding of acetaminophen, 300-μl blood samples were obtained at t = −30 (blank) and 30 min (with a concentration assumed to be approximately 1/2 × C max). After the blood sample at t = 240 min, an additional dose of 15 mg/kg in 10 min was given to be able to determine plasma protein binding at C max (at t = 250 min). All blood samples were temporarily stored in heparin (10 IU)-coated Eppendorf cups. The blank blood samples for the determination of plasma protein binding were spiked with acetaminophen to obtain a blood concentration of 150 ng/ml. The spiked blood samples were then incubated in a shaking water bath at 37°C for 30 min. All blood samples were centrifuged for 15 min at 5,000 rpm and the plasma was pipetted into clean Eppendorf cups and stored at −20°C before analysis. At the end of the experiments, the animals were sacrificed with an overdose of Nembutal (Ceva Sante Animale, Libourne, France).
Plasma and Microdialysate Sample Analysis
Plasma protein binding was determined with Centrifree® ultrafiltration devices (Millipore BV, Etten-Leur, The Netherlands). All procedures were performed according to the user's manual. The ultrafiltrate was diluted ten times with saline before the analysis.
Acetaminophen concentrations in plasma, microdialysate, and ultrafiltrate were determined as described by Stevens et al. (19). In short, acetaminophen concentrations in plasma, microdialysate, and ultrafiltrate were determined using high-pressure liquid chromotography with electrochemical detection (HPLC-ECD).
To 50 μl of the plasma samples, 50 μl purified Millipore water (resistivity 18.2 MΩ cm, Millipore B.V., Amsterdam, The Netherlands) and 25 μl internal standard (IS), containing 150 ng/ml DHBA, were added. Proteins were then precipitated by adding 100 μl 6% perchloric acid, followed by vortexing and centrifugation (10 min at 4,000 rpm). The supernatant was then transferred into a clean glass tube, after which, 150 μl of sodium acetate (1 M) was added. After vortexing, the samples were injected into the HPLC-ECD. To 20 μl of the microdialysate or diluted ultrafiltrate samples, 20 μl IS was added, followed by vortexing before being directly injected into the HPLC-ECD system.
Data acquisition and processing were performed using Empower® data acquisition software (Waters, Etten-Leur, The Netherlands). For constructing the calibration curve, linear regression analysis was applied using weight factor 1/(y)2. Data analysis, statistical analysis, and plotting were performed using Microsoft® Office Excel 2003 (Microsoft Corporation, USA).
Pharmacokinetic Data Analysis
All plasma concentrations were converted to unbound plasma concentrations, by correction for plasma protein binding. All microdialysate concentrations from striatum, lateral ventricle, and cisterna magna were converted into brain ECF concentrations (C ECF) or CSF concentrations (C CSF) by division of the dialysate concentrations by the average in vivo recovery as determined for each microdialysis probe location (Eq. 2).
Areas under the curve from t = 0 to t = 240 min (AUC0 − 240) were calculated by the trapezoidal rule and tested for differences by single factor ANOVA. The population PK models were developed and fitted to the data by means of nonlinear mixed-effects modeling using the NONMEM software package (version 6.2, Icon Development Solutions, Ellicott City, MD, USA) and analyzed using the statistical software package S-Plus® for Windows (version 6.2 Professional, Insightful Corp., Seattle, USA).
The pharmacokinetic model for acetaminophen plasma and brain concentrations was developed by implementing a physiologically based approach to investigate the exchange between brain ECF and CSF. In order to do so, the volumes of the different brain compartments were fixed to their physiological volumes, which were based on the following considerations:
-
1.
The brain intracellular space (ICS) is approximately 80 % of the brain volume (23). With an average brain weight of 1.8 g in a 250-g rat (own observations), this results in an ICS volume of 1.44 ml.
-
2.
The total CSF volume in a 250-g rat is about 300 μl (24). The volume of the lateral ventricles is about 17% of the total CSF volume (25,26). This results in a volume of the rat lateral ventricles of about 50 μl (0.17 × 300).
-
3.
The volume of the cisterna magna is about 6% of the total CSF volume (27,28). This results in a volume of the rat cisterna magna of about 17 μl (0.06 × 300).
-
4.
The cranial subarachnoid space in rats is estimated to be about 60 % of the total CSF volume (24,29). This results in a volume of the cranial subarachnoid space of about 180 μl (0.60 × 300).
-
5.
The unaccounted for CSF volume (17%; 50 μl) is considered to be mainly located in the third and fourth ventricles (29).
-
6.
The blood–brain transport is restricted by the presence of the BBB and the blood–CSF barrier (BCSFB), which is located at the choroid plexuses of the lateral, third, and fourth ventricles (3), as well as at the cisterna magna.
-
7.
The intra-brain distribution was restricted by the physiological flow paths of brain ECF, in which brain ECF flows towards the CSF compartments at a rate of 0.2 μl/min (30,31), and CSF flows from lateral ventricle, through the third and fourth ventricle, to the cisterna magna and subsequently to the subarachnoid space (cranial and spinal) and back into blood at a rate of 2.2 μl/min (13).
Structural model selections for both the blood and brain PK model were based on the likelihood ratio test (p < 0.01), diagnostic plots (observed concentrations vs. individual and population predicted concentrations, weighted residuals vs. predicted time and concentrations), parameter correlations, and precision in parameter estimates. The inter-animal variability in pharmacokinetic parameters was assumed to be log normally distributed. The residual error, which accounts for unexplained variability (e.g., measurement and experimental error and model misspecification), was best described with a proportional error model.
The validity of the pharmacokinetic models was investigated by means of a visual predictive check (32–34). Using the final PK parameter estimates, 1,000 curves were simulated. Subsequently, the median and the 5th and 95th percentile of the predicted concentrations were calculated, which represent the 90% prediction interval. These were then compared with the observations.
In order to test the ruggedness of the model and estimate the precision of the parameters n = 100, nonparametric (case resampling) bootstraps were performed. To create the bootstrapped datasets, specific rat data (plasma and microdialysate concentrations) were removed randomly from the datasets and replaced with randomly selected rat data from the complete original dataset. Each of these permutations of the original dataset were fitted with the final model determined based on the original dataset. This results in a series of model fits, each with its own set of parameters. These results were displayed graphically and the descriptive statistics of the parameters were compared to parameter estimates of the final model. Only bootstrap runs that successfully minimized were used in this analysis.
For illustrative purposes, we have applied the final physiologically based pharmacokinetic (PBPK) model to predict human brain ECF acetaminophen concentrations on the basis of human CSF concentrations from the spinal subarachnoid space, as presented by Bannwarth et al. (35). This was done by changing the different rat values of the physiological parameters of the PBPK model to the human values.
Results
All results are presented as average values ± standard deviation, unless stated otherwise. The experimental setup allowed for direct comparison of plasma concentrations with brain concentrations on two distinct sites, including the direct comparison of brain ECF concentrations with CSF concentrations within a single rat. Plasma protein binding was linear with concentration, at an extent of 19.5 ± 4.2%. The average in vivo recoveries for the striatum, lateral ventricle, and cisterna magna robes were 12.0 ± 3.3%, 8.1 ± 3.8%, and 8.6 ± 4.7%, respectively. The average unbound acetaminophen concentrations for plasma, brain ECF, CSF from lateral ventricle (CSFLV), and cisterna magna (CSFCM) are shown in Fig. 1.

Observed data from the rat. Average (geometric mean ± SEM) unbound acetaminophen concentration–time profile for plasma (n = 10), brain ECF (n = 10), CSFLV (n = 14), and CSFCM (n = 8)
The unbound plasma concentration–time profile shows a short distribution phase of about 15 min. During the elimination phase (t > 15 min), the unbound plasma concentrations reach an apparent plateau from t = 120 min onward. The concentration–time profiles of brain ECF, CSFLV, and CSFCM show a pattern similar to the unbound plasma concentration–time profile, indicating a relatively rapid distribution. However, brain ECF concentrations are on average fourfold higher than CSF concentrations in both lateral ventricle and cisterna magna. The CSF concentration in both locations is similar.
The concentration–time profile for the brain ECF shows more similarities to the unbound plasma concentration–time profile than the concentration–time profiles of CSFLV or CSFCM. The acetaminophen concentration in brain ECF is significantly higher (p < 0.05) than that in CSF from t = 0 to t = 80 min. As a result, the brain ECF-to-unbound plasma AUC0 − 240 ratio is also higher than the CSF-to-unbound plasma AUC0 − 240 ratio with an average ratio of 121 ± 72%, 28 ± 10%, and 35 ± 17% for brain ECF, CSFLV, and CSFCM, respectively. This indicates that brain ECF-to-CSF exposure ratio is approximately 4.
The compartmental pharmacokinetic analysis showed that the plasma concentrations were best described by a two-compartment model that included inter-compartmental clearance (Q), elimination clearance from the central compartment (CL10), and an additional zero-order drug input into the plasma compartment. This additional, infusion-like, drug input represents the fraction (F abs) of the administered acetaminophen that is recirculated within the study duration (240 min), presumably because of enterohepatic circulation (36–38).
As the next step in the data analysis, to describe the concentrations in each of the brain compartments, a single brain compartment was added. Drug flow between the brain compartment and the plasma compartment was by a clearance in (CLin) and clearance out (CLout), rather than an inter-compartmental clearance (Q).
Subsequently, the addition of a second and third brain compartment was explored for the description of the plasma data, as well as the brain ECF, CSFLV, and CSFCM data (results not shown). As it was our goal to investigate the relationship between brain ECF and CSF pharmacokinetics, we have applied compartmental modeling to describe the relationships between brain ECF and CSF at the two sites.
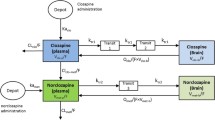
However, this approach was without success, as the model had too much freedom to fit the plasma and brain data correctly. For this reason, and because of the improved value for extrapolation and prediction, we have applied a physiologically based PK model to investigate the exchange between brain ECF and CSF. As CSF flows from lateral ventricle through the third and fourth ventricle, to the cisterna magna, and subsequently to the subarachnoid space and back into blood, we added two CSF compartments that represent the combined third and fourth ventricle (CSFTFV) and the subarachnoid space (CSFSAS) to more adequately describe CSF physiology. Since we have no measurements of the concentrations in the third and fourth ventricle, the transfer clearance between plasma and third and fourth ventricle was assumed to be equal to the transfer clearance between plasma and lateral ventricle. For the correct representation of the brain physiology, we have also included the brain ICS to the model. However, since we also have no measurements of total brain concentrations, it was assumed that brain ICS concentrations are equal to brain ECF concentrations. Therefore, the brain ICS volume is added to the brain ECF volume to account for the total brain volume.
The final PBPK model is shown in Fig. 2. The differential equations of this model can be found in the Appendix. The final estimation of the PK parameters is summarized in Table I. Here, CL10 is the elimination clearance from plasma, Q 12 is the inter-compartmental clearance between plasma and peripheral tissue, CL13 is the clearance from plasma to brain ECF, CL31 is the clearance from brain ECF to plasma, CL14 is the clearance from plasma to CSFLV, CL41 is the clearance from CSFLV to plasma, CL15 is the clearance from plasma to CSFTFV, CL51 is the clearance from CSFTFV to plasma, CL16 is the clearance from plasma to CSFCM, CL51 is the clearance from CSFCM to plasma, Q ECF is the flow rate of brain ECF, Q CSF is the flow rate of CSF, V pl is the plasma volume, V per is the volume of distribution for the peripheral tissue, V ICS is the brain ICS volume, V ECF is the brain ECF volume, V LV is the volume of the lateral ventricle, V TFV is the volume of the third and fourth ventricle combined, V CM is the volume of the cisterna magna, V SAS is the volume of the subarachnoid space, F abs is the fraction of the dose that is reabsorbed over time as a result of enterohepatic circulation, fup is the fraction unbound in plasma, η i is the inter-individual variability of parameter i, and ε j is the residual error on the concentrations in compartment j. In this analysis, the values of the various brain compartments and of the brain ECF and CSF flow were fixed to their physiological values as described above.

Graphical representation of the PBPK model that was used to describe the intra-brain distribution in the rat
The visual predictive check of the final model is given in Fig. 3. It can be seen that the final model describes the data very well within the 90% prediction interval and also can cope with the large inter-individual variation in brain concentrations. This PBPK model was used to predict acetaminophen concentrations in rat CSFSAS. This is shown in Fig. 4. As a result of the CSF flow from lateral ventricle, through the third and fourth ventricle, to the cisterna magna, and subsequently to the subarachnoid space, clear differences in the concentration–time profiles of these CSF compartments can be observed.

The visual predictive check of the final model. The dots represent the individual data points and the gray area represents the 90% prediction confidence interval. The x-axis represents the time in minutes and the y-axis represents the acetaminophen concentration in nanograms per milliliter. The different boxes represent the plasma, brain ECF, CSFLV, and CSFCM data

Predicted acetaminophen concentrations in plasma and the different brain compartments: brain ECF, CSFLV, CSFCM6, and CSFSAS. Predictions are based on a 10-min i.v. infusion of 15 mg/kg acetaminophen in a 250-g rat
By comparing the acetaminophen concentrations in plasma and CSFSAS between rats and humans [as presented by Bannwarth et al. (35)], it can be seen that the kinetics in humans are slower compared to rats (Fig. 5). However, the peak concentrations in plasma appear to be similar, whereas the peak concentrations in CSFSAS in humans are ~eightfold higher compared to rats.

Observed and predicted total plasma and CSFSAS acetaminophen concentrations in rat and human plasma and CSFSAS. Predictions are based on a 10-min i.v. infusion of 15 mg/kg acetaminophen in a 250-g rat and a 70-kg human
The final PBPK model was used for the prediction of human brain ECF concentrations on the basis of human CSFSAS concentrations. However, since Bannwarth and colleagues only measured acetaminophen concentrations in plasma and spinal CSFSAS (35), there are no data available on acetaminophen concentrations in human brain ECF, CSFLV, CSFTFV, and CSFCM. Therefore, we were not able to separately estimate the PK parameters for distribution between plasma and brain ECF, CSFLV, CSFTFV, and CSFCM. However, by fixing the different plasma to brain and brain to plasma clearances to be a certain factor of the clearance from plasma to brain ECF and the clearance from brain ECF to plasma, respectively (e.g., the clearance from plasma to CSFLV is 57-fold smaller than the clearance from plasma to brain ECF, whereas the clearance from CSFLV to plasma is 40-fold smaller than the clearance from brain ECF to plasma) and by assuming that the relationship between the different brain clearances was the same in both rats and humans, we were able to estimate the PK parameters for distribution between plasma and brain ECF, CSFLV, CSFTFV, and CSFCM in humans (Table I). Predictions indicate that brain ECF concentrations in humans are ~threefold higher than plasma concentrations (Fig. 6).

Observed and predicted human acetaminophen concentrations in plasma, brain ECF, CSFLV, CSFCM, and CSFSAS. Predictions are based on a 10-min i.v. infusion of 15 mg/kg acetaminophen in a 70-kg human
Discussion
In the development of CNS-targeted drugs, the prediction of human CNS target exposure is a big challenge. Often, it is assumed that CSF concentrations are more or less equal to unbound brain concentrations, and the most common method for collecting a CSF sample from humans is by a lumbar puncture (39). However, the brain is a dynamic multi-compartmental system, in which all processes of entry, diffusion, metabolism, binding, and elimination determine local CNS concentrations. Not much is currently known about the impact of these processes on brain disposition of different drugs. Here, we present a methodology to investigate brain disposition in rats using a physiologically based modeling approach aiming at improving the prediction of human brain exposure.
In this study, we compared the unbound concentration–time profiles of acetaminophen, as a paradigm compound for passive transport, obtained from the brain striatum and different sites of CSF. To that end, we have used the multiple probe approach, placed into different sites of the brain. Thus, the PK information is obtained by the same technique, without the need of taking CSF samples that influence brain physiology. Our hypothesis was that acetaminophen would distribute homogeneously over the brain.
Since the inter-individual variation in brain concentrations is reasonably large, the statistical significance of the results could be questioned. However, the variability in brain concentrations is most likely the result of the large variation in in vivo recovery, which is the sum of the intra-individual variation, the analytical error, and the true inter-individual variability.
With an assumed log-normal distribution of the inter-individual variability and the residual error, it can be assumed that the observed differences between brain ECF and CSF concentrations are the result of actual differences, rather than a result of the variability in the in vivo recoveries.
Preferably, the in vivo recovery should have been determined in the same animals in which the brain disposition experiments were performed to eliminate the inter-individual variability. However, we decided to perform these experiments in a separate group of animals to keep the duration of experiments within realistic limits.
With in vivo recoveries of approximately 10%, we are working at the limit of the methodology. Therefore, in retrospect, the level of in vivo recovery could have been increased by decreasing the microdialysis perfusion flow rate. However, as our analytical method required a certain sample volume, we decided to use a flow rate of 2 μl/min. Furthermore, we have used 10-min sample intervals to reveal the initial rate of brain distribution, which is crucial information for estimating clearance values into and out of the different brain compartments.
Even though the final resulting variability of the brain disposition data was high, the modeling does take into consideration this variability by means of the inter-individual variability in plasma to brain ECF, plasma to CSFLV, and plasma to CSFCM clearance, as well as a residual error on the brain ECF, CSFLV, and CSFCM data.
Acetaminophen PK in plasma, together with brain ECF PK or CSF PK, has been studied before (18,19,36–38,40–42), but brain ECF PK and CSF PK have never been directly related to each other. The plasma concentration–time profile obtained in this study shows great similarities with those reported previously. For example, Morrison et al. showed a distribution phase of approximately 20 min, after an i.v. bolus dose of 15 mg/kg, followed by an elimination phase until the end of the experiment at t = 120 min (40). Watari et al. reported a distribution phase of 15 min and that the disposition of acetaminophen was best described by a two-compartment model (36). As in previous studies from our group (18,19,42), we observed in this study a plateau in plasma concentrations at time points later than 120 min. The plateau of the plasma concentrations is presumably caused by enterohepatic circulation of acetaminophen. This has been described by Watari et al. (36), Siegers et al. (37), and Hjelle and Klaassen (38). Acetaminophen is metabolized/biotransformed in the liver into acetaminophen sulfate and acetaminophen glucuronide. Acetaminophen sulfate is subsequently transported both into the bile and into the bloodstream, after which, it is mainly excreted via urine. Acetaminophen glucuronide is mainly transported into bile, which is then released in the intestines. After the release in the small intestine, the acetaminophen sulfate and acetaminophen glucuronide can be hydrolyzed into acetaminophen, which is then reabsorbed. The time course of the reuptake of acetaminophen spans several hours (36–38), which therefore could explain the plateau in plasma concentrations from t = 120 min onward.
It is often assumed that CSF concentrations equilibrate readily with brain ECF concentrations due to the lack of a physical barrier between the two (10–12). The rate of equilibration between CSF concentrations and brain ECF concentrations by passive diffusion through brain tissue is dependent on the lipophilicity of the compound (41). For acetaminophen, which is a moderately lipophilic compound with known fast equilibrium between blood and brain, and no indication of active transport at the BBB or BCSFB, we expected the brain ECF and CSF concentrations to be similar. However, we have observed that brain ECF concentrations are approximately fourfold higher than the CSF concentrations. This can probably be explained by the relatively high turnover rate of CSF. With a total CSF volume of 300 μl, and a flow rate of 2.2 μl/min, the total CSF volume is replaced every 2.5 h. However, the volumes of the lateral ventricle and cisterna magna are calculated to be 50 and 17 μl, respectively. This indicates that the CSF in the lateral ventricle and cisterna magna is replaced every 23 and 8 min, respectively. Due to the slow distribution from the brain tissue to the CSF, the CSF acts as a sink, causing the observed concentration gradient within the brain. This has been observed for other passively transported compounds as well (43).
Several studies have reported acetaminophen concentration–time profiles in brain ECF (18,19,40) and CSF (42). We have found more or less similar unbound plasma and brain ECF concentration–time profiles, while in literature, brain ECF concentrations have been reported that were several fold lower than plasma concentrations. Importantly, in the previous studies, brain ECF concentrations were estimated on the basis of in vitro recovery, while in this study, it was based on in vivo recovery correction. We found an in vitro recovery value of acetaminophen of 31.9 ± 5.2%, for a 4-mm microdialysis membrane, which is ~threefold higher than the in vivo recovery of 12.0 ± 3.3% estimated in this study. This might be an explanation for the discrepancy between earlier reports and the current one. Assuming the same ratio between in vitro and in vivo recoveries (~factor 3), it would indicate a concentration–time profile in brain ECF that is more similar to unbound plasma concentrations than to CSF.
On the other hand, using serial CSF sampling, from the cisterna magna, van Bree et al. (42) have found that acetaminophen was slowly distributed into and out of CSF, resulting in a delayed maximum concentration in CSF and a slower elimination. We think that the sampling of CSF jeopardizes brain fluid homeostasis, as it causes a decrease in CSF pressure that normally serves as a trigger for elimination of CSF into blood via the basal cisterns and subarachnoid spaces (44).
Calculations of brain distribution are often performed by modeling the brain compartment as an effect compartment (15,45). Here, the plasma concentration is the driving force for brain concentrations, without the uptake into or elimination from the brain influencing the concentration–time profile in blood. However, for a more realistic approach in describing the data, we have included the brain compartment in the mass balance of acetaminophen disposition, resulting in a physiologically based model. Since the physiological volumes of the different brain compartments are over 35-fold lower than that of plasma, only a small fraction of the amount of acetaminophen in plasma is transported into brain tissue.
Interestingly, the clearance values between plasma and brain ECF are over 40-fold higher than the clearance values between plasma and CSF. This indicates that the transport across the BBB is more substantial compared to transport across the BCSFB.
Given that CSF concentrations are considered to be the best available surrogate for brain ECF concentrations in humans (3,10–12), we have focused on the prediction of human brain ECF concentrations based on human CSF concentrations. Bannwarth and colleagues have measured acetaminophen concentrations in plasma and CSF following a 3-min i.v. infusion of 2 g propacetamol, a prodrug which is hydrolysed to acetaminophen within 7 min (35). The dosage used corresponded to ~15 mg/kg acetaminophen in 10 min, which is equal to the dose used in this study. The observed plasma and spinal CSFSAS concentration–time profile in humans are similarly shaped as the observed acetaminophen plasma concentration–time profile and the predicted CSFSAS concentration–time profile in rat. However, the kinetics in humans are different from that in rats.
For the prediction of human brain ECF exposure on the basis of human CSF concentrations, the PBPK modeling approach is important, as it allowed the extrapolation of our rat data. This was done by changing the different physiological parameters of the rat to the human values and assuming that the relationship between the different brain clearances was the same in both rats and humans. A similar approach has been applied by Kielbasa and Stratford, where brain compartmental volumes were fixed to their physiological volumes and human brain PK parameters were predicted on the basis of allometric scaling of rat brain PK parameters (43). These predictions were then compared to clinical PK parameters based on human plasma or CSF concentrations.
As the possibility to validate human predictions of brain ECF exposure on the basis of direct measurement of brain ECF concentrations in human is highly restricted, there is the need for more mechanistic understanding of the processes involved in the causal path from drug dosing to CNS drug effects, to validate the predictions on the basis of appropriate biomarkers in human CSF or plasma samples. To that end, the physiological brain disposition model presented here will be extended on the basis of brain disposition measurements of a series of other drugs with different physicochemical properties in order to fully characterize the processes involved in brain disposition. For drugs that are known to be actively transported into or out of the brain, a cross-compare designed study with or without the inhibition of the specific transporter will be informative on the contribution of the specific transporter in blood–brain transport. By adding this information to the current PBPK model, together with quantitative information on the transporter expression levels in animals and humans, the PBPK model will serve as a basis for extrapolation of preclinical findings and ultimately only in vitro determinations of physicochemical properties of drugs, to the human situation.
Conclusion
Even for acetaminophen, a drug without active transport processes into, within, and out of the brain, significant differences exist between the striatal ECF and the CSF concentrations after acute dosing. With the ultimate goal of being able to predict CNS drug effects in humans, a more mechanistic understanding of the relative importance of the processes involved in brain target site distribution can be obtained in vivo by applying the intracerebral microdialysis technique in combination with physiologically based pharmacokinetic modeling.
References
World Health Organization. In: Neurological disorders: public health challenges. 2007. http://www.who.int/mental_health/neurology/neurodiso/en/. Accessed 19 Dec 2011.
Pangalos MN, Schechter LE, Hurko O. Drug development for CNS disorders: strategies for balancing risk and reducing attrition. Nat Rev Drug Discov. 2007;6:521–32.
Westerhout J, Danhof M, de Lange ECM. Preclinical prediction of human brain target site concentrations: considerations in extrapolating to the clinical setting. J Pharm Sci. 2011;100:3577–93.
Feng MR. Assessment of blood–brain barrier penetration: in silico, in vitro and in vivo. Curr Drug Metab. 2002;3:647–57.
Bickel U. How to measure drug transport across the blood–brain barrier. NeuroRx. 2005;2:15–26.
Abbott NJ, Dolman DEM, Patabendige AK. Assays to predict drug permeation across the blood–brain barrier, and distribution to brain. Curr Drug Metab. 2008;9:901–10.
Hammarlund-Udenaes M, Fridén M, Syvänen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25:1737–50.
Liu X, Chen C, Smith BJ. Progress in brain penetration evaluation in drug discovery and development. J Pharmacol Exp Ther. 2008;325:349–56.
Jeffrey P, Summerfield S. Assessment of the blood–brain barrier in CNS drug discovery. Neurobiol Dis. 2010;37:33–7.
de Lange ECM, Danhof M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target site concentrations in the clinical setting. Implications of the barriers between blood and brain. Clin Pharmacokinet. 2002;41:691–703.
Shen DD, Artru AA, Adkison KK. Principles and applicability of CSF sampling for the assessment of CNS drug delivery and pharmacodynamics. Adv Drug Deliv Rev. 2004;56:1825–57.
Lin JH. CSF as a surrogate for assessing CNS exposure: an industrial perspective. Curr Drug Metab. 2008;9:46–59.
Cserr HF. Potassium exchange between cerebrospinal fluid, plasma, and brain. Am J Physiol. 1965;209:1219–26.
Cserr HF. Physiology of the choroid plexus. Physiol Rev. 1971;51:273–311.
Bouman HJ. van Wimersma Greidanus TB. A rapid and simple cannulation technique for repeated sampling of cerebrospinal fluid in freely moving rats. Brain Res Bull. 1979;4:575–7.
Nielsen JA, Fossom LH, Sparber SB. Metabolism of 3H-dopamine continuously perfused through push-pull cannulas in rats’ brains: modification by amphetamine or prostaglandin F2α. Pharmacol Biochem Behav. 1980;13:235–42.
de Lange ECM, Danhof M, de Boer AG, Breimer DD. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood–brain barrier. Brain Res Behav. 1997;25:27–49.
de Lange ECM, Danhof M, de Boer AG, Breimer DD. Critical factors of intracerebral microdialysis as a technique to determine the pharmacokinetics of drugs in rat brain. Brain Res. 1994;666:1–8.
Stevens J, Suidgeest E, van der Graaf PH, Danhof M, de Lange ECM. A new minimal-stress freely-moving rat model for preclinical studies on intranasal administration of CNS drugs. Pharm Res. 2009;26:1911–7.
Ståhle L, Segersvärd S, Ungerstedt U. A comparison between three methods for estimation of extracellular concentrations of exogenous and endogenous compounds by microdialysis. J Pharmacol Methods. 1991;25:41–52.
Le Quellec A, Dupin S, Genissel P, Saivin S, Marchand B, Houin G. Microdialysis probes calibration: gradient and tissue dependent changes in no net flux and reverse dialysis methods. J Pharmacol Toxicol Methods. 1995;33:11–6.
Scheller D, Kolb J. The internal reference technique in microdialysis: a practical approach to monitoring dialysis efficiency and to calculating tissue concentration from dialysate samples. J Neurosci Methods. 1991;40:31–8.
Thorne RG, Hrabĕtová S, Nicholson C. Diffusion of epidermal growth factor in rat brain extracellular space measured by integrative optical imaging. J Neurophysiol. 2004;92:3471–81.
Bass NH, Lundborg P. Postnatal development of bulk flow in the cerebrospinal fluid system of the albino rat: clearance of carboxyl-[14C]inulin after intrathecal infusion. Brain Res. 1973;52:323–32.
Condon P, Patterson J, Wyper D, Hadley D, Grant R, Teasdale G, et al. Use of magnetic resonance imaging to measure intracranial cerebrospinal fluid volume. Lancet. 1986;327:1355–7.
Kohn MI, Tanna NK, Herman GT, Resnick SM, Mozley PD, Gur RE, et al. Analysis of brain and cerebrospinal fluid volumes with MR imaging. Part I. Methods, reliability, and validation. Radiology. 1991;178:115–22.
Robertson EG. Developmental defects of the cisterna magna and dura mater. J Neurol Neurosurg Psychiatry. 1949;12:39–51.
Adam R, Greenberg JO. The mega cisterna magna. J Neurosurg. 1978;48:190–2.
Levinger IM. The cerebral ventricles of the rat. J Anat. 1971;108:447–51.
Cserr HF, Cooper DN, Suri PK, Patlak CS. Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am J Physiol. 1981;240:F319–28.
Abbott NJ. Evidence for bulk flow of brain interstitial fluid: significance for physiology and pathology. Neurochem Int. 2004;45:545–52.
Cox EH, Veyrat-Follet C, Beal SL, Fuseau E, Kenkare S, Sheiner LB. A population pharmacokinetic-pharmacodynamic analysis of repeated measures time-to-event pharmacodynamic responses: the antiemetic effect of ondansetron. J Pharmacokinet Biopharm. 1999;27:625–44.
Duffull SB, Aarons L. Development of a sequential linked pharmacokinetic and pharmacodynamic simulation model for ivabradine in healthy volunteers. Eur J Pharm Sci. 2000;10:275–84.
Yano Y, Beal SL, Sheiner LB. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28:171–92.
Bannwarth B, Netter P, Lapicque F, Gillet P, Péré P, Boccard E, et al. Plasma and cerebrospinal fluid concentrations of paracetamol after a single intravenous dose of propacetamol. Br J Clin Pharmacol. 1992;34:79–81.
Watari N, Iwai M, Kaneniwa N. Pharmacokinetic study of the fate of acetaminophen and its conjugates in rats. J Pharmacokinet Biopharm. 1983;11:245–72.
Siegers C-P, Rozman K, Klaassen CD. Biliary excretion and enterohepatic circulation of paracetamol in the rat. Xenobiotica. 1983;13:591–6.
Hjelle JJ, Klaassen CD. GLucuronidation and biliary excretion of acetaminophen in rats. J Pharmacol Exp Ther. 1984;228:407–13.
Hill KK, West SA, Ekhator NN, Bruce AB, Wortman MD, Baker DG, et al. The effect of lumbar puncture stress on dopamine and serotonin metabolites in human cerebrospinal fluid. Neurosci Lett. 1999;276:25–8.
Morrison PF, Bungay PM, Hsiao JK, Ball BA, Mefford IN, Dedrick RL. Quantitative microdialysis: analysis of transients and application to pharmacokinetics in brain. J Neurochem. 1991;57:103–19.
de Lange ECM, Bouw MR, Mandema JW, Danhof M, de Boer AG, Breimer DD. Application of intracerebral microdialysis to study regional distribution kinetics of drugs in rat brain. Br J Pharmacol. 1995;116:2538–44.
van Bree JBMM, Baljet AV, van Geyt A, de Boer AG, Danhof M, Breimer DD. The unit impulse response procedure for the pharmacokinetic evaluation of drug entry into the central nervous system. J Pharmacokinet Biopharm. 1989;17:441–62.
Kielbasa W, Stratford Jr RE. Exploratory translational modeling approach in drug development to predict human brain pharmacokinetics and pharmacologically relevant clinical doses. Drug Metab Dispos. 2012. doi:10.1124/dmd.111.043554.
Kazama S, Masaki Y, Maruyama S, Ishihara A. Effect of altering cerebrospinal fluid pressure on spinal cord blood flow. Ann Thorac Surg. 1994;58:112–5.
Hammarlund-Udenaes M, Paalzow LK, de Lange ECM. Drug equilibration across the blood–brain barrier—pharmacokinetic considerations based on the microdialysis method. Pharm Res. 1997;14:128–34.
Begley DJ. Transport to the brain. In: Begley DJ, Bradbury MW, Kreuter J, editors. The blood–brain barrier and drug delivery to the CNS. New York: Dekker; 2000. p. 93–108.
Kimelberg HK. Water homeostasis in the brain: basic concepts. Neuroscience. 2004;129:851–60.
Nilsson C, Stahlberg F, Thomsen C, Henriksen O, Herning M, Owman C. Circadian variation in human cerebrospinal fluid production measured by magnetic resonance imaging. Am J Physiol. 1992;262:R20–4.
Lee HB, Blaufox MD. Blood volume in the rat. J Nucl Med. 1985;26:72–6.
Frank H, Gray SJ. The determination of plasma volume in man with radioactive chromic chloride. J Clin Invest. 1953;32:991–9.
Dickey CC, Shenton ME, Hirayasu Y, Fischer I, Voglmaier MM, Niznikiewicz MA, et al. Large CSF volume not attributable to ventricular volume in schizotypical personality disorder. Am J Psychiatry. 2000;157:48–54.
Pardridge WM. Drug transport in brain via the cerebrospinal fluid. Fluids Barriers CNS. 2011;8:7.
Acknowledgments
The authors would like to thank Dirk-Jan van den Berg and Robin Hartman for their help and guidance with the surgical procedure, experimental setup, and the plasma and microdialysate sample concentration analysis. This work was supported by Top Institute Pharma, Project T5-105-1: Nanoscience as a tool for improving bioavailability and blood–brain barrier penetration.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Differential Equations
The mass balance equations describing the final PBPK model were expressed as follows:
Plasma:
Periphery:
Brain ECF:
CSFLV:
CSFTFV:
CSFCM:
CSFSAS:
Where:
-
k 12 = Q 12/V pl
-
k 21 = Q 12/V per
-
k 13 = CL13/V pl
-
k 31 = CL31/V ECF
-
k 14 = CL14/V pl
-
k 41 = CL41/V LV
-
k 15 = CL15/V pl
-
k 51 = CL51/V TFV
-
k 16 = CL16/V pl
-
k 61 = CL61/V CM
-
k 10 = CL10/V pl
- A i :
-
Amount of acetaminophen in compartment i (in nanograms)
- C i :
-
Concentration of acetaminophen in compartment i (in nanograms per milliliter)
- k :
-
Rate constant (in minutes)
- Q :
-
Flow rate (in milliliters per minute)
- CL:
-
Clearance (in milliliters per minute)
- V :
-
Volume (in milliliters)
- F abs :
-
The fraction of the dose that is reabsorbed over time (in percent)
- pl:
-
Plasma
- pl,u:
-
Unbound acetaminophen in plasma
- per:
-
Peripheral compartment
- ECF:
-
Brain ECF
- CSF:
-
CSF
- LV:
-
Lateral ventricle
- TFV:
-
Third and fourth ventricle
- CM:
-
Cisterna magna
- SAS:
-
Subarachnoid space
- 12:
-
From plasma to peripheral compartment
- 21:
-
From peripheral to plasma compartment
- 13:
-
From plasma to brain ECF compartment
- 31:
-
From brain ECF to plasma compartment
- 14:
-
From plasma to CSFLV compartment
- 41:
-
From CSFLV to plasma compartment
- 15:
-
From plasma to CSFTFV compartment
- 51:
-
From CSFTFV to plasma compartment
- 16:
-
From plasma to CSFCM compartment
- 61:
-
From CSFCM to plasma compartment
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Westerhout, J., Ploeger, B., Smeets, J. et al. Physiologically Based Pharmacokinetic Modeling to Investigate Regional Brain Distribution Kinetics in Rats. AAPS J 14, 543–553 (2012). https://doi.org/10.1208/s12248-012-9366-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-012-9366-1