
Abstract
The leveraged use of biomarkers presents an opportunity in understanding target engagement and disease impact while accelerating drug development. For effective integration in drug development, it is essential for biomarkers to aid in the elucidation of mechanisms of action and disease progression. The recent years have witnessed significant progress in biomarker selection, validation, and qualification, while enabling surrogate and clinical endpoint qualification and application. Biomarkers play a central role in target validation for novel mechanisms. They also play a central role in the learning/confirming paradigm, particularly when utilized in concert with pharmacokinetic/pharmacodynamic modeling. Clearly, these attributes make biomarker integration attractive for scientific and regulatory applications to new drug development. In this review, applications of proximal, or target engagement, and distal, or disease-related, biomarkers are highlighted using the example of the recent development of sitagliptin for type 2 diabetes, wherein elucidation of target engagement and disease-related biomarkers significantly accelerated sitagliptin drug development. Importantly, use of biomarkers as tools facilitated design of clinical efficacy trials while streamlining dose focus and optimization, the net impact of which reduced overall cycle time to filing as compared to the industry average.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Drug development attrition and the declining number of new chemical entities have been subjects of major scientific and legislative debate (1–6). Importantly, this debate has called into question the productiveness and effectiveness of traditional drug development given the limited probabilities of success of any new chemical entity. Notably, two regulatory initiatives one in the United States, and one in the European Union have addressed the so-called ‘pipeline problem’, and these include the Food and Drug Administration (FDA) critical path to new medicines initiative in the US (7) and the European Technology Platform for Innovative Medicines in the Europe (8). These initiatives were launched to enhance and accelerate the development of new chemical entities, as well as to insure the rapid application of scientific breakthroughs to approval and use. Both of these worldwide initiatives emphasize the application of biomarkers and disease progression modeling as a centerpiece to effective drug development.

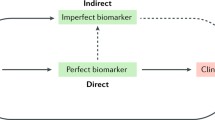
An idealized model for biomarkers, illustrating proximal or target engagement and distal or disease-related attributes
A particularly worrisome statistic in new drug development is that only ~11% of all new mechanisms are pharmacologically viable (1) and enter clinical development, highlighting the need for efficient use of biomarkers in translational medicine. To mitigate this issue, a number of initiatives have been developed to insure the effective integration of biomarkers in early drug development. These have included the FDA/National Institutes of Health (NIH) Consensus Conference of 1999 (9,10), the biomarker consortium in 2006 (9,10), and several efforts within Pharmaceutical Research and Manufacturers of America (PhRMA), FDA, NIH, and professional scientific organizations such as American Association of Pharmaceutical Scientists, American College of Clinical Pharmacology, and American Society for Clinical Pharmacology and Therapeutics. Declining number of new chemical entities, late stage attrition due to safety and efficacy, and suboptimal risk/benefit of marketed drugs all have prompted an increasing attention to the utility of biomarkers (1).
The discipline of experimental medicine engages in clinical research with a focus on biomarkers, the development of experimental models, and clinical target validation in support of drug development programs (2,9,10). Specifically, research is designed to provide an early and often preliminary assessment of beneficial pharmacologic activity or efficacy, and/or safety of new compounds in early clinical development in order to facilitate decision making. These approaches have the potential to provide a bridge from results in preclinical models to clinical results. This may provide an early read on pharmacodynamics and the pharmacokinetic-pharmacodynamic relationship. The use of experimental medicine has a clear charter: increase efficiency of drug development process by providing needed information on mechanisms of action, dose-response, early reads of efficacy and safety, thereby improving the quality and speed of decision-making (de-prioritize failures and accelerate winners). In some settings, experimental medicine may aid in regulatory evaluation and approval.
While experimental medicine leverages a range of tools that include biomarkers, imaging, molecular profiling and pharmacogenomic approaches, the focus of the present review is on markers for target engagement (i.e., proximal markers or how robustly a drug interacts with the mechanism) and disease (i.e. distal markers or indicators of drug efficacy). Further, while biomarkers reflect both safety and efficacy attributes, the focus of the present article is on markers related to efficacy.
There are consensus guidelines on the definitions and nomenclature of biomarkers and these have been outlined by the Biomarkers Definitions Working Group and is summarized in a publication (11). This review focuses on biomarkers which include measurements that suggest the etiology of, the susceptibility to, or the progress of disease; measurements related to the mechanism of response to treatments; and measurements linked to established clinical endpoints in response to therapeutic interventions. In this review, applications of distal and proximal biomarkers to assess pharmacological activity are presented. Some distal biomarkers have been clearly demonstrated to reflect therapeutic response, and others less clearly validated. Proximal biomarkers reflect the drug mode of action, but do not necessarily have an established or direct relationship to therapeutic response; examples include markers of enzyme inhibition or receptor binding. When such proximal biomarkers have a close, validated relationship to therapeutic response, efficacy information can be derived from extent of change in the biomarker, but such information must generally be viewed cautiously, as the tight relationship between extent of change in a proximal biomarker and impact on a distal biomarker in one model may not necessarily apply in other settings.
Regulatory guidance indicates that a distal biomarker is not considered an acceptable surrogate endpoint for ascertaining efficacy of a new drug unless it has been demonstrated to function as a valid indicator of clinical benefit (i.e., is a valid surrogate). Examining the relationship between drug concentration and the change in a biomarker as a function of time using exposure/response relationships can provide important and useful information. Such PK/PD relationships aid in the development of clinical trial designs, thereby accelerating drug development.
The area of biomarker qualification has been discussed in a recent review (9,10). Briefly, qualification is the process of establishing the link between a biomarker and the clinical/therapeutic endpoints that the biomarker is intended to relate. A fit-for-purpose biomarker qualification is a graded evidentiary process linking a biomarker with biology and clinical endpoints and dependent on the intended application.
Ideally, biomarker development needs to initiate as early as possible in drug discovery and development. The biomarker strategy and concomitant exposure/response strategy needs to be initiated as early as a novel mechanism or scientifically qualified target is identified.
Biomarkers and pharmacodynamic models can aid in making decisions about moving a compound forward in development, guiding early clinical development strategy. In the sitagliptin type 2 diabetes development program, plasma dipeptidyl-peptidase IV (DPP4) activity was implemented as a proximal or target engagement biomarker. Evidence of target engagement at a well tolerated dose will enhance the level of confidence that a given dose/concentration is adequate for proof-of-concept purposes and potentially allow decisions made about a compound to be generalized to the target. In the sitagliptin program, measures of serum glucose, insulin (reflecting β cell response), glucagon (reflecting α cell response), etc were used as distal or disease related biomarkers since they are intimately linked to clinical outcomes.
For a biomarker to aid in decision making, it is imperative that there is a defined relationship (preferably quantitative) between a target engagement promixal marker and a disease related distal biomarker (Fig. 1). If such links are established, then the opportunities to engage biomarkers as decision making tools are greatly enhanced. Specifically, the interplay between proximal target engagement biomarkers and distal disease related biomarkers may aid in a better understanding of the likelihood of success a new mechanism of disease might have, and as such an inference on proof-of-concept can be made. A positive proof-of-concept can be considered achieved if both target engagement and disease related biomarkers are positive, whereas when both target engagement and disease related biomarkers are negative, it may suggest that the mechanism may not have been tested to its potential. On the other hand, if target engagement biomarker is positive and there were no associated effect on disease related biomarker, then an inference that proof-of-concept has not been attained (or that the probability of doing so is low) with either the compound and the mechanism could be drawn. Inherent in such assessments is the quantitative modeling of proximal and distal measures to support proof-of-concept as well as role of the systems biology in quantitative fingerprinting of disease pathways. The greater the uncertainty in a new mechanism, the lower the confidence there is in terminating compounds or arresting development necessary to achieve quick-kill and redirecting resources.
In this review, the specific aspects of the important interplay of proximal and distal biomarkers are highlighted for a new mechanism for diabetes. The case study highlights how biomarkers can accelerate early clinical development and streamline late development time to filing.
CASE STUDY: SITAGLIPTIN, A NOVEL DPP4 INHIBITOR
Background on Type 2 Diabetes—Epidemiology and Pathophysiology
The worldwide prevalence of type 2 diabetes is growing by 3% per annum, and 90% of all patients with diabetes have type 2 (previously referred to as adult-onset) diabetes (12,13). Underlying this marked increase in prevalence are sedentary lifestyles and the consumption of diets rich in saturated fat and calories leading to higher prevalence of obesity. Obesity, especially central or visceral obesity, leads to insulin resistance, a key underlying factor in the pathogenesis of T2D (14).
There are three key pathogenic defects contributing to hyperglycemia in patients with T2D which are generally present in all populations, albeit the extent that each may contribute can differ between individuals and across populations. These defects include insulin resistance, loss of insulin secretion, and hepatic glucose overproduction (15). Insulin resistance is characterized by a decrease in the ability of insulin to stimulate tissue glucose uptake and utilization, and represents an early defect in patients who ultimately develop T2D. To counter insulin resistance, the pancreatic β-cell increases the release of insulin both in fasting and post-meal states. This maintains euglycemia (normal glucose levels) but leads to a stress on the β-cell. In patients destined to develop T2D, β-cell compensation begins to fail, insulin secretion diminishes, and decompensation leads to frank hyperglycemia—and onset of diabetes. Thus, the loss of β-cell function reflected by a decrease in insulin secretion underlies the progression from normal glucose tolerance to impaired glucose tolerance and then to frank diabetes (16,17). This reduction in β-cell function is thought to occur both because of the loss of pancreatic islet β-cell mass (18) and because of the loss of glucose-stimulated insulin secretion. Not only does the deterioration of β-cell function lead to the onset of diabetes, continued deterioration after the onset of diabetes underlies the need for progressively more treatments. Initially patients may respond to monotherapy but most patients require combination therapy, and eventually parenteral insulin therapy.
A third pathogenetic defect in patients with T2D is hepatic glucose overproduction. This defect correlates with the rise in fasting plasma glucose levels, and in part reflects inappropriate elevations in glucagon due to excessive pancreatic islet α-cell glucagon release. Glucagon is a key glucoregulatory hormone that stimulates liver glucose production. In non-diabetic individuals, glucagon rises in the fasting state helping to maintain glucose production and normal glucose levels, and is suppressed after a meal (with a carbohydrate load) thereby helping to avoid a marked rise in post-meal glucose concentrations. In patients with T2D, fasting glucagon levels are elevated, and glucagon levels are inadequately suppressed—or paradoxically increased—after a meal. These alterations in glucagon lead to higher fasting and post-meal hepatic glucose production, contributing to hyperglycemia (19).
The Incretin Axis and DPP-4 Inhibition
The mechanisms underlying the loss in pancreatic β-cell function with progression to T2D and subsequent worsening of diabetes remain poorly understood. However, defects in the incretin hormones—gut-derived peptides that stimulate insulin secretion—have been recognized and may contribute to diminished β-cell function. Incretins are released into the circulation after consumption of a meal. Glucagon-Like Peptide-1 (GLP-1) and Glucose-Dependent Insulinotropic Peptide (GIP) account for the majority of the incretin response. Incretins increase insulin gene expression, insulin biosynthesis and insulin release in a glucose-dependent manner—that is, these effects only occur with normal or elevated glucose levels, but are not seen when glucose levels fall below normal levels. Impairments in the incretin hormones have been observed in patients with T2D, and even in patients with pre-diabetic conditions. These defects include a decrease in GLP-1 concentrations—with a relatively normal response to GLP-1, and normal GIP levels—with diminished response to GIP (20,21). GLP-1 and glucose-dependent insulinotropic release was significantly reduced, but infusion of GLP-1 appropriately stimulated insulin secretion (20). A reduced response to GIP infusion was also observed (21). Since GLP-1 signaling is intact in patients with T2D, agents that mimic the effect of GLP-1 (GLP-1 analogs) have been developed. GLP-1 itself is difficult to use clinically given the very short half life in the systemic circulation. An early proof-of-concept was shown with subcutaneously administered GLP-1 in patients with T2D; a sub-chronic (6-week) continuous infusion resulted in clinically significant decreases in fasting plasma glucose (14.1 to 10.1 mM) and HbA1c (from 9.2 to 7.9%) as compared to placebo (22). To avoid the need for continuous subcutaneous infusion, analogs with extended half-lives (because they are more resistant to metabolism by the enzyme DPP4) have been developed. Exenatide (Byetta™) is now approved for patients with T2D and liraglutide (NN2211) is in late stage development (23,24). Both agents are subcutaneously administered and have been shown to improve glycemic control by reducing fasting and postprandial glucose concentrations via glucose-dependent insulin secretion, restoring first-phase insulin response, and β-cell responsiveness to glucose (25, 26). In addition, GLP-1 receptor agonists suppress glucagon secretion, delay gastric emptying, and reduce food intake (25).
A second strategy targeting the incretin axis is the pharmacological inhibition of the enzyme DPP4, as illustrated in Fig. 2. Both GLP-1 and GIP are rapidly inactivated by the DPP-4; therefore, inhibition of DPP-4 stabilizes and thereby increases the concentration of the active forms of these incretins.

Target engagement (DPP4, GLP) and distal (insulin, glucose, glucagon) biomarkers for glucose control via the DPP4 pathway
Pharmacology of DPP4 Inhibition
Dipeptidyl peptidase-IV (DPP4) inhibitors represent a new therapeutic approach to the treatment of type 2 diabetes as illustrated in Fig. 2 (27,28). Of the many compounds in this class in development, sitagliptin (Merck & Co., Inc.) is the first approved by the US FDA. Late stage development compounds include vildagliptin (Novartis, Inc) and saxagliptin (Bristol-Myers Squibb, Inc.).
Sitagliptin is an orally active, potent, and selective DPP4 inhibitor that targets the incretin axis in the treatment of patients with T2D (29). This pharmacological approach has been supported by data obtained with DPP4 deficient mice (30), and in studies using DPP4 inhibitors in humans (31–38). For example, mice that are bred to be genetically deficient in DPP4 demonstrate improved glucose tolerance, with approximately threefold elevated active GLP-1 levels, increased insulin secretion, and decreased circulating glucagon (30). Selective inhibitors of DPP-4, such as sitagliptin, have shown minimal toxicities in animal safety studies – however agents that are not as selective, inhibiting both DPP-4 and other related proteases such as DPP-8/9, have been shown to exhibit significant preclinical toxicities (39). In clinical studies of patients with T2D treated with sitagliptin, sustained DPP4 inhibition has been observed, leading to increased active GLP-1 and GIP levels, decreased post-glucose load glucose excursion, increased insulin and C-peptide levels, and decreased glucagon concentrations (33, 36–38). Glucose lowering with DPP4 inhibition occurs via increased insulin release and reduced glucagon levels (31,40,41).
A DPP4 inhibitor, such as sitagliptin, may also have several advantages over currently available insulin secretagogues such as sulfonylurea (SU) agents; these include a neutral effect on body weight (vs weight gain with an SU agent) and an incidence of hypoglycemia that is generally similar to placebo (vs an higher rate of hypoglycemia with an SU) (42). This effect appears to be consistent with the observation that GLP-1 stimulates insulin secretion in a glucose-dependent manner (43).
A potential advantage of DPP4 inhibitors is based on possible trophic effects of GLP-1 and other incretins on pancreatic beta-cells. DPP4 inhibitors have been shown to improve pancreatic β-cell function and mass in chronic rodent models of diabetes (44,45) and in patients with T2D (46,47). However, only with long-term clinical studies will it be understood if this beneficial effect in animal models translates to a benefit in patients with T2D.
The following sections highlight the prospective use of biomarkers in early clinical development for sitagliptin. Specifically, the sections highlight application of target engagement or proximal biomarkers such as DPP4 enzyme activity and active GLP-1 and GIP levels, as well as those that are more distal or disease-based, such as insulin, c-peptide, glucose, and glucagon. Importantly, integration of these biomarkers occurred in the first-in-human study in healthy normoglycemic volunteers as well as subsequent studies in patients with T2D and obese patients.
Application of Biomarkers in Sitagliptin’s Early Clinical Development Strategy
Preclinical Studies and Translational Research
An extensive preclinical investigation with sitagliptin provided an effective guidance for translational development. During preclinical development, it was critical to assess the degree of DPP4 inhibition that is likely to elicit sustained and clinically meaningful pharmacodynamic effects in humans. Studies in mice examined the decrease in the blood glucose profile (AUC) after a glucose load with a range of doses of sitagliptin. Sitagliptin decreased the glucose AUC in a dose-dependent manner achieving maximum efficacy at 1 mg/kg (which approximated to 46% inhibition) (29). Experiments included measurements of plasma DPP4 inhibition, active GLP-1, and drug concentrations. Maximal efficacy was found to approximately correspond to plasma DPP4 inhibition >80% (note that assay values did not correct for dilution and that in vivo values are higher) and to plasma concentrations ≥100 nM, which resulted in a two- to threefold increase in active GLP-1, analogous to that observed upon glucose challenge in DPP4 deficient mice (Nancy Thornberry, data on file). This enabled the selection of primary endpoints in clinical studies with healthy subjects and patients with T2D including plasma pharmacokinetics of sitagliptin, plasma DPP4 enzyme activity, and levels of active GLP-1 fasting and following a meal or glucose challenge. A Clinical Development Lab (CDL) played an integral role in developing validated assays for active GLP-1 and plasma DPP4 inhibition in human samples, streamlining the transfer of the assays from basic (preclinical) research to clinical research.
To provide direct evidence of the pharmacologic effects of sitagliptin in type 2 diabetics, a single-dose study examining reductions in glucose post-OGTT/meal challenge was performed immediately following completion of the single-dose healthy volunteer studies.
Early Clinical Development—Single Dose Clinical Studies
The early development program for sitagliptin had two objectives: first to confirm proof-of-concept for sitagliptin, and second, to provide dose focus information for later stage drug development. Proof-of-concept, as defined earlier in the manuscript, was to achieve positive target engagement of the proximal biomarkers (exemplified here as DPP4 activity and active GLP-1 augmentation) and disease related disease biomarker (such as blood glucose) responses. If this were to be met and dose/response adequately delineated, then acceleration of the development program could be achieved. A central premise behind this objective was that the pharmacodynamic profile (DPP-4 inhibition, GLP-1 stabilization) observed after acute dosing would be predictive of glucose-lowering efficacy following chronic dosing. Thus, plasma DPP4 activity was incorporated in the first human study for sitagliptin in normoglycemic healthy subjects (33). Sitagliptin demonstrated marked inhibition of plasma DPP4 activity in a dose dependent manner (Fig. 3) (33). Specifically, sitagliptin at doses of 100 mg or higher produced ~80% inhibition over 24 h. Analysis of the pharmacokinetic-pharmacodynamic relationship between plasma sitagliptin concentration and inhibition of plasma DPP4 activity further revealed that inhibition of plasma DPP4 activity of 80% or greater occurred when sitagliptin plasma concentrations were at least 100 nM (33). It was later discerned that the extent of inhibition of DPP4 activity in vivo would be higher (e.g., 80% inhibition in this assay would be estimated to represent more than 95% inhibition in vivo if the Michaelis-Menton equation is applied) since no dilution corrections during assay procedures had been employed (33). These initial data were used to predict dosing regimens for patients with T2D, based on the assumption that DPP-4 inhibition in animal models that maximally lowered glucose levels would also provide effective glucose lowering in the clinical setting in diabetic patients. Based on studies in healthy subjects, this predicted that a dose of 100-mg or higher once-daily would be associated with near-maximal glucose lowering.

Time course of inhibition of plasma DPP4 activity after administration of placebo (open circles) or single oral doses of sitagliptin—1.5 (solid circles), 12.5 (open squares), 50 (solid squares), and 200 (open triangles) mg (A) and 5 (solid circles), 25 (fed [solid squares] and fasted [open squares]), and 100 (open triangles) mg (B) to healthy, young male subjects. [Reprinted with permission from Macmillan Publishers Ltd: Clinical Pharmacology and Therapeutics (78(6), copyright Dec 2005]
Several doses of sitagliptin produced postmeal increases in GLP-1 that were approximately two to threefold higher than corresponding values for placebo. This increase is also consistent with near maximal acute glucose lowering efficacy in preclinical studies (Nancy Thornberry, data on file). Inhibition of sitagliptin further appeared to increase incretin levels by stabilizing the active form of GLP-1 rather than increasing secretion, this hypothesis being supported by the finding that sitagliptin increased post meal active GLP-1 levels as well as the ratio of active to total GLP-1 levels but had no impact on total GLP-1 levels. There was no clinically meaningful effect of sitagliptin on fasting and postmeal levels of glucose, insulin, glucagon, and C-peptide, an expected finding for healthy normoglycemic subjects. The observation that increase in GLP-1 and GIP with sitagliptin in healthy subjects did not lead to hypoglycemia was an important one. This supported the glucose-dependency of insulin secretion, reviewed above. Presumably, in normal individuals the increase in active incretin levels with sitagliptin that enhances insulin release likely leads to an initial very small decrease in glucose—and as soon as glucose levels fall below normal, no further insulin release occurs. In normal subjects, no meaningful reduction in glucose concentrations—and no episodes of hypoglycemia—are observed. In contrast, as reviewed above, in patients with diabetes who have elevated glucose levels, sitagliptin increases active GLP-1 levels, leading to insulin release and lowering of glucose from elevated concentrations to more normal concentrations. These data supported the continued clinical development of sitagliptin, as assessed by safety, pharmacokinetics, and pharmacodynamics. Importantly, these data from single dose studies confirmed the observations in preclinical models of diabetes that DPP-4 inhibition increased active incretin levels and improved glucose concentrations, with maximum response at >80% DPP-4 inhibition.
Understanding the dynamic aspects of how DPP4 inhibition modulated incretin response was crucial in facilitating drug development acceleration. Since both GLP-1 and GIP are released in response to a meal and help achieve glycemic control, the degree to which DPP-4 inhibition could influence the level of active incretin augmentation necessary for glucose lowering efficacy after an oral glucose tolerance test (OGTT) could be assessed after single doses. Sitagliptin reduced glycemic excursion following a glucose challenge in patients with T2D (Fig. 4) (37). Sitagliptin also dose dependently inhibited plasma DPP4 activity which correlated well with the two time points of OGTT, wherein a more dose related separation was evident (37). Sitagliptin significantly enhanced post-OGTT levels of active GLP-1 and GIP levels as well as the ratio of active to total GLP-1 and active to total GIP levels. Total GLP-1 and GIP levels were slightly reduced by about 11 to 20% after an OGTT following treatment with sitagliptin as compared to placebo (37). Sitagliptin significantly increased post-glucose challenge insulin and C-peptide levels, and reduced post-glucose challenge glucagon levels (Fig. 4). Near-maximal post-OGTT glucose lowering appears to be roughly associated with plasma sitagliptin concentrations of approximately 100 nM or greater, inhibition of plasma DPP4 activity of approximately 80% or greater and approximately 2-fold or greater enhancement of active GLP-1 and GIP levels. Exploratory E max models, assessing the relationship between sitagliptin plasma concentrations and the reduction of postchallenge glucose as well as with increases in active GLP-1 and GIP levels, further supported that estimated EC75 values (i.e., 75% of a maximal effect, assuming a near maximal response) for sitagliptin plasma concentrations of approximately 100 nM may be necessary for efficacy.

Plasma glucose (a), serum insulin (b), serum C-peptide (c), and plasma glucagon (d) concentrations after administration of single oral doses of sitagliptin 25 (white circles) or 200 mg (black triangles) or placebo (black circles) and an OGTT at 2 h postdose. Plasma glucose concentrations are also displayed for the 2 h after a standardized meal at 6 h postdose and an OGTT at 24 h postdose. Data are expressed as geometric mean ± SE. [Reprinted with permission from The Journal of Clinical Endocrinology & Metabolism Vol. 91, No. 11 4612–4619, Copyright 2006, The Endocrine Society]
Early Clinical Development—Multiple Dose Studies
Both proximal and distal biomarkers of DPP4 pathway were incorporated in a multiple dose study in healthy normoglycemic subjects (38). Sitagliptin produced dose- and concentration-dependent inhibition of plasma DPP4 activity (38), the level of inhibition being consistent those observed in prior clinical studies and in pre-clinical rodent models (see Section on translational research above). The relationship between sitagliptin plasma concentrations and inhibition of plasma DPP-4 activity was not altered after multiple doses, given no substantial differences in EC50 values.
Sitagliptin, at doses of 25-mg and higher demonstrated post-meal increases in active GLP-1 levels (vs. placebo) of approximately two- threefold or higher, a value that correlated with near maximal acute glucose lowering efficacy in pre-clinical rodent models (Fig. 5). The ratio of active to total post-meal GLP-1 levels generally increased approximately two- to threefold following sitagliptin treatment, as compared to placebo and an obvious dose response beyond this level of stabilization was not evident. DPP4 inhibition by sitagliptin appears to enhance incretin levels by stabilizing the active form of GLP-1 rather than stimulating incretin secretion, since sitaglitpin increases post-meal active GLP-1 levels as well as the ratio of active to total GLP-1 levels, but does not appear to increase levels of total GLP-1.

Effect of sitagliptin (25, 50, 100, 200, 400, and 600 mg once daily and 300 mg twice daily) or placebo on geometric mean of weighted average active GLP-1 concentrations through 2 h following standardized meals at 4, 10, and 24 h postdose in healthy male subjects. [Reprinted from Clinical Therapeutics, 28(1), Jan 2006, pages 55–72, copyright (2006), with permission from Excerpta Medica, Inc.]
The results with sitagliptin were found to be consistent with those observed by vildagliptin, wherein a 4-week study in subjects with mild T2D supported similar inhibition of DPP4 activity in conjunction with reduced glucagon levels and unaltered insulin levels (31). This suggested that acceleration of the program based on first principles elaborated above was justified.
Early Clinical Development—Special Populations (Obese Subjects)
The pharmacodynamics of sitagliptin after multiple dosing was also assessed in obese subjects (36). Treatment with sitagliptin 200 mg b.i.d. for 28 days led to near maximal inhibition of plasma DPP4 activity (~90%, uncorrected). Therefore, DPP4 inhibition was sustained over a longer period of dosing.
Active GLP-1 levels increased, following an OGTT, by approximately two- to three-fold as compared to placebo and to pretreatment baseline active GLP-1 levels. Sitagliptin also increased the ratio of active to total GLP-1 levels by a similar degree. Treatment with sitagliptin also resulted in a statistically significant reduction in post-OGTT glucose excursion on both Days 14 and 28, as compared to placebo. There was no weight gain observed after 28 days of treatment with sitagliptin in obese individuals.
Accelerating Sitagliptin Early Development, the Returns
As described above, the interplay between proximal target engagement biomarkers and distal disease related biomarkers has been effectively delineated for sitagliptin, both from a compound perspective as well as the mechanism of action perspective. A positive proof-of-concept was achieved since both target engagement and disease related biomarkers were positive. This reduced the uncertainty in a new mechanism for the treatment of T2D. The biomarker strategy employed in the sitagliptin program made it amenable to simple exposure/response relationships, which guided early clinical drug development. In the case of the sitagliptin program, it provided an opportunity to proceed to Phase IIB dose range-finding studies following completion of the Phase I program, bypassing a traditional Phase II study. This approach was supported by the observed tolerability at supra-therapeutic doses as high as 800-mg (single dose), 600-mg for 10 days or 400-mg for 28-days.
A PK/PD evaluation indicated that near maximal glucose lowering following an OGTT occurs at approximately 100 nM (corresponding to approximately 80% plasma DPP4 inhibition, uncorrected). In addition, near maximal increases (i.e., approximately two- to threefold) in the levels of active GLP-1 and intact GIP, as compared to placebo, were also reached at plasma concentrations of 100 nM. These data suggested that sustained plasma concentrations of 100 nM or greater over the dosing interval would result in maximal or near maximal efficacy of sitagliptin in clinical practice. These observations were also in general agreement with preclinical studies where near maximal efficacy was observed in animal models with plasma DPP4 inhibition of 80%.
A trough concentration of approximately 100 nM (thereby achieving 80% inhibition of DPP4 activity) was predicted to be achieved with single oral doses of 100 mg once daily. The strategy for dose selection in Phase IIB was to demonstrate that 100 mg once daily was near maximal with respect to reductions in HbA1c. In addition, multiple twice daily doses up to 50 mg twice daily were also tested to assess the relative benefits of maintaining higher plasma trough concentrations of sitagliptin. Subsequently, studies evaluating the efficacy and safety and tolerability of 200 mg once daily and 100 mg once daily were also incorporated in Phase III.
The average time taken for a new molecule to progress from first introduction to man to Phase III dose is approximately 3.5 years (48). For the sitagliptin program, the biomarker strategy effectively employed in early development enabled first-in-man study to Phase III in as little as 2.1 years. The reduced time to filing was supported by a biomarker strategy which was in alignment with a simple exposure/response approach and increased the efficiency of drug development.
References
I. Kola, and J. Landis. Can the pharmaceutical industry reduce attrition rates? Nature Rev. Drug Discov. 3(8):711–5 (2004).
B. H. Littman, and S. A. Williams. The ultimate model organism: progress in experimental medicine. Nature Rev. Drug Discov. 4(8):631–8 (2005).
R. Krishna. Introduction to dose optimization. In R. Krishna (ed.), Dose optimization in drug development, Taylor and Francis, v161, 2006, pp. 1–13.
R. Krishna. Quantitative clinical pharmacology: making paradigm shifts a reality. J. Clin. Pharmacol. 46(9):966–7 (2006).
C. -P. Milne. US and European regulatory initiatives to improve R&D performance. Expert Opinion on Drug Discovery. 1(1):11–4 (2006).
K. I. Kaitin. Decline in drug approvals is driving industry, FDA to enhance efficiency. Tufts Center for the Study of Drug Development Impact Report. 7(1) (2005)Jan/Feb.
Challenge and opportunity on the critical path to new medical products, US Food and Drug Administration, 2004. Available at: http://www.fda.gov/oc/initiatives/criticalpath/whitepaper.html. Accessed December 29, 2006.
Innovative medicines for Europe, EFPIA, 2004. Available at: http://ec.europa.eu/research/fp6/index_en.cfm?p=1_innomed. Accessed December 29, 2006.
J. A. Wagner. Bridging preclinical and clinical development – biomarker validation and qualification. In R. Krishna (ed.), Dose Optimization in Drug Development, Informa Healthcare, New York, 2006, pp. 35–43.
J. A. Wagner, S. A. Williams, and C. J. Webster. Biomarkers and surrogate end points for fit-for-purpose development and regulatory evaluation of new drugs. Clin. Pharmacol. Ther. 81(1):104–7 (2007).
Biomarkers Definitions Working R. Group. Biomarkers and surrogate endpoints in clinical trials: proposed definitions and conceptual framework. Clin. Pharmacol. Ther. 69:89–95 (2001).
P. Zimmet, K. G. Alberti, and J. Shaw. Global and societal implications of the diabetes epidemic. Nature. 414:782–87 (2001).
S. Wild, G. Roglic, A. Green, R. Sicree, and H. King. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 27:1047–53 (2004).
J. R. Sowers. Obesity as a cardiovascular risk factor. Amer. J. Med. 115(Suppl 8A):37S–41S (2003).
R. A. DeFronzo. The triumvirate: beta-cell, muscle, and liver: a collusion responsible for NIDDM. Diabetes. 37:667–87 (1988).
D. Tripathy, K. F. Eriksson, M. Orho-Melander, J. Fredriksson, G. Ahlqvist, and L. Groop. Parallel manifestation of insulin resistance and beta cell decompensation is compatible with a common defect in Type 2 diabetes. Diabetologia. 47:782–93 (2004).
G. C. Weir, and S. Bonner-Weir. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 53(Suppl 3):S16–21 (2004), Dec.
A. Butler, J. Janson, S. Bonner-Weir, R. Ritzel, R. A. Rizza, and P. C. Butler. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 52:102–10 (2003).
G. Jiang, and B. Zhang. Glucagon and regulation of glucose metabolism. Amer. J. Physiol. Endocrinol. Metab. 284:671–8 (2003).
T. Vilsboll, T. Krarup, C. F. Deacon, S. Madsbad, and J. J. Holst. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes. 50:609–13 (2001).
T. Vilsboll, F. K. Knop, T. Krarup, A. Johansen, S. Madsbad, S. Larsen, T. Hansen, O. Pedersen, and J. J. Holst. The pathophysiology of diabetes involves a defective amplification of the late-phase insulin response to glucose by glucose-dependent insulinotropic polypeptide-regardless of etiology and phenotype. J. Clin. Endo. and Metabol. 88:4897–903 (2003).
M. Zander, S. Madsbad, J. L. Madsen, and J. J. Holst. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 359:824–30 (2002).
B. Rolin, M. O. Larsen, C. F. Gotfredsen, C. F. Deacon, R. D. Carr, M. Wilken, and L. B. Knudsen. The long-acting GLP-1 derivative NN2211 ameliorates glycemia and increases beta-cell mass in diabetic mice. Am. J. Physiol. Endocrinol. Metab. 283(4):E745–52 (2002).
M. A. Nauck, M. Hompesch, R. Filipczak, T. D. Le, M. Zdravkovic, and J. Gumprecht. Five weeks of treatment with the GLP-1 analogue liraglutide improves glycaemic control and lowers body weight in subjects with type 2 diabetes. Exp. Clin. Endocrinol. Diabetes. 114(8):417–23 (2006).
M. A. Nauck, S. Duran, D. Kim, D. Johns, J. Northrup, A. Festa, R. Brodows, M. Trautmann. A comparison of twice-daily exenatide and biphasic insulin aspart in patients with type 2 diabetes who were suboptimally controlled with sulfonylurea and metformin: a non-inferiority study. Diabetologia. 50(2):259–267 (2007).
S. A. Doggrell. Recent evidence of sustained benefit with exenatide in Type 2 diabetes. Expert Opin. Pharmacother. 7(14):2003–6 (2006).
D. J. Drucker, and M. A. Nauck. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 368(9548):1696–705 (2006).
M. K. Reimer, J. J. Holst, and B. Ahren. Long-term inhibition of dipeptidyl peptidase IV improves glucose tolerance and preserves islet function in mice. Eur. J. Endocrinol. 146(5):717–27 (2002), May.
D. Kim, L. Wang, M. Beconi, G. J. Eiermann, M. H. Fisher, H. He, G. J. Hickey, J. E. Kowalchick, B. Leiting, K. Lyons, F. Marsilio, M. E. McCann, R. A. Patel, A. Petrov, G. Scapin, S. B. Patel, R. S. Roy, J. K. Wu, M. J. Wyvratt, B. B. Zhang, L. Zhu, N. A. Thornberry, and A. E. Weber. (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 48(1):141–51 (2005), Jan 13.
D. Marguet, L. Baggio, T. Kobayashi, A. M. Bernard, M. Pierres, P. F. Nielsen, U. Ribel, T. Watanabe, D. J. Drucker, and N. Wagtmann. Enhanced insulin secretion and improved glucose tolerance in mice lacking CD26. PNAS. 97:6874–9 (2000).
B. Ahren, M. Landin-Olsson, P. A. Jansson, M. Svensson, D. Holmes, and A. Schweizer. Inhibition of dipeptidyl peptidase-4 reduces glycemia, sustains insulin levels, and reduces glucagon levels in type 2 diabetes. J. Clin. Endocrinol. Metab. 89:2078–2084 (2004).
S. Ristic, S. Byiers, J. Foley, and D. Holmes. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obese Metab. 7(6):692–8 (2005).
G. A. Herman, C. Stevens, K. Van Dyck, A. Bergman, B. Yi, M. De Smet, K. Snyder, D. Hilliard, M. Tanen, W. Tanaka, A. Q. Wang, W. Zeng, D. Musson, G. Winchell, M. J. Davies, S. Ramael, K. Gottesdiener, and J. A. Wagner. Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin. Pharmacol. Ther. 78(6):675–88 (2005).
R. E. Pratley, S. Jauffret-Kamel, E. Galbreath, and D. Holmes. Twelve-week monotherapy with the DPP-4 inhibitor vildagliptin improves glycemic control in subjects with type 2 diabetes. Horm Metab. Res. 38(6):423–8 (2006).
J. Rosenstock, R. Brazg, P. J. Andryuk, K. Lu, and P. Stein. Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin added to ongoing pioglitazone therapy in patients with type 2 diabetes: a 24-week, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Clin. Ther. 28(10):1556–68 (2006).
G. A. Herman, A. Bergman, F. Liu, C. Stevens, A. Q. Wang, W. Zeng, L. Chen, K. Snyder, D. Hilliard, M. Tanen, W. Tanaka, A. G. Meehan, K. Lasseter, S. Dilzer, R. Blum, and J. A. Wagner. Pharmacokinetics and pharmacodynamic effects of the oral DPP-4 inhibitor sitagliptin in middle-aged obese subjects. J. Clin. Pharmacol. 46(8):876–86 (2006).
G. A. Herman, A. Bergman, C. Stevens, P. Kotey, B. Yi, P. Zhao, B. Dietrich, G. Golor, A. Schrodter, B. Keymeulen, K. C. Lasseter, M. S. Kipnes, K. Snyder, D. Hilliard, M. Tanen, C. Cilissen, M. De Smet, I. de Lepeleire, K. Van Dyck, A. Q. Wang, W. Zeng, M. J. Davies, W. Tanaka, J. J. Holst, C. F. Deacon, K. M. Gottesdiener, and J. A. Wagner. Effect of single oral doses of sitagliptin, a dipeptidyl peptidase-4 inhibitor, on incretin and plasma glucose levels after an oral glucose tolerance test in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 91(11):4612–9 (2006).
A. Bergman, C. Stevens, Y. Zhou, B. Yi, M. De Smet, K. Snyder, D. Hilliard, M. Tanen, W. Tanaka, A. Q. Wang, W. Zeng, D. Musson, G. Winchell, M. J. Davies, S. Ramael, K. Gottesdiener, J. A. Wagner, and G. A. Herman. Pharmacokinetic and pharmacodynamic properties of multiple oral doses of sitagliptin, a dipeptidyl peptidase-IV inhibitor: a double-blind, randomized, placebo-controlled study in healthy male volunteers. Clin. Ther. 28(1):55–72 (2006).
G. R. Lankas, B. Leiting, R. S. Roy, G. J. Eiermann, M. G. Beconi, T. Biftu, C. C. Chan, S. Edmondson, W. P. Feeney, H. He, D. E. Ippolito, D. Kim, K. A. Lyons, H. O. Ok, R. A. Patel, A. N. Petrov, K. A. Pryor, X. Qian, L. Reigle, A. Woods, J. K. Wu, D. Zaller, X. Zhang, L. Zhu, A. E. Weber, and N. A. Thornberry. Dipeptidyl peptidase IV inhibition for the treatment of type 2 diabetes: potential importance of selectivity over dipeptidyl peptidases 8 and 9. Diabetes. 54(10):2988–94 (2005).
B. Ahren, E. Simonsson, H. Larsson, M. Landin-Olsson, H. Torgeirsson, P. A. Jansson, M. Sandqvist, P. Bavenholm, S. Efendic, J. W. Eriksson, S. Dickinson, and D. Holmes. Inhibition of dipeptidyl peptidase IV improves metabolic control over a 4-week study period in type 2 diabetes. Diabetes Care. 25:869–875 (2002).
B. Ahren, R. Gomis, E. Standl, D. Mills, and A. Schweizer. Twelve- and 52-week efficacy of the dipeptidyl peptidase IV inhibitor LAF237 in metformin-treated patients with type 2 diabetes. Diabetes Care. 27:2874–2880 (2004).
R. Scott, M. Wu, M. Sanchez, P. Stein. Efficacy and tolerability of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy over 12 weeks in patients with type 2 diabetes. Int. J. Clin. Pract. 2006 Dec 5.
M. A. Nauck, M. M. Heimesaat, K. Behle, J. J. Holst, M. S. Nauck, R. Ritzel, M. Hufner, and W. H. Schmiegel. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J. Clin. Endo. Metab. 87:1239–46 (2002).
B. Sudre, P. Broqua, R. B. White, D. Ashworth, D. M. Evans, R. Haigh, J. L. Junien, and M. L. Aubert. Chronic inhibition of circulating dipeptidyl peptidase IV by FE 999011 delays the occurrence of diabetes in male Zucker diabetic fatty rats. Diabetes. 51:1461–69 (2002).
J. Mu, J. Woods, Y. P. Zhou, R. S. Roy, Z. Li, E. Zycband, Y. Feng, L. Zhu, C. Li, A. C. Howard, D. E. Moller, N. A. Thornberry, and B. B. Zhang. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic {beta}-cell mass and function in a rodent model of Type 2 diabetes. Diabetes. 55(6):1695–704 (2006).
B. Ahren, G. Pacini, J. E. Foley, and A. Schweizer. Improved meal-related beta-cell function and insulin sensitivity by the dipeptidyl peptidase-IV inhibitor vildagliptin in metformin-treated patients with type 2 diabetes over 1 year. Diabetes Care. 28(8):1936–40 (2005).
A. Mari, W. M. Sallas, Y. L. He, C. Watson, M. Ligueros-Saylan, B. E. Dunning, C. F. Deacon, J. J. Holst, and J. E. Foley. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 90(8):4888–94 (2005).
K. I. Kaitin. Fastest drug developers consistently best peers on key performance metrics. Tufts Center for the Study of Drug Development Impact Report 2006 Sep/Oct;8(5).
Acknowledgements
The authors acknowledge the input Peter Stein and Arthur Bergman provided in various aspects of the original contributions, portions of which have been referred to in this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Krishna, R., Herman, G. & Wagner, J.A. Accelerating Drug Development Using Biomarkers: A Case Study with Sitagliptin, A Novel DPP4 Inhibitor for Type 2 Diabetes. AAPS J 10, 401–409 (2008). https://doi.org/10.1208/s12248-008-9041-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-008-9041-8