
Abstract
The American Association of Pharmaceutical Scientists (AAPS) biosimilar focus group on nonclinical and clinical assays has developed this manuscript to guide the industry on best practices and testing strategies when developing neutralizing antibody (NAb) assays for biosimilar programs. The immunogenicity assessment to biosimilar and originator drug products is one of the key aspects of clinical programs for biosimilars to demonstrate biosimilarity. Establishing that there are no clinically meaningful differences in immune response between a proposed product and the originator product is a key element in the demonstration of biosimilarity. It is critical to collect, evaluate, and compare the safety and immunogenicity data from the clinical pharmacology, safety, and/or efficacy studies especially when the originator drug product is known to have potential for immune-mediated toxicity. This manuscript aims to provide a comprehensive review and recommendations on assay formats, critical reagents, approaches to method development, and validation of the neutralizing antibody assays in extrapolation within the scope of biosimilar drug development programs. Even if there are multiple options on the development and validation of NAb assays for biosimilar programs, the type of drug and its MoA will help determine the assay format and technical platform for NAb assessment (e.g., cell-based or non-cell-based assay). We recommend to always perform a one-assay approach as it is better to confirm the biosimilarity using one-assay for NAb. If a one-assay approach is not feasible, then a two-assay format may be used. This manuscript will provide all the details necessary to develop NAb assays for biosimilars.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
As per the Food and Drug Administration (FDA) draft guidance, “Quality Considerations in Demonstrating Biosimilarity to a Reference Product” (1), Biosimilarity is defined to mean that “the biological product is highly similar to an originator/reference product notwithstanding minor differences in clinically inactive components, and for which there are no clinically meaningful differences between the biological product and the originator/reference product in terms of the safety, purity, and potency of the product.” Additionally, the FDA in its guidance on “Scientific Considerations in Demonstrating Biosimilarity to a Reference Product” (2) recommends a stepwise approach to demonstrating biosimilarity, which can include a comparison of the proposed product and the reference (“originator”) product with respect to structure, function, animal toxicity, human pharmacokinetics (PK) and pharmacodynamics (PD), clinical immunogenicity (anti-drug antibodies, i.e., ADAs), and clinical safety and effectiveness. As a scientific matter, FDA expects a sponsor to conduct comparative human PK and PD studies (if there is a relevant PD measure(s)) and a clinical immunogenicity assessment. The agency opines that the nature and scope of the clinical study or studies will depend on the nature and extent of residual uncertainty about biosimilarity after conducting structural and functional characterization and, where relevant, animal studies. Clinical pharmacology studies and originating data are normally a critical part of demonstrating biosimilarity and for clinical extrapolation. These drug exposure studies are conducted to demonstrate that no clinically meaningful differences between the biosimilar and the originator product exist. They also include PD endpoints and pharmacometric analysis (pharmacometric analysis is the assessment of clinical relevance of the PD endpoints); one of the most critical parts of clinical pharmacology studies is to demonstrate comparable rates of immunogenicity between the originator and the biosimilar.
Although animal immunogenicity assessments are also conducted to assist in the interpretation of the animal study, these results generally do not predict potential immune responses to protein products in humans. Differences observed in animal immunogenicity assessments may reflect potential structural or functional differences between the two products not captured by other analytical methods.
The importance of the immunogenicity data in Biosimilar assessment has been confirmed by the FDA in its guidance document (2) stating, “The goal of the clinical immunogenicity assessment is to evaluate potential differences between the proposed product and the originator product in the incidence and severity of human immune response. Thus, establishing that there are no clinically meaningful differences of the immune response between a proposed product and the originator product. This is a key element in the demonstration of biosimilarity.”
The European Medicines Agency also provide a similar focus as above. The EMA in its regulations (3) states, “Immunogenicity testing of the biosimilar and the originator products should be conducted within the comparability exercise by using the same assay format and sampling schedule which must meet all current standards. Analytical assays should be performed with both the originator and biosimilar molecules in parallel (in a blinded fashion) to measure the immune response against the product received by each patient. The analytical assays should preferably be capable of detecting antibodies against both the biosimilar and the originator e molecule but should at least be able to detect all antibodies developed against the biosimilar molecule.” It is therefore critical to collect, evaluate, and compare the safety and immunogenicity data from the clinical pharmacology, safety, and/or efficacy studies. This is even more critical when the originator product is known to have the potential for immune-mediated toxicity.
Considering the importance and criticality of biosimilar therapeutics, a biosimilar subcommittee focus group has been formed within the AAPS biosimilar focus group. The goal of this biosimilar subcommittee is to combine key industry leaders’ expertise in an effort to reach consensus on issues surrounding the assays used to quantify biosimilar and originator drug products. The subcommittee for biosimilars also recognized the importance of assessing comparable immunogenicity of the biosimilar products to their originator products. In 2014, a white paper from the AAPS Ligand Binding Assay Bioanalytical Focus Group (LBABFG) Action Program Committee (APC) subcommittee was published with a focus on PK assays to support biosimilar comparability studies (4).
The objective of this paper from the focus group is to recommended approaches to create harmonization of global biosimilar bioanalytical assessment practices among industry sponsors and regulatory authorities in the area of Neutralizing antibody assessment.
Immunogenicity assessments typically use a tiered approach, which requires validated methods for the detection of binding as well as neutralizing ADAs (NAb). The complexities reside in the fact that some NAb assays are generally qualitative. Additionally, NAb assays are highly susceptible to drug interference and sometimes difficult to set up in the presence of matrices (cell-based assays in particular), as also stated by the FDA (1).
Immune responses to biological therapeutic protein drugs may affect patient safety and product efficacy and can neutralize their biological activities with adverse events. Adverse events caused by immunological response, such as anaphylaxis, cytokine-release syndrome, or cross-reactive neutralization to non-redundant endogenous protein may potentially lead to termination of a biological therapeutic drug product development program.
The potential clinical outcome of immune responses may be as follows:
-
Consequences on efficacy: NAbs may block the efficacy of drug products by targeting epitopes critical for efficacy. Neutralizing antibodies may also cross-react with a non-redundant endogenous counterpart. The range of safety consequences varies and is often unpredictable (5).
-
Anaphylaxis: acute allergic reaction characterized by certain clinical features. The definition currently accepted by the agency relies on clinical diagnostic criteria and does not specify a particular immunologic mechanism (6).
-
Cytokine release syndrome: reactions caused by the rapid release of proinflammatory cytokines (7,8,9).
-
Infusion reactions: range of acute effects, from symptomatic discomfort to sudden, fatal reactions ( (10)).
-
Non-acute reactions: delayed hypersensitivity (i.e., serum sickness) and immune responses secondary to immune complex formation (11).
Not only would NAb inhibit the efficacy but also cross-react with the endogenous protein counterpart and thereby blocking the biological function of the endogenous protein. (e.g., neutralizing antibodies to erythropoietin cause pure red cell aplasia by also neutralizing the non-redundant endogenous protein) (11,12,13).
Both neutralizing and non-neutralizing antibodies may alter the PK of the product by enhancing clearance (and thereby shortening serum half-life) or, conversely, by prolonging serum half-life and product activity. In the context of biosimilar development, in order to establish comparability, it is necessary to determine both neutralizing and non-neutralizing antibody incidence for the originator drugs and the biosimilar. The obtained data is critical to link the immunogenicity to clinical results (impact on PK results and association with adverse events). Depending on the risk associated with the drug and its mechanism of action (MoA), the choice of the format for NAb assay should be assessed. The next section will describe the different type of formats as well as the recommended platforms that can be used.
NEUTRALIZATION ASSAY FORMAT
The NAb assay format selection should be a scientifically driven approach that relies on a combination of three key factors: (i) the therapeutic MoA, (ii) the evidence of desirable assay performance characteristics, and (iii) risk of immunogenicity (14). Typically, Nab format selection using the key factors above is made to decide if a cell-based or a non-cell-based assay is to be developed for an immunogenicity program. Cell-based assays provide a functional readout using relevant cell lines, which is in close relation to the biological function of the drug and thus may more closely reflect the in vivo situation. However, cell-based NAb assays are difficult to establish, as suitably relevant cell lines and a proper assay endpoint have to be identified. An additional challenge with cell-based assay is assay sensitivity and consistency across programs. This is the most critical part as the cellular response has to be robust and specific. In addition, the cellular response may be impaired by many factors such as matrix interference and the presence of drug product.
In this context, non-cell-based or competitive ligand-binding (CLB) assays are relatively easier to set up with higher assay precision and better sensitivity, as well as having the option to use different detection systems (e.g., ELISA and ECL). There are different principles on how a CLB assay may be designed. An appropriate format may be determined on a case by case scenario. One possibility is to immobilize the target on a microtiter plate to capture the labeled drug, resulting in an assay signal. Another way is to use labeled drug as the capture molecule while the labeled target is used as the detection molecule (15,16,17).
While cell-based assays reflect the actual functional and biological activity of the drug, these assays not only are more complex compared to non-cell-based assays, but also are more time consuming and often not as sensitive and robust as CLB assays. Therefore, if the nature of the drug and/or physiological conditions result in roadblocks, then a non-cell-based assay must be developed.
In some other cases, there are also other critical factors influencing the selection of a CLB assay, e.g., the format might be preferable for detecting ADAs that are potentially neutralizing because they inhibit binding to the cognate antigen. Although EMA and FDA immunogenicity guidance recommends the use of cell-based bioassays for the detection of NAbs, non-cell-based assays are considered appropriate when the drug’s MoA is to block a soluble target or cellular receptor (3,11,18). For example, if the target for a biological therapeutic drug is a cell membrane bound receptor, the development of a CLB assay can be justified, as binding to the target receptor would be the first step in the MoA of the biological therapeutic drug. The CLB assay can be used to detect clinically relevant NAbs since the CLB assay sensitivity, drug tolerance, and other parameters can be optimized relatively easier when compared to cell-based assays. Several recent studies have compared both cell-based assays and CLB assays for the detection of NAbs and have found comparable assay performance (15,16,17,19).
This manuscript reviews the two types of NAb assays for biosimilar drugs and aids in the decision of whether to use a cell-based functional assay or non-cell-based assays.
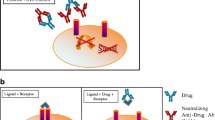
In general, cell-based assays can be further classified as direct and indirect assays based on the drug’s MoA and tied to the choice of assay endpoint (Diagram 1) (20).
Direct assays are generated by drug products that exert their effect directly on a cell, stimulating responses. In the absence of NAbs, the drug product binds to a ligand-specific receptor on the cell surface and elicits a cellular response. In the presence of NAbs, that response is either decreased or abrogated. Indirect assays are used when the drug product (usually a monoclonal antibody or a soluble receptor) works by blocking the binding of a ligand to a specific cell-surface receptor. The desired result is a reduction or absence of a cellular response. When NAbs are present, they bind to the drug product, preventing it from attaching to the cellular receptor. As a result, the ligand can bind to its cognate receptor and trigger a specific cellular response (21,22,23,24).
Figure 1a represents the direct cell-based activation by the drug. Based on the results of this activation, a constant drug concentration is selected for the NAb assay. The antibody positive control (APC) with neutralizing activity will show inhibition of the activation in Fig. 1b. Figure 1c represents the indirect cell-based assay showing inhibition of the activity by the drug. Based on the results of this inhibition, a constant drug concentration is selected for the NAb assay. The APC will show restoration of drug-mediated activity inhibition (increase of activity) in Fig. 1d.

a Represents the direct cell-based activation by the drug. b The antibody positive control (APC) with neutralizing activity will show inhibition of the activation. c Represents the indirect cell-based assay showing inhibition of the activity by the drug. d The APC will show restoration of drug-mediated activity inhibition (increase of activity)
For identifying a suitable NAb assay format for biosimilar products, there are a number of aspects that may be considered. Assay performance characteristic is an important determinant for NAb assay format selection.
Due to technology advancements over time, there is no mandate from the regulatory agencies that the assay platform chosen for the biosimilar has to be identical to the assay platform used for its originator during development and regulatory filing. However, the same assay format should be used to evaluate both the biosimilar and originator NAbs during the current assay development in order to support non-clinical or clinical comparability studies (usually it is not necessary to establish NAb assays during pre-clinical drug development phase, unless it is warranted due to high-safety risk).
METHOD DEVELOPMENT: ONE-ASSAY APPROACH VERSUS MULTIPLE-ASSAY APPROACH
The bioanalytical industry recognizes that it is very challenging to develop and validate immunogenicity assays to support comparability studies for biosimilar drug development.
Unlike the consensus view point for biosimilar PK assays, where a single ligand-binding assay to quantify both biosimilar and originator compounds is recommended to support pre-clinical and clinical comparability studies (4); different opinions exist for biosimilar immunogenicity assays. With regard to using a one-assay approach (usually using biosimilar reagents) or a two-assay approach (i.e., each assay optimized for the respective drug) for immunogenicity assays (2,4,25,26), the industry trend is now leaning towards the one-assay approach for immunogenicity assays (communications among multiple AAPS biosimilar subcommittees and regulatory agencies). The AAPS Biosimilar committee recommends using one assay for Biosimilar NAb evaluation (Table I).
The main advantage of using a one-assay approach is that there will be no “between-two-assays” variability, therefore minimizing the potential impact of immunogenicity differences due to assay bias. In addition, it will simplify sample analysis logistics and allow for “blinded” sample testing to maximize unbiased immunogenicity data interpretation for the comparability studies.
One common argument of using the one-assay approach (one biosimilar and one originator) or three-assay approach (one biosimilar and two originators, i.e., US and EU) over a one-assay approach is that due to potential subtle differences between the biosimilar and originator drug molecules (e.g., different glycosylation or other post-translational modifications), the ability to detect ADA (or NAb) against the originator drug may be limited if only one set of reagents (i.e., reagents for the biosimilar drug) is used. One important explanation to this point is that since the regulatory agencies (FDA and EMA) only require that the biosimilar product is “no more immunogenic” than its originator (1,3), using biosimilar reagents in a one assay will be the most conservative approach ensuring the most optimal detection of immunogenicity (4,25,26) to the Biosimilar product.
If a one-assay approach is used, it is recommended to use the biosimilar set of reagents to conduct assay development work (e.g., cut-point, sensitivity, selectivity, and precision). In a case that two- or multiple-assay approach should be used, additional assay development work will need to be conducted for two or more independent assays, which will be more challenging since additional assay parameters (e.g., cut-point, sensitivity, selectivity, precision, etc.) from two or more assays would need to be evaluated for their “comparability” or “similarity” using specific reagent pairs.
For an effective assay development, it is recommended to start with a one-assay approach to assess important assay parameters (discussed in the following paragraphs). If the development of a one-assay approach for both the biosimilar and originator is not possible due to confounding factors in the assay, the two- or three-assay approach may be used with adequate scientific justification.
Regardless of the assay approach that a laboratory chooses to use, evaluation of key assay attributes must be conducted during the assay development stage to ensure the assay(s) can detect the ADAs (or NAbs) against the biosimilar or the originator “similarly” (or equivalently).
Specifics for Biosimilar NAb Assays and Critical Reagent Requirements
As extensively discussed in literature as well as in the sections above, NAbs may potentially impact the PK and pharmacological properties of biological therapeutic drugs and therefore need to be appropriately monitored (23,24,27). The methods should be specific, sensitive, with good precision, and capable of tolerating a relevant amount of circulating drug product (24). These requirements are expected for methods for the detection of NAbs in general and are therefore also true for NAb methods that will support the development of biosimilar products. However, in addition, there are several unique considerations associated with developing and validating a biosimilar NAb assay.
In case of Biosimilars, product characteristics such as protein concentration and potency comparisons are key to assuring that comparability assessments reflect actual product similarities and are not due to differences in the product attributes. Often there are concentration differences related to product labeling. The “actual” concentrations are used for the biosimilar, whereas “nominal” concentrations are used for originator product. “Nominal” concentrations span the “high to low” allowable limits of the label specification, and these concentration differences between products may be large enough to add bias to the comparability assessment (1). As a result, verification of protein concentration is required prior to conducting development of bioanalytical assays for a biosimilar program. Another area contributing to potential concentration variability is in the reconstitution of the lyophilized drug product. Verification of protein concentration after reconstitution is also required to assure that bias is not introduced. There can be post-translational modifications resulting in structural differences between the two products, such as glycosylation patterns with high mannose, which have a potential impact not only on PK profiles, but also on assay-binding kinetics and the development of ADAs specific to these regions. By the time a Biosimilar program comes to the immunogenicity assessment phase, it is assumed that extensive CMC characterizations have already been conducted for different biosimilar drug batches, as well as originator batches (e.g., EU vs. US). It is also assumed that the analytical similarity of the physical attributes of the drug products/proteins has been established within acceptable specification ranges. It is recommended to use one representative batch of the biosimilar or originator (preferably the same batches which are to be used for the clinical comparability study if available) for assay development, validation, and sample assessment studies.
Another requirement in developing a biosimilar NAb assays is the need to understand test products that have multiple MoAs. During an originator product licensure, it is likely that a NAb assay was developed based on the major contributor of the MoAs and technology limitations at the time. With significant advancements in technology available to measure and assess proteins and an increasing understanding of biology, it is accepted that different NAb assays can be developed for biosimilar products based on distinct MoAs of a test product. Therefore, it is important to select an assay based on a MoA that is not only a major contributor to the therapeutic efficacy, but also one that is most sensitive in detecting potential differences between the originator drug and the biosimilar.
Critical reagent consideration is yet another important aspect in developing NAb methods to support biosimilar product development. Critical reagents include both the originator product and the biosimilar as well as assay positive controls (APCs) that are specific to both originator and biosimilar products and can neutralize both the biosimilar and the originator(s) drugs with suitable sensitivity. The identification of a NAb APC early on is equally important to the assay design. With such high demand in biosimilars, many reagent manufactures have created reagents such as ADA and NAb positive controls that can be used in the assay. In some instances where the reagent is not available, development of these reagents will require adequate lead time to assure they have the characteristics necessary to evaluate neutralizing potential with a significant amount of data demonstrating the reagent(s) recognize both products comparably. Additional reagents may be considered critical reagents depending on the assay strategy and specifics of the biologic.
Depending on the NAb assay strategy, specific experiments need to be carried out to demonstrate that there is bioanalytical equivalence of the biosimilar and originator products. “Equivalence” is a bioanalytical comparability assessment which should have a statistical basis for the conclusion. If one assay is selected, it is important to show that both originator product and biosimilar give comparable qualification results within the assay; if two assays are selected, then it is important to show that both assays have similar sensitivity and specificity and can yield comparable results. The steps needed for the different approaches are described in the next sections. Requirement for biosimilar ADA assay development and validation will follow the AAPS LBABFG BMV APC focus group white paper (4), and this article will only focus on the specific requirements for NAb assays.
Assessment of Biosimilar and Originator Drugs in the NAb Assay System Using Appropriate MRD (Minimum Required Dilution)
NAb assays are based on the ability of ADA to neutralize a drugs’ biological function. Therefore, it is critical to find an optimal drug concentration in the assay system which allows for sufficient bioactivity signal and the concentration selected allows optimum neutralizing effect by the APC. Both biosimilar and originator drugs should be used in the experiments to ensure that they are comparable in the assay. MRD evaluation is very critical and should be a part of initial assay development experiments while studying bioactivity and neutralization potential of the drug and the APC, respectively. MRD must be carefully selected to ensure that the integrity of the matrix is conserved as well as to demonstrate that the matrix does not interfere in the assay. In the case of cell-based assays, it is especially helpful to evaluate the optimum MRD against a buffer curve to ensure no matrix interference exist. In order to achieve this balance, multiple concentrations of the matrix should be tested in the assay. Note that the sensitivity of the assay must be reported after factoring the MRD or matrix dilution factor. Sensitivity results should be compared to demonstrate that the drugs behave similarly.
Drug-dose response curve should be tested using serially diluted drugs (biosimilar and originator(s)) to check for a similar and consistent range of response. The drug concentration which gives a strong enough signal over background (recommend to be two to fivefold (or higher) over “no-drug” baseline), but not in the plateau region of the response curve should be chosen for further assay development work. Signal/noise (S/N) of less than twofold often does not provide enough “window” for NAb APC to work, whereas too high of a S/N will diminish the overall assay sensitivity because more NAb would be needed to neutralize the drug effect. Although it may be challenging to achieve, it would be best if the same concentration of biosimilar and innovator drugs can have similar drug-dose response curves. In this scenario, it should be noted that defining “comparability” or “similarity” between biosimilar and originator compounds in these tests could be challenging since the assay is non-quantitative. The next step is to select and determine the level of APC for the assay.
Positive NAb Controls
To assess if a NAb assay can detect NAb against biosimilar and originator drugs, there are multiple strategies that may be employed. Whichever platform and methods are used, an APC needs to be generated. The APC(s) should be affinity purified (against the drug used for hyperimmunization) and quantified so that assay sensitivity can be evaluated. If the therapeutic drug is a monoclonal antibody (mAb), it is also important to enrich/purify the ADA which targets complementarity determining region (CDR) for optimum performance. The APC(s) can be generated either against the biosimilar or the originator(s) or both drugs. If the use of one APC is acceptable, it will be then recommended to generate the APC against the biosimilar drug; however, the APC can also be commercially purchased or generated to the originator drug and used with scientific justification of reactivity. It is also possible that the polyclonal APCs may not have adequate neutralizing effect. In this case, a monoclonal APC can also be used as assay positive control.
If multiple APCs are used in an assay (against the biosimilar and originator(s)), all the APCs should have “similar” sensitivity. The NAb-dose response curve should be performed for both APC using either drug compound at the optimal concentration (discussed in above section). This “similarity” is difficult to achieve as the immune responses from the hyperimmunization are highly variable. Therefore, it is not recommended to use this approach. It should be noted that enough APC for the entire program should be generated or purchased if possible to avoid lot changes during the course of the program (expiration of reagents will also need to be taken under consideration). If lot changes are required, then critical reagent qualification NAb assays would need to be conducted to evaluate the performance of the new APC in the assay.
Even though two NAb APCs may be used in the assay development phase for the one- or multiple-assay approach, once a one-assay approach is deemed fit; upon careful assessments of the critical assay parameters discussed in the paper, one APC (usually the one against biosimilar drug) should be used for subsequent assay validation and samples analysis to support comparability studies.
It is important to keep in mind that these APCs should not be taken as true reflection of the immune responses in clinical studies. APCs should only be considered as surrogates to allow for laboratory quality control of the NAb assay for validation and sample analysis as well as to assess the validity of each experiment. In addition, the purpose and intent of including the NAb APC are to ensure that the assay performance is observed and trended consistently over time. These NAb APCs may be evaluated by trending the data from run to run and over multiple days and studies. While the APC is an assay performance control for consistency from time to time, it should be noted that the APC performance data should not be part of the final comparability assessment between the originator and biosimilar.
The different approaches to the choice of a NAb-positive control can be evaluated during the method development assessment to ensure the APC is reflective of the suitability of the assay.
Sensitivity Assessment
The sensitivity of a typical NAb assay of at least 0.5 μg/mL should be acceptable as per the USP Chapter (18). Due to a variety of reasons, there may be circumstances where the APCs may not reach abovementioned sensitivity. In this case, sound scientific justifications should be provided for acceptance of the obtained sensitivity, allowing for a better assay development and comparability exercise. Sensitivity assessments are performed in method development with the chosen product(s) at concentration demonstrated in section [III-2] which is inhibited with serial diluted APC.
In case the two- or multiple-assay approach is chosen and different APCs are used, it should be noted that these positive controls may not generate “similar” results due to the fact that these antibody controls are often polyclonal in nature and usually are produced by hyper-immunizing animals with drug compounds. The difference in this test may not reflect any true difference between the biosimilar and originator compounds themselves.
Assay Sensitivity should be determined by titration of the APC in the matrix of the study with multiple runs by more than one analyst. The titration curves may be prepared by serial dilution of NAb control and data be fitted by the best 4- or 5-parameter logistic curve fit. The screening cut-point established for the assay could be interpolated on the APC titration curves. Sensitivity could be calculated by the following equation, sensitivity = mean interpolated concentration + (t0.05, df X SD) where the mean interpolated concentration is the mean of the interpolated cut-point of the titration curves, t0.05, df is the critical value determined from the one-sided t-distribution corresponding to a 5% failure rate, and “df” is the degrees of freedom that depends on the number of values used in the calculation (20).
In a case that two assays are used, then a comparison of sensitivity for the two assays is required. Depending on the sensitivity of the assay (under or above 500 ng/mL), the acceptance criteria to determine the similarity in assay sensitivity for the biosimilar and the innovator will be different. The two to threefold difference may be considered acceptable for assays with sensitivity less than 250 ng/mL, whereas other statistical approaches may be used for assays with sensitivity > 250 ng/mL (26).
Drug Tolerance Test
It is important to evaluate drug tolerance for both biosimilar and originator compounds in the NAb assay. It is well recognized that drug tolerance for NAb assays is often poor, and typical assays are unable to tolerate the desired efficacious drug concentration in study samples. This is especially true for mAb therapeutics where very high concentration of drug may be present in circulation for long period of time. Nevertheless, significant number of attempts and trials should be made during assay development to increase drug tolerance of the assay as much as possible. It is also important that both drugs demonstrate similar drug tolerance. However, it should be noted that due to non-quantitative nature of NAb assays, the difference of two to threefold maybe acceptable (28).
METHOD VALIDATION: ONE-ASSAY APPROACH VERSUS MULTIPLE-ASSAY APPROACH
Regardless of whether on-assay approach or multiple assay approach is taken, NAb assay validation should always follow the same regulatory guidance and industry white papers (11,18,20,24,29). The same validation parameters (e.g., assay cut-point, sensitivity, selectivity, drug tolerance, assay precision, bench top and freeze/thaw stability, etc.) should be assessed. The assessment should be made as per an approved assay validation plan/protocol with a defined target assay acceptance criterion for each assessment. We recommend using the anti-biosimilar antibody control and biosimilar drug as the assay reagent for validation to be most conservative as discussed in the Bioanalysis journal editorial (26). However, if the multiple assay approach is taken, in addition to validating two or more independent methods, using one APC for both assays is preferable. If respective APC for each drug Nab assay validation, then additional assay criteria to evaluate “similarity” for each parameter between the assays should be included in the validation protocol so it can (1) guide validation outcome and (2) provide useful information for downstream data interpretation for the comparability studies.
CONFIRMATORY ANALYSIS AND TITRATION
As of publication, no specific guidance from regulators or white papers have been issued on how to conduct confirmatory and NAb-tittering assays in biosimilar programs.
In recent years, it has been widely accepted that confirmatory steps in NAb assays are not routinely conducted. Given the rationale that the NAb assay has an established screening cut point and is a further characterization of the ADA assay, which already includes the confirmatory test, it is recommended that the confirmatory and NAb-tittering assays are not necessary for the initial comparability NAb studies of biosimilars. This suggestion is valid if there is no pre-requisite of a clinical protocol design and/or end points. However, if samples generate an unexpected or non-specific assay response, the reactive samples need to undergo further testing including confirmatory assay or titration, the following points may be taken in consideration.
If a confirmatory step is indeed required for a study, a variation of techniques, such as crosslinking proteins A, G, and/or L to beaded agarose, resins, chromatography, or usage of agonist molecule, may be used. A different approach must be used since some of the drug products tested in a Nab assay are therapeutic monoclonal antibodies that bind to agarose beads. For this class of biosimilars, a different strategy must be used in the confirmatory assay. For instance, samples may be tested neat, as in not adding any drug or ligand.
NAb tittering is not regularly recommended for Biosimilar NAb assays and should be handled on a case to case basis. When qualitative NAb assay is conducted, no titer is reported and only positive or negative results are reported based on the screening NAb assay. However, in some biosimilar programs, there could be some clinical advantages for the biosimilarity data interpretation to perform NAb tittering. For example, some clinicians have found it helpful to have the result of the NAb assay expressed as a titer to evaluate not just incidence but also amplitude of the response. However, if NAb tittering is performed, evaluation of the titer data would need to include statistical strategies. This would help negate any bias from other factors, such as inter-patient variability on the interpretation of the similarity of the response from the biosimilar and the originator for example is the approval of Remsima (30), that a NAb assay titer does not add value. In this case, the ADAs are directed to immunodominant epitopes located in the CDRs, so the NAb-screening assay format is likely to be measuring the same population of ADAs, probably with higher specificity and sensitivity, and the drug is an antagonist, rather than an agonist thus reducing the value of a confirmatory or a titer step. For drugs that are categorized as high-risk agonists, such as cytokines or growth factors, the reporting of titers could be important to evaluate the potential for clinical risk to patients’ due to neutralization of the endogenous counterpart (31).
If done, the NAb titer should be reported by performing twofold, threefold, or higher serial dilution of the sample till the NAb-response signal is below the cut point.
Depending on different methods and their respective capabilities, the titer can be reported as below:
-
a.
Determine a titer value based on a reciprocal value of the highest dilution of the sample that results in a signal above the cut-point value.
-
b.
Report an endpoint titer as the reciprocal of the interpolated dilution of the sample at which the response would be equal to the cut point value
CONCLUSION
Table II summarizes the recommended assay parameters:
In conclusion, there are multiple options on the development and validation of NAb assays for biosimilar programs. To select the right option, the type of drug and its MoA will help determine the format for the assay (cell-based functional assay, CLB assay, or others). We recommend performing a one-assay approach first as it is easier to confirm the biosimilarity using one assay for NAb. If a one-assay approach is not feasible, then a two-assay format may be used considering the points and parameters described above.
For majority of the proposed biosimilar products in regulated markets, it is likely that at minimum the following datasets will be required by regulators in support of a marketing application: (a) analytical similarity, (b) nonclinical toxicology, (c) human PK/PD similarity, and potentially (d) comparative clinical efficacy and safety study.
Along with comparative immunogenicity testing in animal models, a key part of both the PK/PD similarity study (single-dose in healthy subjects or repeat-dose in patients) and comparative clinical trial (repeat-dose in patients) will be to compare the incidence of ADA in subjects receiving the biosimilar candidate versus in subjects receiving the innovator/originator product.
If the originator molecule is approved for multiple indications, the biosimilar developer will need to provide a detailed “mechanism of action” based scientific rationale for extrapolation across all conditions of use not directly tested in the comparative clinical trial. A key secondary endpoint from this study will be the comparative ADA responses and secondarily the results of any NAb assessments. Comparative NAb responses and frequency between the biosimilar and innovator product will be an essential component of any dataset supporting extrapolation.
Change history
27 February 2018
In the published article, the author B. Babbitt was cited as affiliation 9, but should have been cited as affiliation 2. In addition, there are 2 errors in the affiliations. The correct affiliations are shown in this erratum.
References
U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER). Quality considerations in demonstrating biosimilarity to a reference protein product. April 2015.
U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER). Scientific considerations in demonstrating biosimilarity to a reference product. April 2015.
European Medicines Agency. EMEA/CHMP/BMWP/42832/2005 Rev1. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical.
Marini JC, Anderson M, Cai X-Y, Chappell J, Coffey T, Gouty D, et al. systematic verification of bioanalytical similarity between a biosimilar and a reference biotherapeutic: committee recommendations for the development and validation of a single ligand-binding assay to support pharmacokinetic assessments. AAPS J. 2014;16(6):1149–58.
Mok CC, van der Kleij D, Wolbink GJ. Drug levels, anti-drug antibodies, and clinical efficacy of the anti-TNFα biologics in rheumatic diseases. Clin Rheumaatol. 2013;32(10):1429–35. https://doi.org/10.1007/s10067-013-2336-x.
Sampson HA, Muñoz-Furlong A, Campbell RL, Adkinson NF Jr, Bock SA, Branum A, et al. Second symposium on the definition and management of anaphylaxis: summary report--second National Institute of allergy and infectious disease/Food Allergy and Anaphylaxis Network symposium. J Allergy Clin Immunol. 2006;117(2):391–7. https://doi.org/10.1016/j.jaci.2005.12.1303.
Stebbings R, Findlay L, Edwards C, Eastwood D, Bird C, North D, et al. “Cytokine storm” in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179(5):3325–31. https://doi.org/10.4049/jimmunol.179.5.3325.
Stebbings R, Eastwood D, Poole S, Thorpe R. After TGN1412: recent developments in cytokine release assays. J Immunotoxicol. 2013;10(1):75–82. https://doi.org/10.3109/1547691X.2012.711783.
Finco D, Grimaldi C, Fort M, Walker M, Kiessling A, Wolf B, et al. Cytokine release assays: current practices and future directions. Cytokine. 2014;66(2):143–55. https://doi.org/10.1016/j.cyto.2013.12.009.
Doessengger L, Banholzer ML. Clinical development methodology for infusion-related reactions with monoclonal antibodies. Clin Transl Immunol. 2015;4(7):e39. https://doi.org/10.1038/cti.2015.14. eCollection 2015 Jul. Review
U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Center for Biologics Evaluation and Research (CBER). Assay development and validation for immunogenicity testing of therapeutic protein products. April 2016.
Prabhakar SS, Muhlfelder T. Antibodies to recombinant human erythropoietin causing pure red cell aplasia. Clin Nephrol. 1997;47(5):331–5.
McKoy JM . et al.. Epoetin-associated pure red cell aplasia: past, present and future considerations Transfusion 2008; 48(8): 1754–1762.
Bonnie W, et al. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J. 2008;18(6)
Hu J, Wala I, Han H, Nagatani J, Barger T, Civoli F, et al. Comparison of cell-based and non-cell-based assay platforms for the detection of clinically relevant anti-drug neutralizing antibodies for immunogenicity assessment of therapeutic proteins. J Immunol Methods. 2015;419:1–8. https://doi.org/10.1016/j.jim.2015.02.006.
Finco D, Baltrukonis D, Clements-Egan A, Delaria K, Gunn GR III, Lowe J, et al. Comparison of competitive ligand-binding assay and bioassay formats for the measurement of neutralizing antibodies to protein therapeutics. J Pharm Biomed Anal. 2011;54(2):351–8. https://doi.org/10.1016/j.jpba.2010.08.029.
Wu BW, Gunn GR, Shankar G. Competitive ligand-binding assays for the detection of neutralizing antibodies. In: Tovey MG, editor. Detection and quantification of antibodies to biopharmaceuticals: practical and applied considerations. Hoboken: John Wiley & Sons, Inc.; 2011. p. 175–92. https://doi.org/10.1002/9781118075685.ch10.
United States Pharmacopeia. Chapter 1106.1. Immunogenicity assays—design and validation of immunoassays to detect anti-drug neutralizing antibody. Available from: http://www.usp.org.
Cludts I, Meager A, Thorpe R, Wadhwa M. Development and characterization of a non-cell-based assay to assess the presence of neutralizing antibodies to interferon-beta in clinical samples. J Immunol Methods. 2013;395(1–2):37–44. https://doi.org/10.1016/j.jim.2013.06.008.
Shakar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Fionco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81. https://doi.org/10.1016/j.jpba.2008.09.020.
Jolicoeur P, Tacey RL. Development and validation of cell-based assays for the detection of neutralizing antibodies to drug products; a practical approach. Bioanalysis. 2012;4(24):2959–70. https://doi.org/10.4155/bio.12.285.
Diagram from AAPS course “AAPS Immunogenicity 101”, Section 2.2, Assay Strategies for the Detection of Neutralizing Antibodies (NAbs), Renuka C. Pillutla, Director, Bioanalytical Sciences – Biologics Bristol.
Gupta S, Indelicato SR, Jethwa V, Kawabata T, Kelley M, Mire-Sluis AR, et al. Recommendations for the design, optimization, and qualification of cell-based assays used for the detection of neutralizing antibody responses elicited to biological therapeutics. J Immunol Methods. 2007;321(1-2):321,1–18. https://doi.org/10.1016/j.jim.2006.12.004.
Gupta S, Devanarayan V, Finco D, Gunn GR III, Kirshner S, Richards S, et al. Recommendation for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55(5):878–88. https://doi.org/10.1016/j.jpba.2011.03.038.
Chamberlain PD Multidisciplinary approach to evaluating immunogenicity of biosimilars: lessons learnt and open questions based on 10 years’ experience of the European Union regulatory pathway. 2014 2014:423—43. https://doi.org/10.2147/BS.S50012.
Cai X-Y, Wake A, Gouty D. Analytical and bioanalytical assay challenges to support comparability studies for biosimilar drug development. Bioanalysis. 2013;5(5):517–20. https://doi.org/10.4155/bio.13.1.
Tovey MG, Lallemand C. Improved analytical methods for the detection and quantification of neutralizing antibodies to biopharmaceuticals. Bioanalysis. 2012;4(17):2179–90. https://doi.org/10.4155/bio.12.186.
Swanson SJ. Strategies to Improve Drug Tolerance in Nab Assays https://zerista.s3.amazonaws.com/item_files/98bc/attachments/39011/original/119_nbc-15-166.pdf (accessed 10 June 2017).
European Medicines Agency. EMEA/CHMP/BMWP/14327/2006 rev. 1 2 guideline on immunogenicity assessment of biotechnology-derived therapeutic Proteins August 2015.
European Medicines Agency. European public assessment report for Remsima®. Availablefrom:http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002576/human_med_ 001682.jsp&mid=WC0b01ac058001d124. Accessed April 26, 2014.
Wu B, Chung S, Jiang X-R, Mcnally J, Pedras-Vasconcelos J, Pillutla R, et al. Strategies to determine assay format for the assessment of neutralizing antibody responses to biotherapeutics. AAPS J. 2016;18(6):1335–50.
Acknowledgements
We greatly thank Joao Pedras-Vasconcelos at the Food and Drug Administration, as well as Shalini Gupta, Director at Amgen, for their assistance with the review of this article. We would also like to thank Yuanxin Xu, Senior Director at Alnylam Pharmaceuticals, for comments that significantly improved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gouty, D., Cai, C.C., Cai, X.Y. et al. Recommendations for the Development and Validation of Neutralizing Antibody Assays in Support of Biosimilar Assessment. AAPS J 20, 25 (2018). https://doi.org/10.1208/s12248-017-0181-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-017-0181-6