
Abstract
Immunogenicity testing and characterization is an important part of understanding the immune response to administration of a protein therapeutic. Neutralizing antibody (NAb) assays are used to characterize a positive anti-drug antibody (ADA) response. Harmonization of reporting of NAb assay performance and results enables efficient communication and expedient review by industry and health authorities. Herein, a cross-industry group of NAb assay experts have harmonized NAb assay reporting recommendations and provided a bioanalytical report (BAR) submission editable template developed to facilitate agency filings. This document addresses key bioanalytical reporting gaps and provides a report structure for documenting clinical NAb assay performance and results. This publication focuses on the content and presentation of the NAb sample analysis report including essential elements such as the method, critical reagents and equipment, data analysis, study samples, and results. The interpretation of immunogenicity data, including the evaluation of the impact of NAb on safety, exposure, and efficacy, is out of scope of this publication.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The assays used for the detection of neutralizing antibodies (NAb) may range from straightforward ligand binding assays to more complicated cell-based assays. There is a wide range of conventions employed for the presentation of neutralizing antibody assay results, as each type of assay may have different results, each of which is dictated by the mechanism of action for the drug, or specific functional domain in the case of drugs that bind to multiple targets. These results can be further complicated by aspects unique to NAb assays, such as the selection of assay format, platform and read-out, the selection and utilization of critical reagents, the estimation of sensitivity with a semi-quantitative or qualitative assay, and the evaluation of drug and/or target tolerance. To efficiently convey these results in a consistent manner across industry, it is beneficial to present this information in a harmonized report format.
As an ongoing discussion on immunogenicity assay result reporting through American Association of Pharmaceutical Sciences (AAPS), a working group was formed to address key bioanalytical reporting gaps for both anti-drug antibodies (ADA) and neutralizing antibody assay results. Subsequently, working group members across multiple organizations including pharmaceutical, biotechnology, contract research organizations, and regulatory agencies discussed these inconsistencies and developed a harmonized framework for immunogenicity reporting. The results of these discussions are presented here, including an example report template to facilitate filings with health authorities. This is the second of a two-part series on immunogenicity reporting which provides recommendations for essential aspects of reporting results for clinical immunogenicity assays. The first part of this series, which focused on the unique aspects of reporting of anti-drug antibody assay results, was previously published (1) .
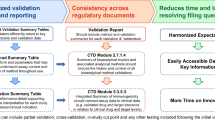
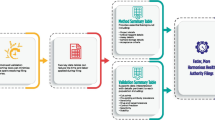
Operational requirements associated with results reporting for study data will vary based on each organization’s reporting conventions, as well as the availability of reportable data. For example, contract research organizations performing sample analysis on behalf of a sponsor may have limited access to sample demographic information, whereas the sponsor may have access to more sample and trial participant information. Blinding of participant data can also limit access to information prior to study conclusion. The availability of information dictates what can be included in a report, and it is recommended that as much relevant data be included to facilitate proper evaluation of the neutralizing antibody results. This publication provides a general guide for standardizing the format of available immunogenicity results presented in the NAb bioanalytical report (BAR) (Fig. 1). The recommendations included in this manuscript are intended to harmonize the presentation of NAb data across organizations and facilitate report review by industry and regulatory agencies.

Summary of bioanalytical reporting format for neutralizing antibody assays
A summary of NAb in context of clinical results, such as incidence, prevalence, or impact (e.g. cross-reactive neutralizing antibody responses, result interpretation if on-board drug tolerance levels are exceeded) is typically provided in documents such as the Clinical Study Report (CSR), or the Integrated Summary of Immunogenicity (ISI); the content of these sections is outside the scope of this paper. It is recommended that the NAb sample analysis reports be provided as appendices to the CSR and the ISI.
To facilitate regulatory filing, the authors have included an editable report template as an appendix for use as a general guide for what may be included in a bioanalytical NAb sample analysis report. Each drug development program will have unique requirements, and therefore not every aspect for result reporting addressed in this publication may apply. It is highly recommended that each user includes those aspects of this guidance that best apply to their drug development program.
Bioanalytical Report Structure and Content
Signature Page and/or Statement of Compliance
Neutralizing antibody bioanalysis is often performed in a somewhat formal or regulated manner, similar to general ADA analysis, and may include review from Quality Assurance (QA). It is recommended to state the principles under which bioanalytical testing was performed (e.g. GLP/GCLP) as well as other associated plans, guidances, or company specific SOPs that were followed. Approvals from study personnel and management (as necessary) is captured with associated signatures and dates. If QA was involved in study oversight and/or report review, the details and dates of audit, as well as QA signature and date, is also included, in accordance with applicable SOPs.
Summary
The summary section provides a concise overview of NAb sample analysis results and may be presented as either text or in a tabular format to facilitate expedient review. The summary may include, but is not limited to, the following:
-
1.
Study related information: drug name, study number, study population, and study title;
-
2.
References to associated documentation for sample analysis: name of analytical facility, brief statement of where samples were collected and the testing strategy (bioanalytical plan/sample analysis plan);
-
3.
Study samples: matrix, total number of study samples received, total number of samples analyzed, samples subjected to repeat analysis;
-
4.
Brief description of the NAb method: status (validated or qualified), assay platform, detection system, assay read out (including units);
-
5.
Result reporting: total number of NAb positive samples as a proportion of total number of NAb samples analyzed or total number of NAb positive samples as a proportion of total number of ADA positive samples analyzed, definition of NAb positive response, the format of reported results (titer, % neutralized, S/N, etc.).
In order to facilitate effective review of positive neutralizing antibody assay results, inclusion of additional participant information, as available, may be useful. For example, the authors recommend included whether NAb positive samples are consistently found in individual participants over several timepoints, which may indicate a potentially clinically relevant NAb response to reviewers, such as regulatory agencies.
Under certain circumstances, a titration tier may be employed for reporting the magnitude of response for NAb positive samples (2). If traditional titration analysis is performed for NAb positive samples, the final calculation of the titer results should include the minimum required dilution (MRD), including all dilutions associated with sample processing steps (3).
Use of a confirmatory tier in NAb sample analysis is not as common, but in some cases, confirmation of drug specific neutralizing response is informative (2, 4, 5). If using, then the assay format and NAb assay readout of the confirmatory tier should be briefly mentioned in this section.
Multi-functional domain biotherapeutic products may pose an increased immunogenicity assessment challenge, as multiple NAb assays may be required to characterize the potential NAb response directed to each functional domain. In this case, all NAb assays used for sample analysis should be individually summarized in this section.
An example Summary statement has been provided in Sect. 4 of the Template (Supplementary Material).
Bioanalytical Method
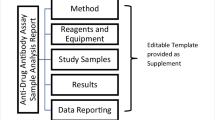
To facilitate the evaluation of reported results, a method summary inclusive of the study drug (biotherapeutic, ADC, bispecific antibody, fusion protein, monoclonal antibody therapy, etc.), a clear description of the sample analysis method, as well as an explanation of the data format (titer or S/N) produced, is provided.
The method summary is provided as a text or tabular format, (a diagram may be helpful if the process is complex) and it is recommended to include descriptions of the assay platform (colorimetric, luminescence, electrochemiluminescence, fluorescence, etc.) and assay format (direct/indirect assay, antibody-dependent cellular cytotoxicity (ADCC), etc.), as well as any pre-treatment of samples such as acid dissociation or immunoadsorption, added preservatives, or soluble protein extraction. Details should be included if samples require any specific handling based on method validation parameters assessments. Any associated data processing required for the method (raw response or normalized response) is listed. An example method summary table is located in Sect. 5 of the report template. This section of the report includes a clear reference or link to the bioanalytical method and/or validation report for cross-referencing as well as a brief description of the study drug and sample matrix. A description of the positive control antibody (polyclonal/monoclonal, species, etc.) as well as system suitability controls such as negative control (NC), positive control (PC) levels, ligand control (LC), and drug or drug control is also described. If sample titration is required, the final reported dilution result (titer or S/N) of the sample should include the MRD (3, 6).
Critical validated/qualified method parameters are summarized including (but not limited to) the NAb screening cut point (CP), titration CP (if applicable), assay sensitivity, drug tolerance, target interference, matrix effect (or selectivity), process stability, precision, and effect of hemolysis/lipemia (see Template Table 5–1). Multi-functional domain therapeutics that require assessment of more than one cut point or domain specificity should be included as a separate table. Updated assay parameters (for example, cut point factor) determined in-study should be noted along with the original validated value (as applicable). As noted, the use of a confirmatory tier or NAb sample titration is not common, but if performed, the validated characteristics of this tier are also listed.
Materials
The materials section includes a description of critical reagents, equipment and software used during sample analysis in tabular format as presented in Sect. 6 of the template and discussed in more detail below.
Critical Reagents
Critical reagents are defined as reagents that are crucial to the performance of the method and may affect assay performance (7, 8). These include, but are not limited to, the study drug, cell line (if applicable), ligand control, positive controls, custom-labeled drug or detection reagent, and negative control matrix. All critical reagents should be listed in a table, along with reagent description, vendor or source, lot and catalog number, preparation date, concentration, expiration and/or retest date, and appropriate storage conditions, (as available). Certificates of analysis for critical reagents are not required to be presented in the bioanalytical report but need to be maintained in the study documentation (9).
Positive Control Antibodies
Similar to ADA assays, NAb assays commonly utilize surrogate positive controls. A purified antibody with a defined concentration is preferable for an accurate and consistent evaluation of the assay performance. A description of the positive control antibody clonality (monoclonal versus polyclonal), host and target species, and storage conditions is included. If a purified antibody is not available, relevant information regarding the source and generation of the positive control antibody is included.
Matrix and Controls
The negative control used in the assay is critical for assay performance. Many interfering factors in the negative control matrix can affect the assay, such as growth factors, pre-existing antibodies, or endogenous targets. For ligand binding NAb assays, sample results are typically evaluated in the context of the cut point, which is calculated using the negative control. Sample results are compared directly to the assay cut point for positive/negative determination.
For cell-based assays, the ligand (if applicable) and drug are prepared in the negative control, and the negative control containing drug (and ligand if applicable) is used to establish the cut point. Therefore, any change observed in sample signal relative to the negative control can be ascribed to binding of the neutralizing antibody.
Therefore, when a negative control pool used in sample analysis differs from that used in validation, it must be included in the sample analysis report. Additionally, a reference is provided to any documentation (e.g. notebook pages, report number) summarizing assay data for bridging new negative control pools to the original validated negative control pool. All negative control pools used in sample analysis are included as noted in supplementary Template Table 6–1. If the negative control pool is prepared from multiple individual matrix lots, the lot numbers of the individual matrix samples are documented and retained in validation or study-related folders with the study records for cross reference, and do not need to be individually listed in Table 6–1.
Equipment & Software
The equipment and software section should include any cell counters (cell-based assays), plate readers, and software used during sample analysis runs. It is recommended to include individual equipment numbers for plate readers, as well as to detail the software used to collect the data, including version number(s). The equipment section does not need to include freezers, incubators or plate washers used during sample analysis, unless these are critical to assay performance. Any software used related to sample storage, chain of custody and temperature monitoring (LIMS, Rees, etc.) are included in the bioanalytical report. See supplementary Template Sect. 6.2 (Supplementary Material).
Data Analysis
The software utilized for NAb data analysis and the process for how data were analyzed should be included in the report. The process of data analysis is described with respect to the assay format, such as a single tier for characterization as neutralizing or non-neutralizing, or in accordance with the validated tiered analysis which may include screening, confirmatory, and/or titer analysis. Descriptions for data transformation or normalization are included, as well as any statistical analysis to evaluate sample data (i.e. outlier evaluation).
If an in-study cut point is employed, a brief explanation is included in this section, such as the samples utilized and any data transformation required, while the “In-Study Cut Point Assessment” section can include the details of the in-study cut point calculation.
Generally, samples are reported as positive or negative relative to the screening cut point, with results including any required tiered analysis. Sample results must be consistently reported in the context of the assay format. For example, for raw ECL values, the data and all statistical evaluations should be consistently reported using relative luminescence units or RLU. For an assay that reports a signal to noise (S/N) ratio, all data is evaluated and reported as S/N, and N is explicitly defined, such as the OD of pooled negative controls minus plate blank. When assessing the total degree of inhibition in signal relative to control, results should be consistently reported as percent inhibition.
To clarify any potential discrepancies between manually calculated results from the reported data, the number of significant figures for reportable data, any clarifications related to rounding of values included in data regression, and a description of data transfer specifications (or reference to a Data Transfer Agreement) between the laboratory responsible for data generation and the sponsor or clinical database are included in the report.
Study Sample Receipt and Storage
The summary of receipt and storage of samples is presented in the supplementary Template Table 8–1. Any supportive documentation, such as shipment temperature data loggers, sample manifests, and sample reconciliation information, are referenced and stored with study records for sample tracking purposes, but does not need to be included within the report.
It is important to include a brief description of the sample receipt condition, storage, and any temperature excursions experienced during the study in the report, noting any problematic sample conditions experienced during the course of the study. If systems are in place to ensure chain of custody and sample records are maintained with study data, additional details such as sample laboratory origin locations do not need to be included in the report.
The sample identification or accession number and time point/visit information (as listed in the study protocol, laboratory manual, or data transfer documentation) are included. If both original and backup vials were analyzed during the course of the study, it is critical to ensure the correct vial identification/accession number is reported.
Results
The Results section provides context and overall assessment for the conduct of sample testing. Minimally, it comprises study aspects such as evaluation of in-study cut point (if required), sample testing overview, control performance and sample results (both qualitative and if appropriate quasi-quantitative). It is recommended, where possible, to include NAb results from multiple functional domain biotherapeutics in a single report, for example, when assessments are performed in the same lab. In this manner, the method performance and results for each functional domain can be reviewed in a single document. Where a single report is not possible, Sponsors should ensure a separate document contains NAb results for each functional domain (via cross-references between documents).
In-Study Cut Point Assessment
As previously recommended in the ADA sample testing and report harmonization white paper (1), we recommend evaluating the need for and, if necessary, establishing an in-study NAb cut point (CP) when appropriate. It is common for the NAb assay CP to be determined during NAb method validation using commercially purchased individual normal and disease samples, which may not represent the study population. As an ADA characterization step, samples are not tested in the NAb assay until they are first confirmed positive in the ADA assay, making it difficult to use the false positive error rate (FPER) to evaluate the suitability of a validated NAb CP, similarly observed by Myler, et al. (3, 6). However, performing an evaluation of baseline samples in the NAb method against the CP established during validation will determine whether the validated CP is suitable for use during NAb sample analysis.
If an in-study cut point is warranted, the number of samples required for the evaluation of in-study cut point comparison depends on several factors, including the number of baseline samples or placebo samples available, timing of sample availability, adequate sample volume, and appropriate consent. When possible, a minimum of 20 in-study baseline samples (10) representative of the population is recommended for evaluation. This number of baseline samples may not be feasible in certain rare indications or populations (e.g., pediatric) with limited enrollment. For these reasons, it is practical to wait until enrollment has been completed to allow assessment of baseline samples in parallel, rather than in series. In the case that at least 20 baseline samples are available, each subject sample is only required to be tested once, but ideally tested across multiple plates and days by at least two analysts. Although these data sets may be too small to incorporate balanced experimental design, the inter-day and inter-analyst variability calculated from all sample data will capture both the analytical and biological variation and will therefore provide a more accurate calculation of cut point. The data assessment includes outlier identification and distribution evaluation prior to the calculation of the cut point from these baseline samples. For smaller sample sizes (n < 20), baseline samples may be analyzed multiple times over a minimum of two days with at least two analysts. Comparison of the in-study and validation cut points can be done with histograms and boxplots, in addition to statistical assessment of the difference of means (ANOVA) and variances (Levene’s test). The false positive error rate (FPER) with both cut points could also be compared. If there is a significant difference in the means, variances, and/or the FPER, an in-study cut point should be considered.
The details of the in-study cut point evaluation (number of samples/analysts/days, statistical analysis method used) is summarized in the NAb BAR with a link or reference to any associated statistical analysis reports that may provide details such as outlier assessment. This section describes how the data were compared and provide justification for which cut point was used in NAb sample testing. If the NAb cut point was re-established, the new cut point is captured in supplementary Template Table 5–1.
Sample Testing Overview
This overview provides a summary of the number of passing runs of those performed per tier (as applicable). This section notes if samples were analyzed within the process stability parameters (i.e., freeze–thaw, bench-top, and refrigerator stability) that were established during assay validation. If exceptions occurred, these are captured in the ‘Sample Results’ section of the report.
It is recommended to include, at a minimum, the following in the results table: the run number, identifying information about the sample (i.e. demographic data), tier results (as applicable), and associated comments as shown in Supplementary Template Table 13–5.
Control Performance and Acceptance Criteria
A description of assay controls and their performance summary applicable to the assay format is included in this section of the report. Negative control (NC), low positive control (LPC), and high positive control (HPC) are commonly used to monitor performance and accept or reject assay runs. Depending on the assay format, other controls such as drug control (DC) or ligand control (LC) may also be needed. Additional controls could also be presented based on the respective analysis tier, for example, performance of a titer positive control (TPC) from each run. For multi-functional domain therapeutics, there may be several sets of controls that are included, which must be clearly reported. The format for the assay readout is also detailed (e.g., absorbance, luminescence, fluorescence). A description of each control is provided along with the number of sets/replicates of each per assay run and the defined acceptance criteria as outlined in the sample analysis plan.
Reportable results for all controls are summarized in raw data units and in calculated reportable units (e.g., S/N, % inhibition). Multiple table options are included in the report template (supplemental appendix) to aid in control reporting and performance assessment. These tables contain the commonly used controls NC, LPC, and HPC, and are modified to include the appropriate number of sets evaluated per run as well other controls necessary to monitor assay performance dependent on assay format. Tables 13–2 and 13–4 summarize control performance over all assay runs including the acceptance criteria and pass/fail status of each control. Template Table 13–2 can be used to document both screen and titer tiers (if performed); a separate Table 13–4 may be used for presentation of titer controls (if performed). As noted earlier, the confirmatory tier is not generally performed in NAb sample testing; however, Table 13–3 is included to document this assessment, if required. Other metrics include a summary of overall control performance such as mean, standard deviation, and %CV. Any additional data interpretation is noted, for example, rejected runs or datapoints that were not included in the overall control performance summary, or if an outlier assessment was applied to the controls and those values that were excluded.
If desired, information on reagent lots that were qualified in the assay can be reported, especially in cases where new lots of reagents demonstrate a clear difference in performance (e.g., raw units). The inclusion of text in the report is recommended to explain any discrepancy in control performance. If an in-study cut point is determined and controls are reassessed in the assay, this information should also be reported.
As antibodies have proven to typically be stable for several years when stored frozen (11, 12), long term stability is not always performed for neutralizing anti-drug antibodies in study samples. Information on any excursions from established process stability (freeze–thaw cycles, benchtop, or refrigerated storage) is described.
Sample Results
In this section of the BAR, all NAb sample analysis results are captured, including results from any tiered analyses. In the case of drugs that bind to multiple targets, assessment of neutralization of multiple domains are included in the same report if performed in the same laboratory. It should be reiterated which cut point was used to assess positive/negative NAb status, or in any tiered analysis, as applicable, where in-study cut point assessment was performed. The results may include a summary of the total number of samples analyzed per tier or further characterized, as applicable. If NAb sample titration was performed, explanatory text is provided to inform how results are reported, e.g., titer tier results are reported as sample dilution x MRD. If any ADA positive samples were not assessed in the NAb assay, the reasons for exclusion are detailed in this section.
In the accompanying NAb BAR supplemental template, Table 13–5 contains a recommended layout of minimal sample demographics and results including, but not limited to, the following: run number, subject number, sample ID, visit, sample collection date, result (e.g., positive, negative, or reportable titer value), and comments. The demographics included in the table may largely be driven by an agreed upon data transfer specification/agreement. However, sufficient information must be provided to allow for traceability of samples and results. If any confirmatatory tier, characterization or domain-specific analysis was performed, it is included in the sample results table. If the sample condition (e.g., freeze–thaw stability, lipemic, or hemolyzed) has potential to impact data interpretation, the reported results for these samples are noted in the comments column of this table. Similarly, if it is known that the drug tolerance level of the NAb assay was exceeded, the corresponding sample results may be noted. This section in the body of the report details how any deviations from the method or study plan impacted individual results.
Results can be captured as described in supplemental template Table 13–6 for samples where NAb repeat analysis was performed in analytically acceptable runs. This table is intended to capture samples where repeat analysis was due to assignable causes (e.g., %CV), and samples inadvertently repeated in error. This table should not capture samples where repeat analysis was performed due to analytically unacceptable runs.
Finally, laboratories and Sponsors should be aware that an effort to standardize reporting of immunogenicity tiered data through controlled terminology (standardized data tabulation model, SDTM), under oversight by Clinical Data Interchange Standards Consortium (CDISC) has recently been completed and may impact the manner in which data are reported (13).
Deviations and Investigations
This section details major deviations from either the bioanalytical plan or from the method, as well as an impact assessment of the deviations on the affected runs (as applicable), and overall study impact. Any investigations performed in association with the sample analysis must be documented in the bioanalytical NAb sample analysis report.
Archiving
The archiving or disposition of samples, data, and reports is described in accordance with current archiving policy in a manner that facilitates information retrieval and regulatory submissions.
Discussion
This AAPS working group conducted a cross-industry, anonymous clinical NAb result reporting survey and these results were recently reported in the AAPS Newsmagazine (Industry-wide Survey Responses on Reporting of NAb Results—AAPS Newsmagazine).
The key observations from this study were used to determine the areas of focus for this publication. For example, NAb assay results are most commonly reported in the qualitative format of “positive” or “negative” (88%, 52/59 of survey respondents), while most (60%, 36/60) survey respondents do not report NAb confirmatory results. According to survey respondents, there are different practices across industry for reporting NAb results as titer value: 13% (8/60) always, 38% (23/60) sometimes, and 48% (29/60) never report titer, while most respondents do not report NAb results as S/N. The varied ways to report results from neutralizing antibody assays underscore the need for consistency in the presentation of NAb data. Harmonization in reporting of neutralizing antibody assay results will help benchmark key aspects of assay readouts to facilitate report review.
Provided here are recommendations for the reporting of critical parameters associated with the assessment of neutralizing antibodies, with applicable examples across a wide variety of assays. These recommendations and editable report template are provided as guidance for the reporting of results, with the understanding that each organization may have operational requirements that dictate results reporting. There are cases in which access to information may be limited (as in the case of CROs), or controlled (blinded data sets), and in these cases only the data available can be reported. To provide sufficient information for correct interpretation of results and the reconstruction or reproduction of original assay data, it is recommended that each organization include as much assay-specific information as feasible. This includes sufficient detail for the reviewer to clearly understand 1) how the samples were obtained and processed, 2) how the assay was performed, and 3) how the data was analyzed.
The assays selected for the assessment of neutralizing antibody responses are reflective of the physiological activity of the drug, which may necessitate more than one neutralizing antibody assay in the case of multi-functional drugs. The assay controls selected support accurate detection of physiologically relevant readouts (e.g. upregulation, inhibition, increased or decreased enzymatic activity, etc.), which can vary widely as a function of neutralization of drug modality, and may necessitate a confirmatory tier of testing. A brief description of the readouts and data format is included to facilitate results interpretation. The determination of what needs to be reported should be directed by program needs, which is based on the level of immunogenic risk associated with the therapeutic.
While interpretation of NAb results is out of the scope of this guidance, NAb sample analysis results form the foundation for evaluating the clinical impact of NAb on pharmacokinetics (PK), pharmacodynamics (PD), safety, and efficacy that may inform treatment decisions in the clinical setting (14, 15). Therefore, we included several references as examples of how to assess neutralizing antibody activity in the context of safety, PK, and PD, which is critical for the study CSR or ISI (16, 17). The NAb report is intended to be included as an appendix in the CSR or ISI. Final NAb results need to be clearly presented in the Sample Analysis BAR, with sufficient detail to allow for proper interpretation of results, and reconstruction of original assay results, if required.
References
Jani D, Marsden R, Gunsior M, Hay LS, Ward B, Cowan K, et al. Anti-drug Antibody Sample Testing and Reporting Harmonization. AAPS J. 2022;24(6):113. https://doi.org/10.1208/s12248-022-00762-6.
Gupta S, Devanarayan V, Finco D, Gunn GR 3rd, Kirshner S, Richards S, et al. Recommendations for the validation of cell-based assays used for the detection of neutralizing antibody immune responses elicited against biological therapeutics. J Pharm Biomed Anal. 2011;55(5):878–88. https://doi.org/10.1016/j.jpba.2011.03.038.
Myler H, Pedras-Vasconcelos J, Lester T, Civoli F, Xu W, Wu B, et al. Neutralizing Antibody Validation Testing and Reporting Harmonization. AAPS J. 2023;25(4):69. https://doi.org/10.1208/s12248-023-00830-5.
Immunogenicity Testing of Therapeutic Protein Products - Developing and Validating Assay for Anti-Drug Antibody Detection: Guidance for Industry. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER); 2019.
Guideline on immunogenicity assessment of therapeutic proteins. European Medicines Agency, EMEA/CHMP/BMWP/14327/2006 Rev 1; 2017.
Myler H, Pedras-Vasconcelos J, Phillips K, Hottenstein CS, Chamberlain P, Devanarayan V, et al. Anti-drug Antibody Validation Testing and Reporting Harmonization. AAPS J. 2021;24(1):4. https://doi.org/10.1208/s12248-021-00649-y.
Azadeh M, Sondag P, Wang Y, Raines M, Sailstad J. Quality Controls in Ligand Binding Assays: Recommendations and Best Practices for Preparation, Qualification, Maintenance of Lot to Lot Consistency, and Prevention of Assay Drift. AAPS J. 2019;21(5):89. https://doi.org/10.1208/s12248-019-0354-6.
King L, Farley E, Imazato M, Keefe J, Khan M, Ma M, et al. Ligand binding assay critical reagents and their stability: recommendations and best practices from the global bioanalysis consortium harmonization team. AAPS J. 2014;16(3):504–15. https://doi.org/10.1208/s12248-014-9583-x.
United States Pharmacopeia. General chapter <1106> Immunogenicity assays – design and validation of immunoassays to detect anti-drug antibodies. USP. Rockville, MD. 2014.
Devanarayan V, Smith W, Brunell RL, Seger ME, Krug K, Bowsher RR. Recommendations for Systematic Statistical Computation of Immunogenicity Cut Points. AAPS J. 2017;19(5):1487–98. https://doi.org/10.1208/s12248-017-0107-3.
Michaut L, Laurent N, Kentsch K, Spindeldreher S, Deckert-Salva F. Stability of anti-immunotherapeutic antibodies in frozen human serum samples. Bioanalysis. 2014;6(10):1395–407. https://doi.org/10.4155/bio.14.97.
Correia IR. Stability of IgG isotypes in serum. MAbs. 2010;2(3):221–32. https://doi.org/10.4161/mabs.2.3.11788.
CDISC Knowledge Base. IS Domain Scope Update for the SDTMIG V3.4: A development history and the difficulties of standardizing complicated biological processes. Clinical Data Standards Consortium. Austin, TX. https://www.cdisc.org/kb/articles/domain-scope-update-sdtmig-v3-4-development-history-and-difficulties-standardizing. Accessed 28 May 2024.
Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267–81. https://doi.org/10.1016/j.jpba.2008.09.020.
Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–73. https://doi.org/10.1208/s12248-014-9599-2.
Shakhnovich V, Meibohm B, Rosenberg A, Kierzek AM, Hasenkamp R, Funk RS, et al. Immunogenicity in Clinical Practice and Drug Development: When is it Significant? Clin Transl Sci. 2020;13(2):219–23. https://doi.org/10.1111/cts.12717.
Gunn GR 3rd, Sealey DCF, Jamali F, Meibohm B, Ghosh S, Shankar G. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals. Clin Exp Immunol. 2016;184(2):137–46. https://doi.org/10.1111/cei.12742.
Acknowledgements
AAPS TPIFG team members:
Boris Gorovits, Ph.D., Regeneron Pharmaceuticals
Heather Myler, Ph.D. Takeda Biosciences
FDA:
OBP Office of Biotechnology Products
OSIS Office of study Integrity and surveillance
OCP Office of Clinical Pharmacology
Funding
Apart from the time investment, no other funding was involved in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Darshana Jani: She led and coordinated the project from its inception including co-writing the manuscript. Lead the scientific and technical discussion for all the topics.
* Michele Gunsior and Robin Marsden have equal contribution in the preparation of the manuscript. Co-led the discussions for manuscript topics and creating the example template.
Laura Schild Hay and Bethany Ward assisted in organizing the meetings, maintaining the meeting minutes, and participated in scientific and technical discussion of the key topics.
All authors participated in key discussions about various components of the manuscript, provided expert opinions, and reviewed the manuscript.
Corresponding author
Ethics declarations
Disclaimer
The views expressed in this article are those of the authors and do not reflect official policy of the respective companies or US FDA.
Conflict of Interest
All authors are affiliated with the companies as they are listed in the authors’ section.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jani, D., Gunsior, M., Marsden, R. et al. Neutralizing Antibody Sample Testing and Report Harmonization. AAPS J 26, 80 (2024). https://doi.org/10.1208/s12248-024-00955-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-024-00955-1