
Abstract
Accurate structure elucidation of biologically active molecules is crucial for designing and developing new drugs, as well as for analyzing their pharmacological activity. In this study, density functional theory calculations are applied to explore the electronic structure and properties of phenethylamine derivatives, including Amphetamine, Methamphetamine, and Methylene Dioxy Methamphetamine(MDMA). The investigation encompasses various aspects such as geometry optimization, vibrational analysis, electronic properties, Molecular Electrostatic Potential analysis, and local and global descriptor analysis. Additionally, the study utilizes Natural Bond Orbital analysis and Quantum Theory of Atoms in Molecules to investigate the chemical bonding and charge density distributions of these compounds. Experimental techniques such as Fourier transform infrared (FT-IR) and Raman spectroscopic analysis are employed in the range of 4000-400 \(cm^{-1}\) and 4000-50 \(cm^{-1}\), respectively. Theoretical vibrational analysis with Potential Energy Distribution(PED) assignments is conducted, and the resulting frequencies are compared to experimental spectral data, revealing good agreement. By correlating various structural parameters with the pharmacological activity of each derivative, computational structure elucidation aids in understanding the unique actions of phenethylamine derivatives. The obtained results offer a comprehensive understanding of the molecular behavior and properties of these drugs, facilitating the development of new drugs and therapies for addiction and related disorders.
Similar content being viewed by others


Introduction
The use of psychoactive substances for recreational and other illicit purposes is increasing tremendously. The most popular and the most threatening drugs are phenethyl amines which is a class of substances with documented psychoactive and stimulant effects [1]. According to the World Drug report 2022 of “United Nations Office on Drugs and Crime(UNODC)”, Drug markets were temporarily disrupted in most parts of the world during the first phase of the COVID 19 pandemic, but there was a quick recovery and the drug market of Phenethylamine group is still expanding especially in South-East Asia and North America [2]. Phenethylamines are a large group of structurally similar agents that include amphetamine, methamphetamine, MDMA and the cathinones. A remarkable characteristic of the phenethyl amine group is that a slight change in its structure can make drastic changes in its pharmacodynamics [3]. Amphetamine and its N-alkyl derivatives exert marked excitatory effects on the Central Nervous System [4]. The phenethyl amine group has served as the basic structure for the design and development of hundreds of amphetamine derivatives and each derivative has its own unique action profile even though their basic structure is similar.
The chemical structures of amphetamine, methamphetamine, and methylenedioxy methamphetamine (MDMA) share a common foundation: a phenyl ring with two carbon side chains and an amino group. However, subtle modifications distinguish them. Amphetamine bears an additional methyl group, while methamphetamine boasts an extra methyl group bonded to nitrogen, and MDMA incorporates an additional methylenedioxy group. These alterations significantly impact their biological behavior and interactions within the body [3]. Dopamine, norepinephrine, and serotonin serve as endogenous neurotransmitters, vital for functions such as attention, memory, emotion, reward, and movement. Amphetamine and methamphetamine boost the release and hinder the reuptake of these neurotransmitters, intensifying and prolonging stimulation of their receptors. Notably, methamphetamine surpasses amphetamine in potency and lipophilicity, facilitating easier traversal of the blood-brain barrier and achieving higher brain concentrations. Moreover, methamphetamine exhibits a longer half-life, prolonging its effects within the body. Both drugs act as substrates and inhibitors of the enzyme CYP2D6, crucial for metabolizing numerous drugs and neurotransmitters. MDMA also shares similar mechanisms. Unlike amphetamine and methamphetamine, which lack a methylenedioxy group, rendering them more selective for dopamine and norepinephrine than serotonin, MDMA’s inclusion of a methylenedioxy group makes it more serotonin-selective [5].
Amphetamine, classified as an indirect sympathomimetic amine, induces sensations of alertness and euphoria, making it a common choice for recreational abuse. Despite its popularity in non-medical contexts, amphetamine exhibits therapeutic potential, particularly in treating conditions like obesity and attention deficit hyperactivity disorder (ADHD) . On the other hand, methamphetamine shares similar sympathomimetic properties with amphetamine but exerts more potent effects. Its high addictive potential is associated with numerous serious health complications upon misuse. MDMA, also known as 3,4 methylenedioxy methamphetamine, manifests both stimulant and hallucinogenic effects. As an empathogen entactogen, it fosters feelings of empathy and interpersonal closeness, alongside inducing euphoria and heightened physical energy [6]. However, it’s imperative to exercise caution due to the addictive nature and severe side effects inherent in all three substances.Furthermore, these compounds are believed to interact with the opioid system, potentially augmenting pain relief. Additionally, MDMA’s influence on the hypothalamic-pituitary-adrenal axis leads to increased cortisol and prolactin levels, along with elevated levels of hormones such as oxytocin and vasopressin [7]. Consequently, molecules like amphetamine, methamphetamine, and MDMA occupy a unique space in chemical, medicinal, and pharmacological research, necessitating comprehensive exploration and careful use.
Extensive research has been conducted on the structure-activity relationships of amphetamine, methamphetamine, and MDMA, as evidenced by reports [3, 8, 9]. In 2012, Rolf Wilestone Berg et al. [10] conducted a study on the Raman optical activity and Raman spectra of amphetamine species to identify the different enantiomers of amphetamine derivatives. Similarly, in 2013, Francis Taplin et al. [11] reported spectroscopic analysis of phenethylamines for the same purpose. Recent studies have focused on the detection of amphetamine using quantum mechanical approaches [12, 13]. Despite the extensive research on the structure-activity relationships of amphetamine derivatives, there has been limited investigation using DFT techniques to correlate their quantum chemical properties and related parameters with their pharmacological activity. Density functional theory (DFT) is the most successful and promising quantum chemical approach to calculate the ground-state properties of atoms and molecules which uses a conceptually simple electron density term which depends on just three variables as compared to the more complicated and difficult to interpret electronic wave function. The density functional approach allows the calculation of the entire energy surface, the structure and related properties from knowledge of the electron density distribution alone . In this study, Quantum Theory of Atoms in Molecules(QAIM) [14] is also employed to examine the electron density distributions and reactivity sites of the molecules. This work analyses various vibrational modes of amphetamine derivatives computationally and experimentally with Potential Energy Distribution(PED) assignments. Molecular electrostatic potential (MEP) surface, Frontier molecular orbitals, Natural Bonding Orbitals, Electron Localized function, Non Covalent Interaction, Molecular graph and Reactivity descriptors have been analysed using both Density Functional Theory and Atoms in Molecular Theory using Multiwfn [15]. The above investigation at the molecular level helps to compare the various reactivity parameters of amphetamine, methamphetamine and MDMA and to correlate them with biological mechanism of action like drug-receptor interactions and the binding mechanism.
Method
Experimental
The title molecules amphetamine (1), methamphetamine (2) and 3,4-methylenedioxy-methamphetamine (MDMA)(3) were analysed using FT-IR and FT-Raman techniques. The FT-IR spectra were recorded in the region 4000-400 \(cm^{-1}\) on an IR tracer FT-IR spectrometer(Shimadzu Cooperation, Japan), calibrated using a polystyrene standard. All samples were prepared using the KBr pellet method. The spectra were recorded with a scanning speed of 10 \(cm^{-1}\) per minute at a resolution of 4 \(cm^{-1}\).
Raman spectra were recorded in the region 4000-400 \(cm^{-1}\) using a 532 nm diode laser as excitation wavelength on a Raman Spectrometer (“LabRam HR Evolution, HORIBA, Japan”).
Computational details
All quantum mechanical calculations have been performed with the “Guassian-16 program” [16]. The density functional theory with the exchange correlational functional “B3LYP (three-parameter hybrid functional for the exchange part and the Lee-Yang-Parr correlational functional)” [17, 18] is used for the computational purpose. The “6-311 +G” set supplemented with two sets of diffuse functions and one set of polarization functions and augmented cc-pVTZ set have been utilized for the structural analysis [19,20,21]. The vibrational frequency assignments of the normal modes were carried out based on the PED calculations done with the help of the VEDA program [22, 23]. NBO analysis has been carried out in order to analyse the hyper conjugative and delocalized molecular charge interactions which depicts a clear picture of the intra molecular charge transfer of the molecules.
Quantum theory of Atoms in Molecules was applied to find out the electron density distributions of the molecules. Using Multiwfn software, the electron localized function, NBO Orbitals, Hirshfeld charge, Fukui functions and dual descriptors were analysed [24]. Various global reactivity descriptors such as chemical potential, chemical hardness, softness, electrophilicity and nucleophilicity index were calculated using finite difference approximation. All these functions help to find out the reactive sites of the molecule [25, 26] and to categorize these molecules based on reactivity [27, 28].
Results and discussion
Geometrical optimization
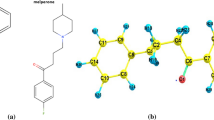
The molecular structure of the compounds 1, 2 and 3 in the ground state have been optimized using “Gaussian-16” utilizing Density functional theory with “B3LYP functional” and “6-311+G(2d,p)/cc-pVTZ basis set”. The optimized geometries are displayed in Fig. 1. The values of bond length, bond angle and dihedral angles were compared with the experimentally reported values and found consistent. The thermodynamic parameters of the compound such as total thermal energy, the rotational constants and the dipole moment values obtained from B3LYP/6-311+ G(2d,p) basic sets for the molecules under study are shown in Table 1.

The optimized structure of amphetamine(1), methamphetamine(2) and MDMA(3)

Frontier orbitals
Frontier molecular orbitals \(\left( FMO\right)\)
The “Frontier molecular Orbitals-HOMO and LUMO” determines the chemical reactivity of the molecules [29]. The energy of “Highest Occupied Molecular Orbital(HOMO)” determines the electron giving capacity which characterizes nucleophilic component and “Lowest Unoccupied Molecular Orbital(LUMO)” energy describes the electron accepting ability which characterizes electrophilic component [30]. The energy difference between HOMO and LUMO is called energy gap which is a critical parameter in determining the chemical stability of a molecule and eventual charge transfer interactions within a molecule.
Figure 2 displays the 3D plots of the frontier orbitals, along with their corresponding energy values. The HOMO of amphetamine shows greater contribution from the states of the \(NH_2\) group compared to the phenyl group, while the opposite is true for the LUMO. In the case of methamphetamine, the phenyl group does not contribute to the HOMO formation, and \(NH_2\) does not contribute to the LUMO. This suggests that, for both amphetamine and methamphetamine, the N atom acts as a favorable site for electron-deficient groups. However, for MDMA, the \(NH_2\) group does not contribute to either the HOMO or LUMO, possibly due to the oxygen substitutions at meta and para positions of the phenyl group.
Furthermore, the HOMO-LUMO energy gap was calculated for the three molecules, resulting in values of 6.2162, 5.760, and 5.3351 eV for amphetamine, methamphetamine, and MDMA, respectively. MDMA has the smallest HOMO-LUMO energy gap, indicating enhanced intra-molecular charge transfer interactions which may potentially influence its biological activities.
Global reactivity descriptors
The global reactivity descriptors such as “chemical hardness(\(\eta\)), softness(S), Chemical potential(\(\mu\)), electronegativity(\(\chi\)) and electrophilicity index(\(\omega\))” are important parameters for understanding the reactivity and stabilty of molecular systems [31] . The global reactivity descriptors of the title molecules were calculated using B3LYP/6-311+G(2d,p) basic set with finite difference approximations. The values were then verified with those obtained through Multiwfn and found to be in agreement.
The “hardness” of the molecule is;
The “softness” of the molecule is;
The “chemical potential” is given as;
The “electronegativity” is expressed as;
The “electrophilicity index” of the molecule is;
where I “ionization potential” and A “electron affinity” which are calculated using delta SCF method [32].
where “\(E\left( N\right) , E\left( N-1\right) and E\left( N+1\right)\)” are the energies of “N, N-1 and N+1” eletron systems.
Electrons flow from regions of high chemical potential to regions of low chemical potential until the electronic chemical potential is constant throughout the molecule. Hence \(\mu\) can be interpreted as a measure of the tendency of electrons to escape from a system. As the chemical potential becomes more negative, it becomes increasingly difficult for an electron to be lost. It is considered as negative of the Mulliken electronegativity [33]. Hardness fundamentally signifies the resistance to deformation of the electron cloud of the atoms, ions or molecules under small perturbation generated during the process of chemical reaction. If the electron cloud is strongly held by the nucleus, the chemical species is ‘hard’ but if the electron cloud is loosely held by the nucleus, the system is ‘soft’ [34, 35]. Hardness and softness are a useful concept for understanding the behaviour of chemical systems. Soft molecules will be more polarizable than hard molecules [36, 37]. Electrophilicity index is a measure of the stabilization energy when the system gets saturated by electrons. A reactive nucleophile is characterized by a lower value of \(\omega\), while higher values indicate the presence of a good electrophile [24].
These descriptors have importance in rational drug design. However, biological systems present a collocation of simple interactions within a complex environment. Molecular descriptors are computed within an in silico framework where the dynamic roles of molecules are not fully considered. Notably, in certain instances, the transient metabolic by-products of a drug exert significant influence on its activity. Consequently, the holistic activity of a drug often arises from the cumulative effects of these individual entities. In such cases the overall activity can be predicted only after calculating the descriptors of each molecule , and it makes the system more complex. Though drug design facilitated by molecular descriptors, serves as a valuable tool for scrutinizing vast molecular libraries to identify potential candidates or for enhancing activity of existing molecules [38].
The electron affinity, ionization potential, and global reactivity descriptors of the title molecules were computed, and the results are presented in Table 2. Based on the findings, MDMA is the softest molecule among the three, as confirmed by its lowest energy gap and ionization energy. Additionally, MDMA has the lowest LUMO energy, which makes it the best electron donor. Furthermore, MDMA has the lowest chemical hardness value, making it the most reactive among the three. Therefore, the order of decreasing reactivity among the molecules is MDMA > Methamphetamine > Amphetamine. Moreover, the lower value of electrophilicity index \(\omega\) suggests that the molecules exhibit good nucleophilic behavior. Furthermore, these molecular descriptors have much significance in the pharmacological activity , and they are used in quantitative structure-activity relationship(QSAR) studies and drug design. The dipole moment and polarizability represent information about charge distribution within the molecule which affect solvation and membrane permeability. The ionization potential and electron affinity provide information regarding the molecule’s stability, which could also be reflected in the drug’s metabolism [39]. According to studies in the literature, a decrease in polarity and an increase in polarizability, along with a greater ability for hydrogen bonding, significantly reduce brain penetration [39]. Consequently, Methamphetamine and MDMA exhibit higher brain penetration compared to Amphetamine, consistent with experimental findings. The electron affinity which is used for the investigation of optimal bioavailability and ionization potential which is related to the blood-brain barrier permeation. Among the three, MDMA has the lowest ionization potential, reflecting its high blood-brain barrier penetration ability. Furthermore, dipole moments of these molecules are low and close with each other and they have comparatively small size. Thus they have a good capacity to penetrate the blood brain barrier [40]. Binding to an active pocket of a receptor depends on the electronic structure of the ligand, with a significant contribution from dipole moment and polarizability [41]. This is because the electrostatic field generated by the receptor would strongly interact with molecules that have a higher dipole moment or polarizability.
Electron Localization Function (ELF) and NCI analysis
“Electron Localization Function (ELF)” is an alternative method for mapping “electron pair probability” in many-electron systems. The pair probability is a complex six-dimensional function that is difficult to analyze visually. ELF simplifies this by focusing on the spherically averaged local behavior of the pair probability as a function of a reference point. ELF is a powerful tool that helps to identify the location of atomic shell and core, binding, and lone electron pairs in atomic and molecular systems [42]. It provides insight into the atomic shell structure, type of chemical bonding, and verification of charge-shift bonds. A high ELF value indicates that electrons are highly localized, which suggests the presence of a covalent bond, a lone pair, or inner shells of the atom. The degree of electron localization directly relates to the confinement of electrons in the domain [43].

Electron Localized function and NCI analysis of amphetamine [1(a), 1(b)], methamphetamine [2(a), 2(b)] and MDMA [3(a), 3(b)] plotted using Multiwfn and VMD
“Non-covalent interactions” are a type of interactions that play a crucial role in various chemical processes, such as the chemical interaction between a protein and a drug, and a catalyst and a substrate [44]. Although they are weaker than covalent bonds, their cumulative effect can be significant, resulting in the stabilization of molecules and complex structures. It is essential to understand the nature and strength of non-covalent interactions to develop new drugs and materials that can interact efficiently with their target molecules or systems.
A plot of NCI together with ELF is shown in Fig. 3 which can reveal both covalent and non-covalent interactions simultaneously. Both analysis have been done using Multiwfn and VMD [45]. The red colored region of the ELF map represents highest ELF value which indicates the presence of a covalent bond or a lone pair and the violet region represents lowest ELF. Both maps show the presence of a bond attractor between the core attractors which indicates a shared electron attraction. Hydrogen has no core attractor and large domain around hydrogen indicates the presence of more negative character and high electron localization. In the NCI analysis map, the red colour region at the centre of benzene ring represents strong repulsion region due to steric interaction. In MDMA, there is two steric interaction region corresponding two “ring critical points”. The green colour region represents the vdW interction region where the electron density is minimum. Amphetamine exhibits a single weak interaction region, whereas methamphetamine and MDMA display two weak interaction regions.
Molecular graph and critical points
Quantum theory of atoms in molecules (QTAIM) is a powerful theoretical framework for understanding the electronic structure of molecules. Within this framework, molecular graphs are constructed based on the distribution of electron density within the molecule [14]. Molecular graph refers to the set of bond paths that connect the nuclei of atoms within a molecule’s equilibrium geometry, along with the corresponding critical points. In the context of QTAIM, as referenced by R.F.W Bader [15] in his article, “a critical point (CP) is a point in space where the first derivatives of the electron density vanish.
At critcal points
where the zero vector signifies that each individual derivative in the gradient operator, \(\nabla\) is zero and not just their sum. These critical points include nuclear critical points,bond critical points, ring critical points and cage critical points which are determined using mathematical algorithms that are based on the laws of quantum mechanics. The number and type of critical points that can coexist in a molecule or crystal follow a strict topological relationship which states that:
for isolated molecule where n denotes the number of the sub-scripted type of CP. The equality is known as the Poincare’–Hopf relationship (PH) and applies for isolated finite systems such as a molecule.”

Molecular graph of amphetamine (a), methamphetamine (b) and MDMA (c)
Figure 4 displays the molecular graphs of the title molecules. In the figure, blue circles indicate nuclear critical points, orange circles represent bond critical points, and yellow circles denote ring critical points. Amphetamine comprises 23 nuclear critical points, 23 bond critical points, and one ring critical point. Methamphetamine, on the other hand, features 26 nuclear critical points, 26 bond critical points, and one ring critical point. Finally, MDMA has 29 nuclear critical points, 30 bond critical points, and two ring critical points.
Hirshfeld charge and local reactivity descriptors
“Fukui Function” proposed by Parr and Yang in 1984, is one of the most important local reactivity descriptor to model chemical reactivity and site selectivity. Large values of ‘f’ at a site favor reactivity of that site [33]. “Conceptual Density Functional Theory” is applied to calculate the Hirshfeld Charge and Local reactivity descriptors using Multiwfn. According to Parr et al., “Fukui function is defined as the change of electron density \(\rho (r)\) at each point r when the total number of electrons is changed or as the sensitivity of chemical potential \(\mu\) of a system to an external perturbation at particular point r”.
Because of its derivative discontinuity, Fukui function is often calculated approximately using finite difference method as follows [46]
\(\rho _N(r), \rho _{N-1}(r)\) and \(\rho _{N+1}(r)\) represent separately the electron densities of the system with N, N-1 and N+1 electrons. \(\rho ^{Homo}(r)\) and \(\rho ^{Lumo} (r)\) are the electron densities of HOMO and LUMO, respectively. A large value of fukui function at a site favors reactivity of that site [33] and hence it is used to identify the electrophilic and nucleophilic attack sites of a molecule. When integrating the above equation for individual atoms in a molecule, the resulting “Condensed Fukui Function (CFF)” [47] can provide a more convenient method for predicting reaction sites within the molecule. For a given atom “K” in the molecule, three types of Condensed Fukui Functions can be defined based on the electron transfer involved [33].
For nucleophilic attack,
For electrophilic attack,
For radial attack,
where \(q_k\) is the gross electronic population of atom k in the molecule.
Dual descriptor (DD) was proposed by Morell et al. in 2005 [48] which is a more convenient tool to be used for predicting the reactive sites as it can reveal both types of reactive sites simultaneously.
where \(\eta\) is the chemical hardness of the system. Using the same approximation as in the Fukui function, Condensed Dual Descriptor (CDD) can be expressed as follows:
The dual descriptor \(f^2(r)\) will be positive in electrophilic regions where \(\rho ^{Lumo}(r)\) dominates and negative in nucleophilic regions where \(\rho ^{Homo}(r)\) dominates. The calculated values of Hirshfeld charges, condensed Fukui function and Dual Descriptor are shown in the Tables 3, 4 and 5.
The values of condensed fukui functions and dual descriptor indicates that for all the three molecules,the region around nitrogen are the most reactive site for nucleophilic attack. For amphetamine, the region around 21N, 17C, 20H, 12C, 6C, 16H, 5C, 4C and 1C are the reactive sites for nucleophilic attack and nucleophilicity decreases in the order \(21N>17C>20H>12C>6C>4C>5C>16H>1C\). For methamphetamine, nucleophilicity decreases in the order \(21N>1C>4C>22H>5C>16H>23C>2C>24H>15C>19H\) and for MDMA, the nucleophilicity decreases in the order \(24N>14O>13O>1C>5C>4c>19H>18C>23H>21H>20C\).

Electrostatic potential map of amphetamine (1a, 1b), methamphetamine(2a, 2b) and MDMA (3a, 3b) using B3LYP/6-311+G (2d, p) in the range -4.207e-2 to +4.207e-2
Molecular electrostatic potential analysis
The molecular electrostatic potential map illustrates the charge distribution of a molecule three-dimensionally which is a critical factor for understanding the reactive sites of electrophilic and nucleophilic attack of a molecule. This map also help to understand the different polar regions of a molecule as well as the net electrostatic effect caused due to total charge distribution [26]. As reported by W J Hehre [25], “the molecular electrostatic potential at a point in the space can be expressed as
where \(Z_A\) is the charge on nucleus A located at \(R_A\) and \(\rho (r)\) is the electron density function. The first and second terms represent the contribution by the nuclei and electrons of the molecule to the electrostatic potential produced at the point r”. The electron density surface mapped with electrostatic potential reveals the shape, size, charge density distribution and site of chemical reactivity of a molecule [49,50,51].
A high electrostatic potential represents the relative lack of electrons and a low electrostatic potential indicates the abundance of electrons at that region. The surface is color coded according to the variation in the electrostatic potential. The lowest electrostatic potential is indicated by red, while the highest is shown by blue, and the zero electrostatic potential is represented by green in the figures. The electrostatic potential increases in the order red < orange < yellow < green < blue. In Fig. 5, the total electron density surface mapped with the electrostatic potential and the contour map of the electrostatic potential are shown. For amphetamine and methamphetamine, the lowest electrostatic potential region was located over the nitrogen atom due to the presence of lone pair of electrons, while the positive potential sites were around the hydrogen atoms. For MDMA, the lowest electrostatic potential region was found over the nitrogen and oxygen atoms. The negative potential regions of the molecule were expected to be the sites of nucleophilic attack, while the positive potential regions were expected to be the sites of electrophilic attack. The green area over the aromatic ring represents a zero electrostatic potential region, leaving a more electrophilic region in the plane of the hydrogen atoms.
NBO analysis
NBO analysis is an efficient tool in molecular structure studies for analysing the hybridization, charge transfer and conjugative interaction effects [52, 53]. NBO analysis has been done using NBO program implemented in the Guassian-16 package with basic set B3LYP/6-311+G(2d,p). The NBO program is designed to analyze the many-electron molecular wave function in terms of localized electron-pair bonding units. This analysis is accomplished by transforming the given wave function into a localized form in terms of the electron densities of the molecule. In other words, NBO analysis provides a way to understand the chemical bonding and electronic structure of a molecule in terms of localized electron-pair interactions. The set of high-occupancy NBOs, each taken doubly occupied, is said to represent the “natural Lewis structure” (NLS) of the molecule [54, 55]. Delocalization effects appear as weak departures from this idealized localized picture. According to the NBO program manual [56], energetic analysis of NBO interactions can be conducted using second-order perturbation theory in terms of the “1-electron effective Fock energy operator”.
The stabilization energy associated with electron delocalization between each donor and acceptor NBO’s is estimated as;
where \(F_{ij}^2\) is the Fock matrix element between NBO’s i and j with energies \(\epsilon _\sigma\) and \(\epsilon _\sigma ^*\) and \(n_\sigma\) is the population of the donor orbital. Larger \(E^{\left( 2\right) }\) value indicates the strong interaction between the corresponding donor and acceptor orbitals and a greater tendency for delocalization effects. The NBO analysis of the title molecules have been done using the NBO module installed with the Guassian package. The calculated values of the stabilization energy using second order perturbation theory analysis of Fock matrix in NBO basis is shown in Tables 6, 7 and 8. The donor and acceptor orbitals having stabilization energy above 3 kcal/mol are included in the table.

Highly overlapping natural bonding orbitals (acceptor and donor) of amphetamine [1(a), 1(b)], methamphetamine [2(a), 2(b)] and MDMA [3(a), 3(b)]
Figure 6 shows the first two highly overlapping natural bonding orbitals of the three molecules plotted using Multiwfn software. The NBO plot of amphetamine shows that the donor NBO \(\pi \left( C_1-C_6\right)\) is substantially overlapped with acceptor NBO \(\pi ^*\left( C_4-C_5\right)\) and donor NBO \(\pi \left( C_2-C_3\right)\) is substantially overlapped with acceptor NBO \(\pi ^*\left( C_1-C_6\right)\), which resulted in high electron delocalization and a great tendency of charge transfer between donor and acceptor orbitals. The stabilization energy associated with electron delocalization between these orbitals are 21.41 and 21.20 Kcal/mol. For methamphetamine, the plot of NBO shows that the highest degree of overlapping is for \(\pi \left( C_1-C_6\right)\) with \(\pi ^*\left( C_4-C_5\right)\) and \(\pi ^*\left( C_2-C_3\right)\) and for \(\pi \left( C_4-C_5\right)\) with \(\pi ^*\left( C_2-C_3\right)\) and \(\pi ^*\left( C_1-C_6\right)\). The stabilization energies corresponding to these interactions are 21.05, 19.75, 19.32 and 20.73 kcal/mol respectively. These conjugate effects enhances electron delocalization and charge transfer occurs between donor and acceptor orbitals. The NBO analysis of MDMA reveals that there occurs a strong hyper conjugative interaction between \(LP(2)O_{13}\) and \(\pi ^*\left( C_2-C_3\right)\) and between \(LP(2)O_{14}\) and \(\pi ^*\left( C_1-C_6\right)\) which results in stabilization energies 25.84 and 25.5 kcal/mol. These enhanced anti bonding orbitals again conjugates with acceptor orbital \(\pi ^*\left( C_4-C_5\right)\) resulting in a high stabilization energy of 216.19 and 225.23 kcal/mol. All these strong hyper conjugative interactions are confined within the ring which clearly indicates the biological activity of the molecule.
Vibrational analysis
The molecular structure of these compounds does not exhibit any special symmetry, meaning that all of them belong to the C1 point group. The normal modes of vibration of these molecules belong to the ‘A’ irreducible representation and are both IR and Raman active. Amphetamine has a total of 63 normal modes of vibration, with 22 stretch modes, 21 bend modes, and 20 torsional modes. Methamphetamine has 72 normal modes of vibration, with 25 stretch modes, 24 bend modes, and 23 torsional modes. MDMA has 81 normal modes of vibration, with 28 stretch modes, 27 bend modes, and 26 torsional modes.
The theoretically calculated Raman activities are converted into corresponding Raman intensities using the following relation derived from the basic theory of Raman scattering [57, 58].
where \(\nu _0\) is the exciting frequency in \(cm^{-1}, \nu _i\) is the vibrational wave number of the \(i^{th}\) normal mode and f is the common scaling factor. h, c and K are universal constants.
To improve the agreement with experimental values, the calculated wave numbers were scaled down by a factor of 0.9679 [59]. Discrepancies between the calculated and experimental wave numbers are mainly attributed to the presence of anharmonicity effects in the real system and the various approximations used in the calculation methods. The calculated and experimental IR and Raman frequencies of the title molecules are shown in the Tables 9,10 and 11 along with the PED assignments. The PED assignments are performed with VEDA 4 program [22, 23].
The experimental and calculated IR spectra of the title molecules are shown in Fig. 7 and that of Raman spectra are shown in Fig. 8.

The calculated and experimental IR spectra of amphetamine [1(a), 1(b)], methamphetamine [2(a), 2(b)] and MDMA [3(a), 3(b)]

The calculated and experimental Raman spectra of amphetamine [1(a), 1(b)], methamphetamine [2(a), 2(b)] and MDMA [3(a), 3(b)]
Assignment of spectra
4000-2800 \(cm^{-1}\)
The vibrational wave numbers expected to appear in this region are those correspond to the C-H stretch modes of \(CH_2\), \(CH_3\), and aromatic groups, as well as the N-H stretch mode. For the three molecules, the C-H stretch belonging to \(CH_3\) and \(CH_2\) group vibrations is typically found between 2800-3020 \(cm^{-1}\) [60, 61]. In the vibrational spectrum of amphetamine, the CH stretch band at 2804 \(cm^{-1}\) belonging to the \(CH_3\) group is absent. The aromatic C-H vibrations of all three molecules are observed and calculated between 2900-3090 \(cm^{-1}\) (as shown in Table 9). Both the asymmetric and symmetric N-H stretch bands of amphetamine appear near 3430 and 3345 \(cm^{-1}\) in both the experimental and calculated spectra. For methamphetamine, the N-H band is at 3535 \(cm^{-1}\), and for MDMA, it is at 3390 \(cm^{-1}\), which are pure bands [62, 63].
1700-1000 \(cm^{-1}\)
The ring vibrations, C-H and N-H in-plane and out-plane bending vibrations and N-C stretching vibrations are expected in this region. The ring modes were observed and calculated for amphetamine between 1576-1158 \(cm^{-1}\) and for methamphetamine, the vibrational wave numbers corresponding to the C-C stretch are found between 1271-1602 \(cm^{-1}\) both in theoretical and experimental spectra. For MDMA, four ring modes are observed and calculated between the range 1211- 1608 \(cm^{-1}\) as shown in Table 9. The C-H bending vibrations of the three molecules are identified between 1128-1498/ 1124-1469 \(cm^{-1}\) for IR/Raman and 1136-1473 \(cm^{-1}\) in the theoretical spectrum [61, 64]. The C-H bending vibration observed at 1411 \(cm^{-1}\) due to the \(CH_3\) group for methamphetamine and MDMA are absent in the corresponding amphetamine IR spectra. The strong peak appears at 1598/1606 \(cm^{-1}\) in the experimental IR/Raman spectra of amphetamine is assigned to H-N-H in plane bending mode while the same is absent in the spectra of methamphetamine and MDMA. The week band at 1086 \(cm^{-1}\) in the calculated spectra of amphetamine is assigned to N-C stretching mode which is at 1095 \(cm^{-1}\)/1105 \(cm^{-1}\) for IR/Raman experimental spectra. The N-C stretching vibration appears at 1101/1097 \(cm^{-1}\) in the IR spectra of methamphetamine/amphetamine [65].Some additional peaks are found in the spectra of MDMA near 1000 \(cm^{-1}\) which is due to the stretch and bend modes of O-C. The peak appears at 1388/1379 \(cm^{-1}\) in the experimental/calculated IR spectrum of MDMA is assigned to the torsional mode of\(H_{11}C_7O_{13}C_2\) and that appears at 1346/1344 \(cm^{-1}\) in the experimental/calculated IR spectrum of MDMA is assigned to O-C stretch modes
1000-0 \(cm^{-1}\)
In this region, the stretching, bending, and torsional modes of C-C, N-C, and O-C are predicted. For amphetamine, the torsional modes corresponding to \(H_8C_3C_4C_5\), \(H_9C_4C_3C_2\), \(H_{11}C_6C_1C_{12}\), \(H_{13}C_{12}C_1C_6\), and \(H_7C_2C_3C_4\) appear at 978, 960, 893, 855, and 698 \(cm^{-1}\) in the experimental spectrum and at 958, 939, 889, 864, and 685 \(cm^{-1}\) in the theoretical spectrum. The IR spectrum of methamphetamine shows bands at 970, 923, and 836 \(cm^{-1}\), which are assigned to the torsional modes of \(H_9C_3C_4C_5\), \(H_8C_2C_3C_4\), and \(H_7C_1C_2C_3C_4\), respectively, with corresponding calculated frequencies of 965, 945, and 826 \(cm^{-1}\). In the vibrational spectrum of MDMA, some additional peaks near 1000 \(cm^{-1}\) may be due to the stretch and bend modes of O-C. The ring bends, which are a characteristic feature of benzene-type rings used to distinguish the type of substitution, are observed in all three molecules [66]. The ring bend at 597/638 \(cm^{-1}\) in the IR/Raman experimental spectrum and 589/613 \(cm^{-1}\) in the theoretical spectrum of amphetamine is assigned to the bending modes of \(C_1C_6C_5\) and \(C_2C_3C_4\), respectively. For methamphetamine, the ring bend at 621/618 \(cm^{-1}\) in the experimental/theoretical spectrum is assigned to \(C_2C_1C_6\), and for MDMA, the ring bend corresponding to \(C_3C_2C_1\) appears at 715 \(cm^{-1}\) in the experimental IR spectrum and at 707.55 \(cm^{-1}\) in the theoretical spectrum.
Statistical methods were employed to compare the calculated and experimental spectra quantitatively, utilizing the Pearson correlation coefficient as a measure of the linear correlation between the data sets. This coefficient is a common tool in the analysis of IR and Raman spectra [67]. The correlation results are illustrated in Figs. 9 and 10 demonstrating a robust correlation (\(R > 0.9996\)) between the experimental and calculated spectral values for all three molecules.

Correlation graph of calculated and experimental IR spectra of amphetamine (1(a)), methamphetamine (2(a)), and MDMA (3(a))

Correlation graph of calculated and experimental Raman spectra of amphetamine (1(a)), methamphetamine (2(a)), and MDMA (3(a))
Conclusions
The density functional theory calculations of amphetamine, methamphetamine, and MDMA offered valuable insights into the molecular and electronic properties of these psychoactive compounds. Molecular descriptors such as ionization potential, electron affinity, chemical potential, chemical hardness, chemical softness, electronegativity, electrophilicity index, dipole moment, energy gap, and polarizability were determined using finite difference approximation methods. These descriptors hold significant importance in pharmacological activity and are instrumental in quantitative structure-activity relationship (QSAR) studies and drug design.
The dipole moment and polarizability provide insights into the charge distribution within the molecules, impacting solvation and membrane permeability. Our findings suggest that while the three compounds share similar electronic structures, significant differences exist in their molecular geometries and charge distributions. The calculated values align with the observed properties of Amphetamine, Methamphetamine, and MDMA. Literature suggests that decreased polarity and increased polarizability enhance brain penetration. Consequently, Methamphetamine and MDMA exhibit greater brain penetration compared to Amphetamine, consistent with experimental results. Electron affinity aids in investigating optimal bio availability, while ionization potential is related to blood-brain barrier permeation. Notably, MDMA displays the lowest ionization potential among the three, indicative of its high blood-brain barrier penetration ability. Although the dipole moments of these molecules are low and comparable, their relatively small size suggests a good capacity to penetrate the blood-brain barrier. Ligand binding to an active receptor pocket depends on the ligand’s electronic structure, with contributions from dipole moment and polarizability, owing to the strong interaction with the receptor’s electrostatic field.
Analysis using Electron Localization Function (ELF) and Non-Covalent Interaction (NCI) reveals both covalent and non-covalent interaction regions within the molecules. Natural Bond Orbital (NBO) analysis indicates that strong hyper conjugative interactions are concentrated on the ring, enhancing biological activity. Among these, MDMA’s NBOs demonstrate the highest hyper conjugative interaction, with a stabilization energy of 225 kcal/mol attributed to the methylenedioxy groups. The vibrational analysis (FT-IR and FT-Raman) of these molecules has been conducted both experimentally and theoretically. The obtained wave numbers were analyzed and compared with PED assignments. A quantitative spectral comparison was also performed using linear correlation methods, revealing excellent agreement between the theoretical and experimental data.
Overall, our study highlights the importance of computational methods in understanding the molecular and electronic properties of psychoactive compounds. These findings could have implications for the development of more effective and safer treatments for psychiatric disorders and substance abuse. Further experimental studies are needed to validate our theoretical predictions and to fully understand the complex pharmacological properties of these compounds. The metabolic products of these drugs are also having a distinct role in their net biological activity. Some of them are more potent than the parent molecule itself. Hence, a complete insight of their activity can be obtained only after analyzing the molecular descriptors of their metabolites which is beyond the scope of this study.
Availability of data and materials
Not applicable.
References
United Nations Office on Drug & Crime (2013) The challenge of new psychoactive substances, Global SMART Program. https://www.unodc.org/documents/scientific/NPS_Report.pdf. Accessed 12 Feb 2022
United Nations Office on Drug & Crime (2021) World drug report. https://www.unodc.org/res/wdr2021/field/WDR21_Booklet_4.pdf. Accessed 12 Feb 2022.
Biel J, Bopp B (1978) Amphetamines: structure-activity relationships. In: Leslie L. Iversen, Susan D. Iversen, Solomon H. Snyder (ed) Stimulants. Handbook of psychopharmacology, vol 11. Springer, New York, p 1–39
Sherrill LK, Gulley JM (2018) Effects of amphetamine exposure during adolescence on behavior and prelimbic cortex neuronactivity in adulthood. Brain Res 1694:111–120
Heal DJ, Smith SL, Gosden J, Nutt DJ (2013) Amphetamine, past and present - apharmacological and clinical perspective. J Psychopharmacol 27:479–496
Moszczynska A (2021) Current and emerging treatments for methamphetamine use disorder. Curr Neuropharmacol 19:2077–2091
Sessa LH, Nutt D (2019) A review of 3,4-methylenedioxymethamphetamine (mdma)-assisted psychotherapy. Front Psychiatr 10:138 (1-7). https://doi.org/10.3389/fpsyt.2019.00138
Glennon RA, Young R, Hauck AE, McKenney JD (1984) Structure-activity studies on amphetamine analogs using drug discrimination methodology. Pharmacol Biochem Behav 21:895–901
Kier LB, Hall LH (1977) Structure-activity studies on hallucinogenic amphetamines using molecular connectivity. J Med Chem 20:1631–1636. https://doi.org/10.1021/jm00222a019. PMID: 592329
Rolf Willestofte B, Irene S, Peter Cyril W, Salim A (2012) Raman optical activity and raman spectra of amphetamine species–quantum chemical model calculations and experiments. Am J Anal Chem 3:410–421
Taplin F, O’Donnell D, Kubic T, Leona M, Lombardi J (2013) Application of raman spectroscopy, surface-enhanced raman scattering (sers), and density functional theory for the identification of phenethylamines. Appl Spectrosc 67:1150–5
Ma H, Hou Y, Fang H, Sarkar A (2021) Investigation of the interaction of amphetamine drug with zn12o12 nanocage: a quantum chemical study. J Comput Electron 20
Li X, Jiao X, Li H, Derakhshandeh M (2021) Amphetamine drug detection with inorganic mgo nanotube based on the dft calculations. Appl Biochem Biotechnol 193:3528–3539
Bader R, Bader R (1990) Atoms in Molecules: A Quantum Theory In: International Series of Monographs on Chemistry, vol 22. Clarendon Press, Oxford. https://books.google.co.in/books?id=up1pQgAACAAJ
Lu T, Chen F (2012) Multiwfn: A multifunctional wavefunction analyzer. J Comput Chem 33:580–92
Frisch MJ et al (2016) Gaussian 16 Revision C.01. Gaussian Inc., Wallingford
Hohenberg P, Kohn W (1964) Inhomogeneous electron gas. Phys Rev 136:B864–B871. https://link.aps.org/doi/10.1103/PhysRev.136.B864. Accessed 24 Aug 2022.
Kohn W, Sham LJ (1965) Self consistent equations including exchange and correlation effects. Phys Rev 140:A1133–A1138. https://link.aps.org/doi/10.1103/PhysRev.140.A1133. Accessed 24 Aug 2022.
Lee C, Yang W, Parr RG (1988) Development of the colle-salvetti correlationenergy formula into a functional of the electron density. Phys Rev B 37:785–789. https://link.aps.org/doi/10.1103/PhysRevB.37.785. Accessed 25 Aug 2022.
Becke AD (1993) Density-functional thermochemistry. the role of exact exchange. J Chem Phys 98:5648–5652. https://doi.org/10.1063/1.464913
Parr RG, Yang W (1989) Density-functional theory of atoms and molecules. Oxford University Press, New York
Jamroz MH (2013) Vibrational energy distribution analysis (VEDA): Scopes and limitations. Spectrochim Acta A Mol Biomol Spectrosc 114:220–230. https://doi.org/10.1016/j.saa.2013.05.096
Jamroz MH (2004) Vibrational Energy Distribution Analysis VEDA 4. Warsaw
Parr RG, Lv Szentpály, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924. https://doi.org/10.1021/ja983494x
Hehre J, Radom L, Schleyer PVR, Pople JA (1986) Ab initio Molecular Orbital theory. Wiley Newyork
Jane M, Peter P (2011) The electrostatic potential: an overview. Comput Mol Sci 1:153–163
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874. https://doi.org/10.1021/cr990029p. PMID: 12744694
Fleming I (1976) Frontier orbitals and organic chemical reactions. Wiley
Aihara J-i (1999) Reduced homo lumo gap as an index of kinetic stability for polycyclic aromatic hydrocarbons. J Phys Chem A 103:7487–7495. https://doi.org/10.1021/jp990092i
Pearson RG (1993) The principle of maximum hardness. Acc Chem Res 26:250–255. https://doi.org/10.1021/ar00029a004
Kumar VRL, Kishor S (2013) Understanding the antioxidant behavior of some vitamin molecules: a first-principles density functional approach. J Mol Model 19:3175–86
Parr RG, Yang W (1984) Density functional approach to the frontier-electron theory of chemical reactivity. J Am Chem Soc 106:4049–4050. https://doi.org/10.1021/ja00326a036
Kaya S, Kaya C (2015) A new equation for calculation of chemical hardness of groups and molecules. Mol Phys 113:1311–1319. https://doi.org/10.1080/00268976.2014.991771
Islam N, Ghosh DC (2011) Spectroscopic evaluation of the global hardness of the atoms. Mol Phys 109:1533–1544
Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85:3533–3539. https://doi.org/10.1021/ja00905a001
Pearson RG (1966) Acids and bases: Hard acids prefer to associate with hard bases, and soft acids prefer to associate with soft bases. Science 151:172–177
Hassan Baig M et al (2016) Computer aided drug design: success and limitations. Curr Pharm Des 22:572–581
Semeniuk A, Kalinowska-Tluscik J, Nitek W, Oleksyn BJ (2008) Intermolecular interactions in crystalline hydroxychloroquine sulfate in comparison with those in selected antimalarial drugs. J Chem Crystallogr 38:333–338
Biel J (1970) Structure-activity relationships of amphetamine and derivatives. Amphetamines Relat Compd 3–19
LaPointe SM, Weaver DF (2007) A review of density functional theory quantum mechanics as applied to pharmaceutically relevant systems. Curr Comput Aided Drug Des 3:290–296
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403. https://doi.org/10.1063/1.458517
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–86
Johnson ER et al (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506. https://doi.org/10.1021/ja100936w. (PMID: 20394428)
LU T, CHEN F-W (2011) Meaning and functional form of the electron localization function. Acta Phys Chim Sin 27:2786. http://www.whxb.pku.edu.cn/EN/abstract/article 27788.shtml
Cao J, Ren Q, Chen F, Lu T (2015) Comparative study on the methods for predicting the reactive site of nucleophilic reaction. Sci China Chem 58:1845–1852
Yang W, Mortier WJ (1986) The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J Am Chem Soc 108(19):5708–11. https://doi.org/10.1021/ja00279a008. Accessed 9 Oct 2022.
Morell C, Grand A, Toro-Labbé A (2005) New dual descriptor for chemical reactivity. J Phys Chem A 109:205–12
Politzer P, Laurence PR, Jayasuriya K (1985) Molecular electrostatic potentials: an effective tool for the elucidation of biochemical phenomena. Environ Health Perspect 61:191–202
Scrocco E, Tomasi J (2005) In: The electrostatic molecular potential as a tool for the interpretation of molecular properties. Springer, pp 95-170
Scrocco E, Tomasi J (1978) Electronic molecular structure, reactivity and intermolecular forces: an euristic interpretation by means of electrostatic molecular potentials. Adv Quantum Chem 11:115–193
Glendening ED, Landis CR, Weinhold F (2012) Natural bond orbital methods. Wiley Interdiscip Rev Comput Mol Sci 2:1–42
Weinhold F, Carpenter JE (1988) The Structure of Small Molecules and Ions., Ch. The Natural Bond Orbital Lewis Structure Concept for Molecules, Radicals and Radical Ions. Springer, Boston, pp 227-236
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926
Lionel Goodman RRS (2006) Diffuse functions in natural bond orbital analysis. J Comput Chem 28:269–275
Weinhold F, Glendening ED. NBO 7.0 Program Manual Natural Bond Orbital Analysis Programs
Kalsi PS (2007) Spectroscopy of organic compounds. New Age International
Polavarapu PL (1990) Ab initio vibrational raman and raman optical activity spectra. J Phys Chem 94:8106–8112. https://doi.org/10.1021/j100384a024
Andersson M, Uvdal P (2005) New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis set 6-311+G(d,p). J Phys Chem A 109:2937–41. https://doi.org/10.1021/jp045733a
Varsányi G, Láng L, Kovner MA, Lempert K. Assignments for vibrational spectra of seven hundred benzene derivatives. Akademiai Kiado, Budapest
Socrates G (2004) Infrared and Raman Characteristic Group Frequencies: Tables and Charts, 3rd edn. Wiley
Cain BR, Freeman JM, Henshall T (1969) On the characteristic vibrations of the nh2 group. Can J Chem 47:2947
Bellamy LJ (1975) The Infra-red spectra of complex molecules. Springer
Colthup N et al (1990) Introduction to Infrared and Raman Spectroscopy. Academic Press
Srivastava A et al (2017) Spectroscopic (far or terahertz, mid-infrared and raman) investigation, thermal analysis and biological activity of piplartine. Spectrochim Acta A Mol Biomol Spectrosc 184:368–381. https://www.sciencedirect.com/science/article/pii/S1386142517303748
Smith B (2018) Infrared spectral interpretation: a systematic approach. CRC Press
Henschel H, Andersson AT, Jespers W, Mehdi Ghahremanpour M, Van der Spoel D (2020) Theoretical infrared spectra: quantitative similarity measures and force fields. J Chem Theory Comput 16:3307–3315
Acknowledgements
The authors wish to express their sincere gratitude to the Department of Chemistry, University College, Thiruvananthapuram for providing access to the Gaussian-16 programme. The Authors extend their thanks to Dr. B. Sandhya IPS, Director, Kerala Police Academy and Smt. Latha Devi MA, Director, FSL for their wholehearted support. The authors would also like to acknowledge the State Forensic Science Laboratory, Thiruvananthapuram and Department of Physics, University of Kerala, Kariavattom, Thiruvananthapuram for their assistance in conducting Spectral measurements.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
The concept was introduced by AB and SP and then discussed and further refined by VM. Instrumental analysis was done by SP. Analysis and interpretation of the data was done by JS and DM. AB drafted the article which was then revised and finalised by SP and VM. JS and DM reviewed the article.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bhaskarapillai, A., Parayil, S., Santhamma, J. et al. Exploring the structural and electronic characteristics of phenethylamine derivatives: a density functional theory approach. J. Eng. Appl. Sci. 71, 136 (2024). https://doi.org/10.1186/s44147-024-00461-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s44147-024-00461-3