
Abstract
DNA microarray data sets have been widely explored and used to analyze data without any previous biological background. However, analyzing them becomes challenging if data are missing. Thus, machine learning techniques are applied because microarray technology is promising in genomics, especially in the analysis of gene expression data. Furthermore, gene expression data can describe the transcription and translation processes of each genetic information in detail. In this study, a new system was proposed to impute more realizable values for missing data in a microarray dataset. This system was validated and evaluated on 42 samples of rectal cancer. Several evaluation tests were also conducted to confirm the effectiveness of the new system and compare it with highly known imputing algorithms. The proposed clustering column-mean quantile median technique could predict highly informative missing genes, thereby reducing the difference between the original and imputed datasets and demonstrating its efficiency.
Similar content being viewed by others


Introduction
Microarray technology is an effective tool for advanced biomedical studies. It can be applied to quality expression (GE) profiling, which is used to measure the expression levels of thousands of qualities on a single chip in a trial. However, missing values (MVs) may be encountered during processing because of environmental, specialized, and natural reasons, such as spotting issues, foundation commotion, counting errors, inadequate determination, picture debasement, clean or scratches on a slide, and methodical causes; thus, mechanical strategies should be developed, but applying any feature selection technique on incomplete microarray data poses a problem because most techniques fail. Many studies have shown that microarray data sets can contain up to 10% of missing data and up to 90% of genes with one or more missing data in some cases [1, 2].
Handling missing data is a challenge for researchers classifying cancers because these data should be imputed for information consideration. They are also used to understand the overall data and perform complicated tasks, such as predictive analysis and data protection against distortion.
In this study, a modified clustering column-mean quantile median (CCMQM) algorithm is proposed to overcome the drawbacks of missing gene expression data. It is compared with commonly known imputation methods with the same dataset: singular value decomposition (SVD), Bayesian principal component analysis (BPCA), column-mean, Gene-mean, column-median, gene-median, local least squares (LLS), and k-nearest neighbor (KNN) techniques. These imputation algorithms are compared by determining the likeness between the original and imputed data points. Scoring test methods are used to compare them and form the preferred usage of local, global, and hybrid techniques.
The remaining parts of this paper are organized as follows: “Results and discussion” section briefly reviews some of the recent work related to the imputation of missing microarray gene expression values; “Methods” section introduces and describes the methodology and the general scheme of the proposed combined systems; and “Conclusions” concludes the study.
Microarray imputation data have been explored using several machine learning methods to solve microarray missing data problems and enhance the corresponding solutions. With the accessibility of knowledge about the implementation concepts of each method, studies have been performed to develop an optimal system. In 2014, a hybrid approach called recursive mutual imputation (RMI) was developed to improve imputation accuracy for a high amount of missing data. In RMI, local and global methods (BPCA) and (LLS) are merged, and an effective powerful tool is developed to impute MV more efficiently than other comparative methods by obtaining high normalized root mean square error (NRMSE) ratios with high missing ratios of data [3]. In 2015, H. L. Shashirekha et al. solved a frequent problem in microarray gene data affecting results by presenting a mutual nearest neighbors (MNN) algorithm as an extension of KNN. They found that MNN is comparable with other known techniques when no high NRMSE occurs with increasing missing ratios in a data sample [4]. In 2019, a doubly sparse DCT domain with nuclear norm minimization (DSNN) method was proposed and pathway enrichment experiments were performed to establish the significance of imputation on four blood cancer datasets. In this method, a doubly sparse DCT domain is applied with nuclear norm minimization. Its performance is better and its results are superior to those of other methods in terms of classification and cancer pathways by good significance on the imputed data by DSNN method [5]. Later in 2020, a method with a naive Bayes classifier was integrated to examine the influence of missing ratios on imputation data. When the missing ratio exceeds 20%, the prediction accuracy decreases; thus, choosing the appropriate imputation technique is challenging because the expectation precision decreases when the ratio is > 70% information loss [6]. Furthermore, MVs are identified by using the optimized hybrid of fuzzy C-means and majority vote (opt-FCMMV), where the majority vote (MV) and optimization are achieved by particle swarm optimization (PSO) on ovary and lung cancer datasets with 3 MV mechanisms (MCAR, MAR, and NMAR) and 5 different percentage values (5%, 10%, 30%, 50%, and 80%); the performance of this technique is better than that of FCM and FCMMV because it can improve accuracy, especially in case of high-dimensionality data [7]. Most studies have applied machine learning techniques to reduce the risks of missing genes, which is considering a single gene criterion as per the operation of the chosen technique. Thus, designing a prediction system that can estimate the values of missing data near actual values; this is challenging when a gene subset is compared with the original set of genes. High imputation accuracy may also be unlikely when many microarray datasets are tested. For this purpose, a new gene imputation technique is proposed in this study. In this technique, preprocessing clusters are combined with gene imputation technique followed by a median. This system is then evaluated on rectal cancer microarray datasets by recording the classification accuracy and studying the different parameters for each. Our results reveal that the proposed system achieves excellent and comparable outcomes.
Results and discussion
Each evaluation test is applied to 3 mechanisms with 3 missing ratios on the 9 IMs. In each test, it describes a different side of the algorithms used. The values in the evaluation tests are assigned in an elaborative schedule for each condition in each test such as seen in Table 1 and shown in a bar chart to illustrate the output of each case.
The main findings through Figs. 1, 2, 3, 4, 5, and 6, in NRMSE test, the BPCA algorithm has the best response, whereas Col-Mean has the worst response and the highest value for NRMSE and CCMQM in the medium range between the two algorithms as shown in Fig. 1. For the duration of the test, CCMQM has the shortest processing time, whereas Gene-Mean has the longest processing time as Fig. 2. For the GC test in Fig. 3, the best algorithm is the modified CCMQM because it has the least coefficient score, whereas the worst algorithm is the SVD that consumes the highest score. For the ED test, CCMQM gains the least distance in Fig. 4, so it is considered the most reliable algorithm, whereas the Col-Median is the poorest one because it yields the largest distance. For the SNR test, the best method is the Col-Median with the highest ratio; conversely, the worst method is LLS, but CCMQM is in the moderate range between the best and worst algorithms as shown in Fig. 5. For the Fisher discriminant test, the best methods are LLS and Col-Mean because they have the smallest FC. By contrast, the worst method is Col-Median in Fig. 6. CCMQM is a comparable method in this test.

Normalized root mean square error of the imputation methods

Period taken for each imputation method

Gini coefficient scores of imputation methods

Euclidean distance test values

Signal-to-noise ratio

Fisher discriminant test values
All the imputation methods performance are integrated in Table 2 with comparable results of best √, moderate ↔ and worst ↓evaluate and Table 3 contains our result study with other previous work in the same field.
Generally, all imputation methods depend on multiple conditions as follows:
-
System performance and generation
-
System processor and number of cores
-
System storage
-
System RAM size
Furthermore, all imputation methods use different criteria of calculation and tracking in order to show the calculated results, e.g., all previous imputation methods where calculated using pre-prepared functions which is generic and suitable for any amount of data which use more processing time compared to a dedicated code as what is used in the purposed code
On the other side, the purposed method was inspired from column mean imputation method which uses much more time and kindly accept the following as description:
-
From processing time point of view
-
Column mean need to calculate an infinite number of cells in each column in order to calculate the mean value then loop on all other columns respectively
-
CCMQM method calculate only a sample from a column with respect to an acceptable variance where number of samples = number of missing values in the column and this uses much less time to evaluate.
-
Methods
This study is performed mainly to introduce the modified algorithm CCMQM and compare its results with those of some commonly known imputation methods. The modified system is validated on a rectal cancer microarray dataset, and other known methods are applied to the same data. Different parameters are considered in a trial to reach the highest prediction accuracy and the performance of this system is verified through statistical evaluation tests.
Gene expression data analysis
The spot intensities of TIFF and RAW images are calculated using image analysis programs and then exported from the used program into several text files and Excel spreadsheets, representing raw data. The microarray measurements in these files indirectly represent the target quantity (the gene abundance) by measuring the fluorescence strength of the spots for each fluorescent dye (Cy5 for red and Cy3 for). First, the GenePix Array List (GAL) describes the position and content of each spot on an array from plain text files. Substance name and ID lists are pasted directly into the array setting file to form a GAL file. Second, GenePix results (GPR) are demonstrated in a spreadsheet. One version of GenePix calculates up to 108 different measurements for each spot, including the number of feature and background pixels for each feature at each scan wavelength and the mean, median, and totality of pixel intensities at each scan wavelength for a feature and background pixels. The data matrix of an expressed gene is then constructed from these spreadsheets. The foreground and background intensities of the red and green channels are determined. Then, four quantities, namely, RF, GF, RB, and GB, are used to calculate the log ratio of the net array intensities of the red and green channels. The net intensities are determined with the GenePix output from the changes in the mean of the foreground and the median of the background of both channels. The matrix dimension becomes n × m in which n genes are represented in rows and m samples are represented in columns. Each sample represents data from one array, and log ratios are calculated from the data introduced in one spreadsheet.
Data preprocessing
Data are preprocessed to control the quality of the image data produced by scanning a microarray and converting the image data to a table of expression level values or expression level ratios for the genes on the microarray. Different preprocessing and analysis techniques, such as the limma package in R, are applied using linear models of microarray data for analysis [9]. These procedures are crucial for a spotted array with a two-color method. In this study, preprocessing is an essential part for the input data to suitably analyze and make it valid to enter the model.
It solves many problems in raw data such and figured out them as a checklist before implementing:
-
Removing the categorizing row and column.
-
Eliminating genes with no name.
-
Minimize the high values by apply log transformation.
-
Performing quantile normalization to achieve the same sample distribution at each state.
The capturing and storing of microarray information are not the last steps of the process itself. The amount of information from a single microarray experiment is quite large, thus software apparatuses should be utilized to understand its meaning. GE information analysis is typically applied to (i) discriminate between different known cell natures or conditions, e.g., between normal and tumor tissues or between tumors of different types, or monitor tumors under different treatment schemes; and (ii) identify different and previously unknown cell types or conditions, e.g., new subclasses of an existing class of tumors. The same problems are encountered in genes that are being classified, such as unknown cDNA sequences must be assigned to gene classes, or a kind of genes should be divided into new functional classes based on their expression patterns under many experimental conditions [10]. These double tasks are defined as class prediction and class discovery [11]. In ML literature, they are known as supervised and unsupervised learning; the learning in question in microarrays data analysis being of GE values. If classes are identified with labels, discriminate analysis or supervised learning methods rather than clustering methods are commonly used [12]. Data having a ratio of missingness equal to 1% in represented data are neglected, whereas those with missingness of 1–5% are manageable. However, data with missingness of 5–15% should be subjected to appropriate approaches to achieve good imputation results. When datasets have > 15% MD, choosing IM may strongly influence the results. As such, MV from the original data should be selected from about 5% of all genes randomly and assigned. Then, ignorable (MCAR and MAR) and non-ignorable (NMAR) missingness types are measured at 3 missingness rates (10%, 20%, and 30%).
Ignorable MV is produced by randomly choosing the samples at three missingness rates. Then, they are removed. Furthermore, the upper or lower tails (10%, 20%, and 30%) of data are selected to produce non-ignorable MV. Their values are subsequently removed to ensure that the missing rate depends on the actual GE through data processing for imaginarily produced MV.
Complete workflow
In Fig. 7, having some steps in sequence to achieve the desired output of this system starting with data preprocessing that worked on the raw dataset of 16,156 sample genes of normal and rectal cancer from the GEO database [13] has the genome-based microarray data accession number GSE15781 [14] for rectal cancer, with dataset containing 42 samples from 22 patients with rectal cancer and 20 healthy individuals patients to prepare it for entering the generation stage for applying the 3 mechanisms of missing data to enter the imputation algorithms (9 criteria techniques), and the three categories are local group for KNN and LLS techniques while for the global group are BPCA, SVD, Gene-mean, gene-median, column-mean, and column-median, and the modified global technique is Clustering column-mean quantile median (CCMQM) following up to the evaluation stage that based upon some statistical tests as NRMSE, time consuming, Euclidean distance, signal-to-noise ratio (SNR), Gini coefficient, and Fisher discriminant.

Complete workflow system
Gene statistical tests
Normalized root mean square error
The used imputation methods are compared in terms of NRMSE. The RMSE is a regularly used measure of the variance between values predicted by a model.
NRMSE is calculated using the following formula in Eq. (1):
Where Y-original and Y-imputed represent the original and imputed datasets, respectively; the NRMSE values range from 0 to 1, and the smaller the values are, the better the evaluation performance will be [15].
Time consumption of imputation methods
Another metric of the performance comparison is execution time. In our study, the execution times for statistic-based IM are comparable with changes and proportional increases in MV. Overall, one model can be preferred to others by considering metric time. A considerable trade-off exists between prediction accuracy and time taken for imputation.
Gini coefficient
Gini coefficient (GC) measures the inequality among values of a variable. The higher the index value is, the more distributed the data will be. Alternatively, GC can be considered to be half of the relative mean absolute difference so that Gini coefficient gives us a measure of competitiveness, and therefore a measure of uncertainty.
Coefficient output can take any values from 0 to 1 (0 to 100%).
A GC of 0 indicates perfect equality of distribution of income within a population, whereas a GC of 1 represents a perfect inequality when one person in a population receives all the income, while other people earn nothing. In some cases, if the income of a population is negative, GC exceeds 100%.
GC can be calculated as follows:
Where A and B are the areas above and under the Lorenz curve, respectively
Euclidean distance
First performed in the nineteenth century by Augustin-Louis Cauchy, Euclidean distance (ED) is a nonparametric test through which the distance between each gene p and the ideal gene q is calculated agreeing to the ED and expressed by the Eq. (3). Each GE value in one gene and its corresponding value in the ideal gene are treated as two points in space. ED is calculated as the square root of the sum of the squared differences of the two real value vectors. The distance between two object points is usually defined as the smallest distance among pairs of points from the two objects, the International System of Units (SI) should be used. Equivalents in other units may be given in parentheses.
Where p and q are the two points of the ED difference.
Signal-to-noise ratio
Signal-to-noise ratio (SNR), which was first proposed by Golub et al. in 1999 [11], measures the relative usefulness of the feature by ranking the features. This test is performed by comparing gene correlations with the expected gene correlations. The measure of the relative contribution by a sample to the signal and noise, not the ratio, is examined. It gives each gene a value because of maximal differences in the mean expression between two bunches and a negligible variety of expression inside each accumulate [16]. In this method, genes are first ranked according to their expression levels by using the SNR test statistic. In this test, the signal strength indicates the class conditional means, and noise is categorized as the conditional standard deviation. In microarray data, the features selected for classification can be ranked. SNR is represented as follows:
Where μi1 and μi2 are the mean differences of sample classes 1 and 2, respectively; σi1 and σi2 are the standard deviations of the samples in each class; i = 1 to ng.
Fisher discriminant criterion
Fisher’s linear discriminant analysis, which was first introduced by Duda et al. [17] in 1973, is a mixture of observed or measured variables that best describe the separations between known groups of observations. It mainly aims to classify or predict issues at which the dependent variable appears quantitatively. According to its criteria, higher values are assigned to features that vary significantly among classes (original gene and predicted gene in our example) relative to their variances. Genes are arranged in a descending order by which the first genes are considered the most informative from the Fisher discriminant criterion test end result [18].
The Fisher discriminant ratio is used to evaluate the classes separately by each feature. The Fisher values of the range and variance clarify the degree of overlap between classes in a data set. The higher the value of a given feature is, the greater the number of classes separable by that feature and the lower the overlap and complexity of data will be. Therefore, features with minimum overlaps in data sets and low complexity should be selected. The time complexity of (1) is 0 (m, n, c), where m is the number of samples, n is the number of features, and c is the number of data set classes [19].
The Fisher discriminant ratio is determined as
Two classes can be approximated by N (μi1, σi1) and N (μi2,σi2), respectively, where N (μi,σi) indicates a normal distribution with mean μi and variance σi.
Modified system overflow
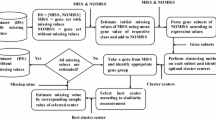
The following block diagram and the pseudo-code simplify the phases of the data route in the modified system (CCMQM) that takes place on a missing data set to impute and evaluate the imputation given for each missing value. Preprocessed data are exposed to decreasing ratios (10%, 20%, and 30%). The data dimensions of rows and columns are determined to take a random sample in the presence of missing spots and another sample without the missing spots with the same number of columns in both samples. The samples are subjected to some statistical calculations to obtain a substitutable persuasive value. This value is then substituted into all the missing spots, and the reformed data are compared with the existing data. Comparable files are attached to Excel files and visualized in bar charts for convenient observation to verify if the system yields the desired output (Fig. 8).

Proposed system overflow
A pesudo-code representing the detialed steps of the system:
Include all needed packages using the following libraries
Crane Library
BioConductor Library
// Generation //
Declare matrixes to store the imputed data from CCMQM method "CCMQM_Matrix"
Declare matrixes to store your ouput results from testing methods showing column and
row names
Read "Data File" matrix to proceed preprocessing procedure
Include "Data File" in a data frame
Declare "Data" to store "Data File" after data framing
Declare number of normal and cancer patients shown in the matrix
Include a separator column between normal and cancer patients for easily tracking
Declare long int to store any number of rows to make your code generic
Declare "Datamiss 10", "Datamiss 20"& "Datamiss 30" to store the amount of missing values refering to the used technique
// Imputation //
Use MCAR function to start first technique
for (i in 1:number of columns in "Data")
{
Use system.time to start counting the time
Store the value in "Start_Time_MCAR_Datamiss10"
Select first column
Count number of "Nan" in the first column
Take a random sample of numbers in the first column = number of
missing "Nan" in the first column
Store this samples in "sample_datamiss10_1st_col"
Declare data frame for "sample_datamiss10_1st_col"
Calculate Column Mean for "sample_datamiss10_1st_col"
Substitute with this value with every missing "Nan"
Save the new Imputed Column in "CCMQM_Matrix" 1st column
Use NMRSE function to calulate Column performance
Store the calculated value in "NMRSE Matrix"
Use Gini Coeffitient function
Store the calculated value in "Gini Matrix"
Calculate End.time from difference bet. system.time
Store the calculated value in "Time Matrix"
}
Use same as previous for Datamiss20 & Datamiss30
Use MAR function to start Second technique
for (i in 1:number of columns in "Data")
{
Use system.time to start counting the time
Store the value in "Start_Time_MAR_Datamiss10"
Select first column
Count number of "Nan" in the first column
Take a random sample of numbers in the first column = number of
missing "Nan" in the first column
Store this samples in "sample_datamiss10_1st_col"
Declare data frame for "sample_datamiss10_1st_col"
Calculate Column Mean for "sample_datamiss10_1st_col"
Substitute with this value with every missing "Nan"
Save the new Imputed Column in "CCMQM_Matrix" 1st column
Use NMRSE function to calulate Column performance
Store the calculated value in "NMRSE Matrix"
Use Gini Coeffitient function
Store the calculated value in "Gini Matrix"
Calculate End.time from difference bet. system.time
Store the calculated value in "Time Matrix"
}
Use same as previous for Datamiss20 & Datamiss30
Use NMAR function to start Third technique
for (i in 1:number of columns in "Data")
{
Use system.time to start counting the time
Store the value in "Start_Time_NMAR_Datamiss10"
Select first column
Count number of "Nan" in the first column
Take a random sample of numbers in the first column = number of
missing "Nan" in the first column
Store this samples in "sample_datamiss10_1st_col"
Declare data frame for "sample_datamiss10_1st_col"
Calculate Column Mean for "sample_datamiss10_1st_col"
Substitute with this value with every missing "Nan"
Save the new Imputed Column in "CCMQM_Matrix" 1st column
Use NMRSE function to calulate Column performance
Store the calculated value in "NMRSE Matrix"
Use Gini Coeffitient function
Store the calculated value in "Gini Matrix"
Calculate End.time from difference bet. system.time
Store the calculated value in "Time Matrix"
}
Use same as previous for Datamiss20 & Datamiss30
// Evaluation //
Declare all output matixes in dataframe
Save Output matrixes as .xlsx files in defined address in your system
use excel to generate evaluated bar charts for resonable output
Use output data to attach them on the main imputation methods code for study
Comparisons.
Conclusions
In this study, microarray data are used because they can reshape molecular biology, especially in substituting MD with acceptable values. They have been widely developed, but they still convey many uncertainties. If these uncertainties are not well resolved, microarray data may be useful for subsequent analysis, but they may be unreliable. Thus, microarray data should be preprocessed before any interpretation.
The modified imputation technique for microarray GE data likely enhances performance and provides two main advantages. Specifically, the time consumed is shorter than other rapid imputation techniques. Additionally, imputation performance improves in most of the evaluation tests performed. Therefore, this technique will help researchers use more computational datasets for the corresponding methods in the future.
CCMQM is included in a system to eliminate MV problems and predict comparable values closely related to truth datasets with less computational time. It is the best method for generating MV based on the Gini coefficient and Euclidean distance values. The KNN method consumes a shorter execution time than other ML-based imputation techniques. Among the imputation techniques, CCMQM is the least time-consuming. Studies on NMCR are still limited. Although many studies have discussed this point, uncertainties regarding the ability of imputation techniques to detect more accurate values at different missing rates remain and should be addressed in future studies.
Availability of data and materials
In our study we downloaded data from the Gene Expression Omnibus at the NCBI known as (GEO) database, provides data in a tab- delimited format [www.ncbi.nlm.nih.gov/geo/]. GEO database has the genome-based microarray data accession number GSE15781 for rectal cancer, with dataset contains 42 samples from 22 patients with rectal cancer and 20 healthy individuals.
Abbreviations
- BPCA:
-
Bayesian principal component analysis
- CCMQM:
-
Clustering column-mean quantile median
- DSNN:
-
Doubly sparse with nuclear norm minimization
- ED:
-
Euclidean distance
- GC:
-
Gini coefficient
- IMs:
-
Imputation methods
- KNN:
-
K-nearest neighbor
- LLS:
-
Local least squares
- MVs:
-
Missing values
- MNN:
-
Mutual nearest neighbors
- NRMSE:
-
Normalized root mean square error
- opt-FCMMV:
-
Optimized hybrid of fuzzy C-means and majority vote
- PSO:
-
Particle swarm optimization
- GE:
-
Quality expression
- RMI:
-
Recursive mutual imputation
- SNR:
-
Signal-to-noise ratio
- SVD:
-
Singular value decomposition
References
Tuikkala J (2006) Improving missing value estimation in microarray data with gene ontology. Bioinformatics 22(5):566–572
Liew AW-C (2011) Missing value imputation for gene expression data: computational techniques to recover missing data from available. Brief Bioinform 12(5):498–513
Li H (2014) A hybrid imputation approach for microarray missing value estimation, I.E.E.E. International Conference on Bioinformatics and Biomedicine.
Shashirekha HL, Analysis of imputation algorithms for microarray gene expression data, International Conference on Applied and Theoretical Computing and Communication Technology, 2015.
Farswan A (2020) Imputation of gene expression data in blood cancer and its significance in inferring biological pathways. Front Oncol 9:1442
Gobi M (2020) An efficient navie BAYES imputation method for missing values. Int Res J Modern Eng Technol Sci 2020;2(7)
Kumaran SR (2020) Estimation of missing values using optimized hybrid fuzzy c-means and majority vote for microarray data. J Inform Commun Technol 19(4):459–482
Rosa A (2017) The ability of different Imputation Methods to Preserve the Significant Genes and Pathways in Cancer
Smyth GK (2021) Limma: linear models for microarray data
Smyth GK (2003) Statistical issues in cDNA microarray data analysis. In: Functional Genomics: Methods and Protocols, vol 224, pp 111–136
Golub TR (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286:531–537
Yin W (2005) Background correction for cDNA microarray images using the TV+L1 model. Bioinformatics 21:2410–2416
Barrett T (2013) NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41(Database issue):D991–D9D5
Snipstad K (2010) New specific molecular targets for radio-chemotherapy of rectal cancer. Mol Oncol 4:52–64
Oba S (2003) A Bayesian missing value estimation method for gene expression profile data. Bioinformatics 19:2088–2096
Mishra D (2011) Feature selection for cancer classification: a Signalto-noise ratio approach. Int J Sci Eng Res 2:1–7
Duda RO (1973) Pattern Classification and scene analysis. Wiley
Hernandez JC (2007) A genetic embedded approach for gene selection and classification of microarray data. In: Proceedings of EvoBIO LNCS, vol 4447, pp 90–101
Seijo-Pardo B (2016) Ensemble feature selection: homogeneous and heterogeneous approaches. Knowl Based Syst 0:1–19
Acknowledgements
There is no acknowledgement based upon authors.
Funding
No funding was obtained for this study.
Author information
Authors and Affiliations
Contributions
NY implemented all the practical work that have been done and analyzed the results, and was responsible for the proposed algorithm. MA supervised and revised the work, shared in the discussion, and obtained the results. MS was a major contributor in writing the manuscript, helped in point selection of study, and supported the data framework. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yehia, N., Wahed, M.A. & Mabrouk, M.S. Clustering column-mean quantile median: a new methodology for imputing missing data. J. Eng. Appl. Sci. 69, 113 (2022). https://doi.org/10.1186/s44147-022-00148-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s44147-022-00148-7