
Abstract
The tumor microenvironment (TME) is crucial to neoplastic processes, fostering proliferation, angiogenesis and metastasis. Epigenetic regulations, primarily including DNA and RNA methylation, histone modification and non-coding RNA, have been generally recognized as an essential feature of tumor malignancy, exceedingly contributing to the dysregulation of the core gene expression in neoplastic cells, bringing about the evasion of immunosurveillance by influencing the immune cells in TME. Recently, compelling evidence have highlighted that clinical therapeutic approaches based on epigenetic machinery modulate carcinogenesis through targeting TME components, including normalizing cells’ phenotype, suppressing cells’ neovascularization and repressing the immunosuppressive components in TME. Therefore, TME components have been nominated as a promising target for epigenetic drugs in clinical cancer management. This review focuses on the mechanisms of epigenetic modifications occurring to the pivotal TME components including the stroma, immune and myeloid cells in various tumors reported in the last five years, concludes the tight correlation between TME reprogramming and tumor progression and immunosuppression, summarizes the current advances in cancer clinical treatments and potential therapeutic targets with reference to epigenetic drugs. Finally, we summarize some of the restrictions in the field of cancer research at the moment, further discuss several interesting epigenetic gene targets with potential strategies to boost antitumor immunity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
As one of the most threatening diseases hazardous to human health with limited therapeutic strategies, tumors are generally composed by a group of abnormally proliferative tumor cells which enmeshed in an extremely intricate ecosystem termed as the tumor microenvironment (TME). The TME is an aggregation of various non-neoplastic cells, extracellular matrix (ECM) that consists of multiple growth factors, chemokines and cytokines, along with the blood and lymphatic vascular networks [1]. Among which, the surrounding cells could be classified according to different phenotypes [2, 3], such as the immune cells including T and B lymphocytes and natural killer cells (NKs), the myeloid cells including dendritic cells (DCs), tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), the stroma cells including cancer-associated fibroblasts (CAFs) as well as some cancer stem cells (CSCs) and so on.
There is increasing evidence indicating that the favorable TME plays a crucial role in modulating the onset of carcinogenesis, tumor progression and metastasis [4]. As an example, it’s been demonstrated that TAMs could maintain an immune-suppressive microenvironment notably via the production of chemokines [5, 6], and the presence of CAF is a strong indicator of the poor clinical prognosis of various cancers [2, 7]. What’s more, the tumor cells could reversibly alter phenotypes of surrounding cells via cell-to-cell contacts, secretion of dissolvible cytokines and exosomes release, in ways to create the suppression of the immunosurveillance in TME [8, 9]. Consequently, developing therapeutic approaches targeting the TME has become one of the most attractive areas in cancer therapy [10].
Epigenetic regulation, identified as a heritable and reversible way to modify gene expression and chromatin structure without changing DNA sequence, is primarily initiated by the diverse profile of covalent modifications to nucleic acids and histone proteins [4, 11]. There are three main ways of epigenetic regulation: DNA methylation, histone modification, and non-coding RNA (ncRNA) alterations [12]. Epigenetic mechanism is widely acknowledged to affect all aspect of tumor progression [11, 12], further regarded as an essential hallmark of malignancy by breaking the balance between multiple oncogenic and tumor suppression gene pathways in cancer cells [13,14,15]. Furthermore, epigenetic modification also takes place in TME, contributing to the evasion of immunosurveillance by influencing the differentiation, infiltration, and activation of immune cells [16], such as switching immunogenic DCs into immunoregulatory DCs (regDCs), and promoting the polarization of TAMs from M1 to M2 phenotype [8]. Accordingly, epigenetic regulation, as a key modulator in the process of cancers, has been nominated as a potential clinic therapeutic approach in cancers.
In this review, we focus on the latest epigenetic events happened to TME, summarize the clinical treatments and potential therapeutic targets towards various cancers based on the epigenetic machinery. We outlook some prospective epigenetic gene targets, also discuss some of the limitations and doubts in the field of cancer research and epi-regulatory studies currently.
Fundamental modes of epigenetics
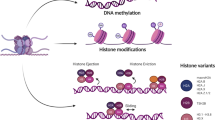
Due to the dramatically developed global proteomic and genomic technologies over the past few years, epigenetics has grown into one of the most active research projects in biology, involving numerous physiological and pathological processes. Epigenetic regulations have been thoroughly studied and summarized in various reviews [4, 13, 16,17,18,19], mainly consisting of the followings (Fig. 1). We’re going to briefly summarize some fundamental modes of epigenetics, especially concentrating on some classical regulatory molecules and pathways.

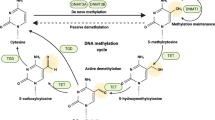
Basic regulatory mechanisms of epigenetic modification. Histone modification, including acetylation and methylation, occurs on the N-terminal tails of H3 and H4 subunits and leads to chromatin configuration alteration and subsequent transcriptional blockade. DNA methylation is a highly dynamic process in which DNMTs transfer methyl groups onto the 5’ position of cytosine in the CpG islands, whereas enzymes in the TET family can remove it. RNA methylation often occurs on the 6’ position of adenosine, and is also reversible under the modulation of the writer complex (METTL3, METTL14 and WTAP) and the erasers (FTO and ALKBH5) Non-coding, including miRNAs, lncRNAs and so on, can interfere the protein translation and participate in the pro-translational modification of proteins
DNA and RNA methylation
DNA methylation commonly happens with the covalent addition of a methyl group and is mostly transferred from S-adenosylmethionine (SAM) onto the C5 position of the cytosine ring (5mC) of DNA [20, 21]. In the mammalian genome, this modification is generally enriched within cytosine- guanine dinucleotide (CpG) site, which is present throughout the genome but majorly situated in gene regulatory regions [22, 23] such as gene promoters or transcription start sites [24]. The methylation of CpG islands, consisted by CpG sites and abundant repetitive sequences, is closely associated with the deregulation of cellular pathways and gene expression, particularly gene silencing, in many biological processes and various diseases including cancers [17]. For example, the hypermethylation of CpG islands in the promoter regions of tumor suppressor genes (TSGs) leads to TSG silencing, eventually result in tumorigenesis [25].
The state of DNA hypermethylation is catalyzed by DNA methyltransferases (DNMTs), including DNMT1, DNMT3A/3B along with DNMT3L. Among them, maintaining methylation on the new DNA strand predominantly during DNA-replication S phase is medicated by DNMT1, while de novo DNA methylation in the unmethylated genomic regions principally during embryonic development is catalyzed by DNMT3A/3B with the aid of DNMT3L [26, 27], the latter one who could expand the methylated pattern on DNA sequences and simultaneously activate the enzymatic ability of DNMT3A/3B [28]. On the contrary, the removal of the methyl group is catalyzed by the ten-eleven translocation (TET) family triggered by either active or passive mechanisms, known as the synergistic activation with the thymine DNA glycosylase (TDG) and the base-excision repair (BER) machinery [29, 30] or the DNA replication-dependent passive pathway [31], respectively. Therefore, as one of the most common epigenetic regulations, DNA methylation on both global and specific sites has been broadly considered as the promising biomarkers to forecast tumor phenotypes or overall survival or patient prognosis [32,33,34,35,36,37,38].
Except for DNA methylation, RNA methylation has gained increasing attention. Although all nucleotides composed RNA could be modified and interact with each other [39], the most abundant target is N6-methyladenosine(m6A), which refers to the methylation at the sixth nitrogen atom of RNA base A. Generally speaking, the m6A modification is enriched near stop codons of transcripts and in conserved regions of the 3′-untranslated regions (3′-UTRs) [40]. This process is catalyzed by m6A-methyltransferases, such as the complex of Methyltransferase-like (METTL)-3/14, Wilms’ tumor 1-associated protein (WTAP) and so on [41, 42], while de-catalyzed by the RNA demethylase α-ketoglutarate-dependent dioxygenase homolog 5 (ALKBH5) [43]. Except for those “writers” and “erasers”, the m6A group should be identified by various “readers” to exert multiple functions, which is mainly undertaken by YT521-B homology (YTH) domain family members including YTHDF1, YTHDF2, YTHDF3, YTHDC1 and so on [44]. The presence of m6A exerts a wide range of effects on RNA stability and function, subsequently modifying cellular processes and altering pathological conditions including the development of cancer [45, 46].
Histone modifications
As the fundamental subunits of chromatin, nucleosome encompasses four subtypes of histone proteins including H2A, H2B, H3 and H4 [47]. The posttranslational regulations of histone, containing acetylation, methylation, phosphorylation, ubiquitination and so on [48], mostly happened to histone H3 followed by H4 [49], could alter the accessibility of genes for the transcriptional machinery and ultimately modulate gene expression by disrupt chromatin structures [50]. Among those histone modifications, histone acetylation and methylation is the most pivotal and common types.
Catalyzed by histone acetyltransferases (HATs), the acetylation of histone lysine residues undergoes the convertible addition of acetyl groups in the lysine-rich N-terminal tails, consequently loosens the DNA-histone bonds and promotes the binding of transcription factor, mostly associated with the activation of transcriptional activities [13, 51]. On the contrary, catalyzed by histone deacetylases (HDACs), the deacetylation of histone facilitates compact wrapping of the DNA duplex around histones, subsequently linked to gene repression [13] and closely correlated with poor clinic prognosis, which has been widely verified in various cancers [52, 53]. According to the homology of sequence and the mechanism of catalyze, so far 18 mammalian HDACs have been identified and preliminarily classified into four classes: class I (HDAC1-3 and 8), class II (HDAC4-7, 9 and 10), class III (Sirt1-7) and class IV (HDAC11) [54].
Except for acetylation, histone methylation, the third major type of epigenetic modification [55], is mediated adversely by two major classes of enzymes respectively called histone methyltransferases (HMTs), which adding the methyl group to specific histone lysine or arginine residues, and histone lysine methyltransferases (HKMTs), which is opposed to this process [56]. Multiple methylation status and dynamic histone loci could elicit different outcomes, either repress or active gene expressions [57], and the dysregulation of which might play the fundamental roles in abnormal cellular proliferation, invasion, and metastasis during disease progression [58,59,60]. As an example, it’s been reported that the downregulation of lysine methyltransferase EZH2 resulted in the reduction of histone H3 lysine 27 (H3K27me3) in lung cancer [61], while the activation of histone H3 lysine 4 (H3K4) and the subsequent elevation of the antioxidant signaling pathway in prostate cancer was discovered owing to the lysine methyltransferase 7 (SetD7) [62].
Non-coding RNAs
Constituted by long non-coding RNAs (lncRNAs), microRNAs (miRNAs) and circular RNAs (circRNAs) [63], ncRNAs are indispensable epigenetic regulators for various physiological and pathological activities via impeding the transcription of messager RNAs (mRNAs) or binding to proteins [64]. As the most thoroughly studied ncRNAs, miRNAs modulate gene expression through repressing translation or promoting degradation of the target mRNA by binding to 3′-UTR of which, thus have been considered as the critical cogs in the onsets and maintenance of cancers [65,66,67] and correlated with poor clinical prognosis [68, 69]. It’s been well demonstrated that miRNAs have engaged in remodeling TME by being encapsulated within extracellular vesicles (EVs) or exosomes [70]. For example, EVs released from CAFs could promote the migration and the invasion of oral squamous cell carcinoma (OSCC) [70, 71]. As a consequence, it’s worth noting that tremendous tumor treatments involving nanoparticle-conjugated miRNA mimics have made significant progress [72], which would be detailly discussed in the following.
Except for miRNA, compelling evidence have displayed that lncRNA have close correlation with epigenetics by taking advantage of chromatin modification or other types of ncRNA, inducing aberrant gene expression and implicating tumorigenesis [73]. Furthermore, it’s been demonstrated that various lncRNAs have ability to regulate chemotherapy via modulating pharmacological process of chemical drugs in several digestive system cancers [74,75,76]. Wu., et al. [77] has verified that bladder cancer associated transcript-1 (BLACAT1), a novel gene regulator lncRNA, could provoke the oxaliplatin(OXA)-resistance in gastric cancer through combining with miR-361, providing a novel insight and a potential therapeutic mechanism towards cancer chemoresistance.
Collectively, diverse epigenetic mechanisms have been considered as a decisive factor in the tumorigenic processes, contributing to tumor initiation and propagation, immune escape, metastatic, angiogenesis and prognosis [78,79,80]. To date, it’s emerging that both individual and combinatorial use of epigenetic drugs have shown prominent clinic therapeutic effect, while it’s believing that a plethora of novel epigenetic strategies could pose a promising future to oncotherapy [81]. The detailed epigenetic modifications and epigenetic drugs targeted multiple components in TME within various tumor types would be discussed and summarized in the following (Fig. 2).

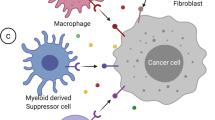
Epigenetic regulation of cell populations in TME. In TAMs, DNMT3A and DNMT3B suppress the secretion of IL-6 and TNF-a whereas HATs can rescue it by activating NF-κB pathway. Similarly, HDAC10 and EZH2 negatively regulate the expression of CCL2 by inhibiting SOCS3i and activating DNMT1, respectively. TGF-β1 signaling, the major force that drives CAF activation, is mediated by DNMTs, HDACs and several miRNAs. Of note, the m6A writer METTL3 can increase Col10A expression and promotes acquirement of myofibroblast characteristics. The cytotoxicity of NK cells is impaired due to EZH2-mediated CXCL10 decrease and miR-29 induced NKG2D deficiency, whereas DCs are activated by the m6A reader YTHDF1 and DNMTs. MDSC expansion and function rely on STAT3 activation, which can be suppressed by DNMTs or promoted by HDAC6/11. EZH2 and DNMTs inhibit MHC-I and CXCL9/10 on CD8+ T cells whereas METTL3/14 can activate IFN-γ signaling
Epigenetic regulations in TME
Epigenetic modulation in tumor cells
Epigenetic phenomena occurring directly in various tumor cells have attracted great attention all along, which could straightly dominate comprehensive aspects of tumor biology via impacting the phenotypes of tumor cells or the interaction with other components in TME. However, due to the multiple epi-regulated modes in various cancer types, it’s going to briefly illustrate some typical examples here (Table 1).
The influences that DNA methylation occur in cancer cells on tumor progression are mainly divided into two modes. Firstly, it’s been widely proven that aberrant methylation happened in specific gene sites may directly promote carcinogenesis via diverse mechanisms, such as in acute myeloid leukemia (AML) [120] and hepatocellular carcinoma (HCC) [121]. Additionally, TME homeostasis is also significantly impacted by abnormal DNA methylation in cancer cells, resulting in disordered interactions among various extracellular components [101, 122].
Except for DNA methylation, RNA methylation also plays an indispensable role in cancer biology, which has been detailly concluded and summarized in a 2018 review [123]. However, thanks to the research boom in recent years, multiple newly-discovered RNA methylation mechanisms have been clearly reflected, especially m6A modification. As a typical example, Fang, J., et al. has proven that almost all m6A methylation regulators were greatly correlated with the grades and stages of kidney renal clear cell carcinoma [90], and numerous research have revealed the nonnegligible role of m6A played in the progression of tumorigenesis via multiple mechanisms [102, 124, 125].
As for histone modification, the carcinogenicity mechanisms of both histone acetylation and methylation existing in cancer cells are similar to DNA epigenetics, which could be classified as the direct impact on oncogenic regulations, or the profound influence on surrounding components, especially the immune cells such as the CD8+ T cell in human ovarian cancers [126]. In addition to the classic epi-regulatory enzymes, more and more modulars in cancer cells have been discovered to show the potential epigenetic regulatory activities. In glioblastoma, for an instance, a gain-of-function screen revealed that the circadian locomotor output cycles kaput (CLOCK), generally enhancing the self-renew activities in the normal cells, exhibited as the potential histone acetyltransferase, subsequently regulating glioma cell proliferation and migration and supporting an immune suppressive TME [127].
Although studies targeting ncRNAs modification towards tumor cells needed further exploration, diverse research have illustrated that ncRNAs, notably miRNAs, appear to have a critical function in cancer cells. According to the landscape of epigenetically dysregulated ncRNAs in three breast cancer subtypes, ncRNAs contributed to biological functions that represent as a hallmark of different subgroups [103], hoping to become the potential biomarkers. Furthermore, upon the thorough insight into pan-cancer models, Cioffi, M., et al. found that miR-93 and miR-106b diminished tumor immunity and metastasis of cancer cells respectively by targeting the programmed death ligand 1 (PD-L1) which is known as the immune checkpoint inhibitors that could inhibit immune response [128], and downregulating the production of the chemoattractant CXCL12 [129], uncovering the strong immunosuppressive and metastatic phenotypes built by ncRNA.
Epigenetic modulation in cancer-associated fibroblasts (CAFs)
Fibroblasts, generally quiescent in normal tissues, could be activated and subsequently turned into myofibroblasts in tissue repair and scar formation [130], therefore playing a prominent role in the progression of various tumors which are classically considered as “wounds that do not heal” [131], becoming the most abundant component in TME and having an essential influence on tumor initiation, angiogenesis, metastasis and anticancer therapy [132].
The functional diversity and phenotypic heterogeneity of CAF has been extended with the help of epigenetics study, and the most in-depth investigation is the conversion of quiescent fibroblasts into activated CAF in various cancers [97, 133,134,135], while one of the most classic down-streaming targets is TGF-β1 signaling and its critical mediator SMAD3. With the recognition that various genes responsible for collagen synthesis and fibroblastic phenotypic transformation are activated by TGF-β1 in Smad3-dependent manner [136], it’s been uncovered that the global hypermethylation especially promoter hypermethylation-associated SMAD3 silencing could be attributed to the neoplastic transformation in lung cancer [104], and DNMT3B-catalyzed hypermethylation at miR-200 s promoter could establish CAF activation by sustaining autocrine TGF-β1 in breast cancer [137]. Furthermore, various miRNA in CAF, such as miR-199a/214 in the pancreatic stellate cells (PSCs) [138], miR-101 in hepatocellular carcinoma [139] and miR-145 in oral cancer [140] could modify TGF-β1 signaling and mediate CAF, contributing to the tumor angiogenesis and migration. Additionally, it’s been newly demonstrated that the expression of COL10A1, one of the abundant collagen families controlled by SMAD3, was increased by m6A modification via elevated METTL3 in CAF, thus accelerating cell proliferation and repressing apoptosis of the lung cancer model both in vitro and in vivo [141]. Moreover, applying an HDACs inhibitor CUDC-907 to lung tumor could notably repress the phosphorylation level of SMAD2/3 and promote histones acetylation, ultimately blocking lung fibrosis [142]. In conclusion, TGF-β1/SMAD3 signaling has become a potential therapeutic target in diverse cancers that should be tested in future studies.
Except for miRNAs mentioned above, there is multiple evidence proving that CAF share complex crosstalk with cancer cells through various miRNAs [143], among which the most abundant and well-studied miR-21 and miR-200 will be summarized here. The miR-21 is regarded as a pivotal prognostic marker in lung adenocarcinoma, whose expression in CAF is inversely correlated with patient survival [144]. The overexpression of miR-21 could activate CAF by driving its trans-differentiation [9] as well as the metabolic alteration [145]. Despite the direct effect on CAF, it’s been extensively studied that miR-21 modulates tumor biology including metastasis and invasion [146], such as the upliftingly proliferative capacity of the pancreatic ductal adenocarcinoma (PDAC) cells caused by upregulating expression of various modulars secreted from CAF which highly expressed miR-21 level, such as matrix metalloproteinases (MMP)-3/9, Platelet-derived growth factor (PDGF), and chemokine (C–C motif) ligand 7 (CCL-7) [147]. Meanwhile, the miR-200 family members also exhibit key roles in tumorigenesis and correlate tightly with disease prognosis, such as the accumulation of CXCL12 was miR-141/200a-dependent and related to the immunosuppressive functions of CAF in high-grade serous ovarian cancers (HGSOC) [148].
Epigenetic modulation in myeloid-derived suppressor cells (MDSCs)
The pathological process of tumors involves a myriad of complicated interactions between cancer and immune cells, where immune cells are responsible for both pro-tumorigenic and anti-tumorigenic regulation [149]. MDSCs, a heterogeneous cluster of immature myeloid cells, mainly exhibit the immunosuppressive role by suppressing the proliferation and anti-tumor function of T cells or NK cells to promote tumor development [150]. Thus, the deep investigation into the activation, recruitment and expansion of MDSCs via the epigenetic regulation might be the key to find out the immunological mechanisms in TME, helping to develop immunotherapies with improved therapeutic outcomes.
It's been discovered that MDSCs present a global DNA hypomethylation profile in ovarian cancer [151], and the repression of genes related to an immunogenic phenotype during MDSCs generation implies the establishment of a function-specific DNA methylation pattern. Among the complex DNA methylated targets, an elevated expression of transcription (STAT) 3, known as a master transcription factor mediating a wide range of cell functions of MDSCs [24], has been widely defined as the result of hypomethylated promoter silencing due to the attenuated DNMT3A/3B in various cancers including lung, pancreas and renal cancer [152,153,154,155]. Intriguingly, it’s been verified that after treating MDSCs with interleukin (IL)-6 in the vitro tumor-bearing mice model, the activation of STAT3 further elevated expression of DNMT1 and DNMT3B, resulting in the decreased downstream signaling pathway such as tumor necrosis factor-α (TNF-α)-induced and receptor interacting serine/threonine kinase 1 (RIP1)-dependent necroptosis, eventually promoting MDSCs survival and accumulation [99].
In addition to DNA methylation, IL-6, a potent inducer in tumor aggression and MDSCs expansion [156], could alter RNA methylation in MDSCs to mediate its function. Shang, W., et al. revealed that IL-6-mediated m6A upregulated a lncRNA pseudogene, Olfr29-ps1, which corrected negatively with miR-214-3p, thus modulated immunosuppressive function and differentiation of MDSCs in the inflammatory tumor environment [157]. What’s more, the tight correction between IL-6 signaling pathways and epigenetic regulations has furthermore disclosed by a clinical investigation of colorectal cancer, displaying that in tumor tissues refraining HDAC activation or neutralizing IL-6 substantially inhibits the expression of genes related to immunosuppression and chemotaxis of MDSCs [158]. Same as IL-6, IL-10 is considered as a key modulator of immune responses in MDSCs, whose expression is notably dependent on histone modification. It’s been widely verified that HDAC11 and HDAC6 were cooperatively recruited to the same region of the IL-10 gene promoter in antigen-presenting cells(APCs), as HDAC11 had been identified as a negative modifier of MDSCs expansion and function in mice model while HDAC6 had a contrary effect [159], thus controlling IL-10 transcription and the subsequent STAT3 expression, which targeted to amplify the immunosuppressive molecular S100A8 and Arginase 1 (Arg1) in MDSCs [160]. Thus, for efficacious immunotherapies, a better and deeper understanding of the relationship between those cytokines and signaling pathways regulated by epigenetics is still required.
Despite the direct-regulator miRNAs in MDSCs previously mentioned, multiple ncRNAs intricately modulate MDSCs through various pathways [161]. Here, we display some recent research. Firstly, the functions of MDSCs could be altered via exosomes containing miRNAs secreted from the cancer cells, such as glioma-derived exosomes can enhance the differentiation and propagation of MDSCs by miR-29a, miR-92a, miR-10a and miR-21 targeting to various proteins [162, 163], while the conversion and immunosuppressive phenotypes of MDSCs could be promoted by melanoma extracellular vesicles (EVs) including a group of miRs (miR-146a, miR-155, miR-125b and so on) [164]. Secondly, as Labib Salem, M., et al. [112] has addressed that by detecting the total RNAs in acute lymphoblastic leukemia (ALL) patients, many miRNAs were shared between MDSCs and regulatory T cells (Treg) and most of them were closely related to the immunoregulatory pathways, which reveals the role of miRNA as the biomarkers for the immunosuppressive TME. For example, the suppression of MDSCs was associated with the restoration of miR-195 and miR-16 by blocking PD-L1 in TME, while the elevated levels of those miRNAs were positively associated with the recurrence-free survival of prostate cancer patients [165].
Epigenetic modulation in Tumor-associated macrophages (TAMs)
Among the most abundant immune cells in TME, macrophages are originally antitumoral owing to the phagocytosis and direct tumor-kill [166], but once a tumor has progressed past an initial stage, they switching to play an exactly contrary role commonly deemed TAMs [167]. Depending on converging signals from the cellular environment, macrophages obtain a serious of activation phenotypes, broadly described as M1 macrophages, normally considered as antitumor, and M2 macrophages who contribute to pro-tumorigenesis via inducing immune evasion as well as supporting tumor cell invasion and expansion [168, 169]. As a result, the recruitment and polarization of TAMs, mediated by epigenetic regulation in both TME and malignant cells, is crucial for further exploration for the immunological therapy and antitumor outcomes.
Recent studies on epigenetic modification relevant to TAMs mainly focus on its regulation of phenotypic transformation and polarization. Inhabitation of DNMT3B has been recognized to play an essential role by repressing Arg1, one of M2 macrophage markers further related to its immunosuppressive functions [170], as well as blocking some inflammatory molecules such as TNF-α and IL-1β [171]. These actions significantly decrease immunosuppressive M2 macrophages populations while increase tumor-killing M1 macrophages and NK cells [172]. What’s more, with respect to RNA methylation, numerous vitro models and mice models have confirmed that by reducing the infiltration of M1 macrophages and Treg cells in TME, Mettl3-deficient mice displayed faster tumor growth and attenuated therapeutic efficacy of PD-1 checkpoint blockade. Following m6A sequencing revealed that YTHDF1-mediated translation of Sprouty-related, EVH1 domain containing 2 (SPRED2), which is responsible for inducing NF-κB and STAT3 through the ERK pathway, was impaired by the loss of METTL3 [173, 174], highlighting the m6A machinery as a potential cancer immunotherapy target.
M2 macrophages abundance in TME is also regulated by histone modification. As an example, USP24, a deubiquitinating enzyme upregulated in M2 macrophages of lung cancer cells, could stabilize HAT p300 thus increase H3 acetylation and NF-κB while decrease DNMT1, subsequently elevating TNF-α and IL-6 transcription, ultimately resulting in cancer malignancy [175]. Furthermore, CCL2, along with its ligand CCR2 expressed by monocytes/macrophages and T cells, exhibits underlying effect on TAM survival and function polarization, particularly responsible for immunosuppression [176]. In pancreatic ductal adenocarcinoma, HDAC5 could repress the suppressor of cytokine signaling 3 (SOCS3), a negative regulator of chemokine CCL2, contributing to elevated CCL2 and following recruitment of M2 macrophages [177]. In lung cancer, CCL2 expression was attenuated by EZH2-mediated H3K27me3 in the enhancer regions and DNMT1-mediated DNA methylation in the promoter regions [178], impeding M2-like phenotype of macrophages thereby facilitating metastasis and infiltration [179]. The same impact of CCL2/CCR2 axis on TAM further on tumor migration and invasion has also been demonstrated in breast cancer [180], constructing as a potential antitumor therapeutic target.
Regarding miRNAs, the regulatory mechanism towards TAM could be broadly divided into two categories. Firstly, it’s been widely reported that macrophages take up the exosomes or EVs contained miRNA which are secreted from malignant cells to alter its infiltration and polarization in TME [175, 181,182,183,184]. For example, excreted from breast cancer, miR-183-5p enhanced NF-κB signaling and promoted IL-1β, IL-6, and TNF-α expression in TAM [184]. Besides, macrophages could release the exosomes or EVs to remodel TME and modulate tumor progression [185,186,187,188,189,190,191], as miR-95 in TAM-derived exosomes could be directly uptaken by prostate cancer (PCa) cells and subsequent bound to JunB, a transcription factor governed cell development, consequently improving the proliferation, invasion and epithelial-mesenchymal transition of cancer cells, related to worse clinicopathological feature [192]. To draw a conclusion, the communication within cancer cells, TAM and other components in TME plays a critical role in tumor development and miRNAs occupy an essential position in this process.
Epigenetic modulation in dendritic cells (DCs)
Being one of the most critical members of antigen presenting cells (APCs) for adaptive immune response, DCs are responsible for the activation and maintenance of tumor cytotoxicity by T cells [193]. Although the function and phenotype of activated DCs is generally determined by co-stimulatory molecules (CD80, CD86) and chemokine receptors such as CCR7 [194, 195], the immunological function of DCs could be influenced by multiple factors within TME, which is partly modulated by epigenetic regulations and being considered as the potential targets to elevate immune surveillance and enhance anti-tumor immunity [196].
As 5-AZA, a classical DNMT inhibitor, conjunct with mice vaccination or DCs fusions could similarly elicit anti-tumor immunity via elevating the prefoliation of CD8+ T cells and increasing overall survival in diverse tumor models [197, 198], the strong connection between DNA methylation and immune-related genes in DCs has been uncovered [199]. Additionally, m6A modification in CD80 and CD40 in DCs could be recognized by Ythdf1, the m6A readers, therefore enhancing the translation of those essential immune transcripts that promotes DC activation and DC-induced T cell response in the physiological condition [200, 201]. In vitro model, CCR7 stimulation could promote a lncRNA named lnc-Dpf3 by removing m6A modification, therefore decreasing CCR7-mediated DCs migration along with attenuated adaptive immune responses [202, 203]. Moreover, genome-wide analyses of melanoma models revealed that H3K4me3 was increased on DC maturation genes such as CCR7 and CD86 [204], while De Beck, L., et al. [205] fund that a dual inhibitor of histone methyltransferase G9a and DNMTs could jointly increase the therapeutic efficacy of DC vaccination by arresting tumor cell cycle and delaying tumor growth of tumor-bearing mice, indicating the critical role of the cellular epigenetic regulation in modulating DC phenotypes, maturation and antitumor responses.
Same to the regulatory mechanism of miRNAs towards TAM, DCs also take up the exosomes or EVs secreted from malignant cells. It’s reported that after treating DCs with the pancreatic cancer-derived exosomes, ENST00000560647 was differentially expressed, which predicted to interact with miR-323-3p to directly suppress the invasion and metastasis of cancer cells [206]. Further investigation is required to explore the epigenetic regulatory function of ncRNAs in DCs.
Epigenetic modulation in tumor-infiltrating lymphocytes (TILs)
Regarding as one of the most essential components of the adaptive immune system, TILs are surrounding the cancer cells and become the foundation of current immunotherapy and the determination of the antitumor outcomes [207]. Within the complicated phenotypes and functional properties of TILs, NK cells and T cells, including CD8+ T cells, CD4+ T cells and Treg cells, are most common and closely correlated with the oncological prognosis [208]. Among them, NK cells could rapidly mediate cytotoxicity and involve in various tumor progression, metastasis and survival [209, 210]. CD8+ T cells are capable of producing antitumor cytokines and cytotoxic ligands including interferon-γ (IFN-γ) and TNF-α maintain their antiviral and antitumor functions [211], while Treg cells, whose high content is usually correlated with worse survival in multiple solid tumors, are renowned for their immunosuppressive role by modifying APCs and producing suppressive cytokines including IL-10, IL-35, and TGF-β to counteract CD8+ T cells [212]. Therefore, the in-depth investigation into the epigenetic regulatory mechanisms of TILs is critical for establishing effective antitumor immunotherapy.
As TME could be qualitatively characterized by gene expression-based methods, methylation data are now emergingly applied for molecular diagnosis, for example, the estimation of TILs abundance, infiltration and anti-tumor immune response have been successfully established by analyzing the global differential methylation in the central nervous system (CNS) tumors and glioblastomas [83, 95, 213]. Despite the global DNA methylation, gene expression of Th1 chemokines CXCL9 and CXCL10 was mediated by EZH2 and DNMT1, dampening CD8+ T cell infiltration in ovarian cancers [126], and the demethylation state of promoters in Th1 was proven as a significant mediator to IFN-γ production in colon cancer lymphocytes [214]. In addition to T cells, it’s been verified that hypermethylated CpG at the promoter locus induced the less mature subtype of tyrosine kinase 2 (TYK2), modifying the maturation and antitumor activity of NK cells in vitro model [198]. Those studies jointly indicate that DNA methylation role as a potential biomarker to significantly impact the infiltration and function of immune cells.
As a pro-inflammatory cytokine abundantly produced by TILs and in turn mediates their functions, IFN-γ is also controlled by RNA methylation. Inhibition of m6A in colorectal cancer via METTL3/4 deficiency resulted in the upregulation of IFN-γ-STAT1-IRF1 signaling through stabilizing the STAT1 and IRF1 mRNA, increasing secretion of IFN-γ, CXCL9, and CXCL10 further promoting infiltrating CD8+ T cells, contributing to the enhanced response to anti-PD-1 treatment [119]. Meanwhile, decreased m6A modification in Treg cells blocked SOCS mRNA, inducing the suppression of IL-2-STAT5 signaling pathway and ultimately losing the suppressive function of Treg cells over T cell proliferation which is beneficial to cancer immunotherapy [117]. What’s more, it’s been illustrated that RNA methylation also gives rise to phenotypic stability [89] or glycolytic-epigenetic reprogramming [96] in TILs.
With reference to histone methylation, EZH2 along with its target, H3k29me3, is playing an essential role in modulating the immunological functions of TCLs. The recruitment of T cells in human colon cancer tissue was impacted by CXCL9 and CXCL10, whose expression is regulated by H3K27me3 repression marks [126, 215]. In consistent to those studies, disrupting EZH2 activity in Tregs could selectively deplete T cells and alter their functions in murine colorectal tumor, enhancing the recruitment of CD8+ and CD4+ T cells and remodeling the TME [118], whilst high expression of EZH2 was shown to improve the escape from immunotherapy in neuroblastoma through transcriptional downregulation of major histocompatibility complex (MHC)-1 genes by H3K27me3 modification [216]. Besides, the cellular cytotoxicity and immunoregulatory function of NK cells are profoundly impacted by EZH2/CXCL10 axis as well [217,218,219]. As an example, Bugide, S., et al. [220] demonstrated that in hepatic tumor cells, HDAC10 was sufficient for EZH2 recruitment to the CXCL10 promoter, resulting in CXCL10 transcriptional repression and subsequent suppression of NK cells migration. Except for regulations in CXCL9/CXCL10, histone modification also controls immunological functions of TILs through various mechanisms, for example, Forkhead box P3 (Foxp3), a master lineage-specific phenotypic protein of Treg cells, was proven to play a new role in the TME by combined with HAT1 to control CC chemokine receptor 4 (CCR4) expression via providing provocative (K23 and K27) or repressive (K14 and K18) acetylation to H3, thus influencing the infiltration of Treg cells and anti-tumor immunity [221]. Moreover, the expression of Foxp3 itself was stabilized by acetylated histones at its promoter [222], indicating that histone epigenetic regulation has broad utility for modulating immune responses in TME.
The activation and function of TCLs is typically modulated by exosomes or EVs enriched with miRNA released from malignant cells. For instance, TNF- and CD45-targeting miRNAs such as miR-498, miR-181a/b, and miR-3187-3p could be released from melanoma cells to suppress the immunological function of CD8+ T cells [223], while delivering miR-142-5p from cervical squamous cell carcinoma (CSCC) cells to lymphatic endothelial cells (LECs) was considered as an integral component of the immune checkpoint, which could suppress and exhaust CD8+ T cells through AT-rich interactive domain-containing protein 2 (ARID2)-DNMT1-IFN-γ signaling and the elevated expression of indoleamine 2,3-dioxygenase (IDO) in the downstream [224]. What’s more, Zhou, J., et al. [225] found that TAM could also secrete exosomes contained miRNA including miR-29a-3p and miR-21-5p, which could be transfected into CD4+ T cells, leading to the direct suppression of STAT3 and an imbalance in Treg/Th17 cells, generating an immune-suppressive TME and associated with poor prognosis of Epithelial Ovarian Cancer (EOC) patients. The miR-29 is also identified being downregulated in NK cells in patients with T-cell acute lymphoblastic leukemia (T-ALL), with a reduction in IFN-γ production as well as NK group 2D (NKG2D) expression, who is one of the major activating receptors of NK cells binding to its ligands that are expressed on malignant cells, accompanied by accelerated ALL progression and decreased survival time [226, 227]. In conclusion, biological functions of miRNAs and their potential roles in modulating the differentiation, proliferation and activation of TLCs in TME along with remodeling immune homeostasis may profoundly influence tumor biology, as well as being potentially explored as therapeutic targets.
Epigenetic modulation in other components in TME
Except for the epigenetic regulation towards multiple malignant cells and non-neoplastic cells in TME mentioned above, numerous other components, conjointly with their dynamic interrelationships within TME, substantially participate in tumorigenic processes and immunotherapy prognosis. Here, we primarily focus and draw brief conclusions on B lymphocytes cells and cancer stem cells (CSC).
Being one of the most decisive members in adaptive immune cells, B cells are majorly distributed at the infiltrative boundaries of tumors and the lymphatic structures near TME. Presenting a dual role in tumor biology [228], B cells produce antibodies to directly kill tumor cells and generate pro-tumorigenic cytokines to activate immunosuppressive cells [229], whose abundance has been confirmed to predict disease progression and patient survival [230, 231]. Although limited research focus on epigenetic modification towards B cells in TME, several evidence proved that the differentiation and malignance of B cells partially controlled by epigenetics. As an example, engaged in the pathogenesis of Burkitt lymphoma (BL), Epstein-Barr virus (EBV) infection would lead to the considerably suppression of histone methylation including H3K9me3 and H3K27me3, following the elevated transcriptional competence of most genomic regions, which might possibly explain the active immune response triggered by B cells [232]. Similarly, represented by mutation of the EZH2 protein, epigenetic disorders have been widely observed in acute lymphoblastic leukemia (ALL), non-Hodgkin lymphomas (NHLs) and various cancers originating from B cells malignances [233], while epigenetic mechanisms further participant in the high variability of CD20, the general surface marker expressed by the majority of B cells, which have been broadly demonstrated by treating different malignant B lymphocytes with either DNA methyltransferase inhibitor (DNMTis) or histone deacetylase inhibitors (HDACis) [234,235,236]. Further investigation in the pathology and immune function altered by epigenetics towards B cells in various tumors is needed.
During the past decades, the profound role of CSCs in tumor initiation, progression and regression has become the hottest topic in the field of oncology, and epigenetics has been esteemed as the key mechanism. Characterized as self-renewal and multi-directional differentiation capacity [237], CSCs are a subset of small cells owning the potential to strongly induce tumorigenesis and reproduce cells types in cancers [238]. There are compelling evidence verifying that epigenetic regulations play an extremely crucial role in governing the stemness and self-renew ability of CSCs, along with the abundance and distribution of which in TME. For example, DNA demethylation has been certified as the vital factor to CSCs generation, differentiation and pluripotency [239,240,241,242], and histone modifications enzymes such as EZE2 and HDACs have extensively involved in regulating key signaling in CSCs, aberrantly activating Wnt signaling pathway [243, 244], Hedgehog (Hh) signaling pathway [245] and Notch signaling pathway [239, 246]. Furthermore, thorough explorations into miRNAs have revealed the complicated effect they have on tumor biology including sphere formation and chemoresistance. For example, miR-519d stimulated mitochondrial apoptotic pathway therefore enhanced the therapeutic effect of chemotherapy drug cisplatin [247], while ectopic miR-181b equivalently sensitized cisplatin-resistance cancers by targeting to Notch2 signaling pathway [248]. Diverse epigenetic mechanisms modifying the characteristics and functions of CSCs attributed to the oncogenesis in different cancers have been exhaustively explained and concluded in numerous reviews [238, 249,250,251].
Clinic trials and drugs targets epigenetic modifications in TME
Epigenetic (Epi)-drugs generally refer to artificial compounds that reverse the tumor-favoring epigenetic landscape in the molecular level, mainly by suppressing the essential enzymes for its establishment and maintenance [252]. Those inhibitors include DNMTis, HDACis, histone methyltransferase inhibitor (HMTis) and so on. Of note, some ncRNA-based drugs, especially small interfering RNAs (siRNAs), which refer to the 20–25-base-pairs oligonucleotides capable of arousing the degradation of mRNAs and subsequent gene silence [253], can directly involve in epigenetically regulating the expression level of target genes. Therefore, the siRNA-based drugs were also discussed as an exogenous epigenetic regulator that has therapeutic potential in cancer management (Fig. 3). Emerging epi-drugs have demonstrated inspiring therapeutic efficacy in hematological malignancies. However, most epi-drugs failed to achieve satisfying clinical outcomes in solid tumors due to blocked drug delivery caused by ECM deposition and competitive absorption by other components in TME. In contrast, the stromal cells and infiltrated immune cells in TME are directly exposed to the tissue fluid, making them more susceptible to the effect of epi-drugs. Therefore, TME components would be a promising target for epi-drugs in clinical cancer management (Table 2).

Pharmacological effects of epi-drugs on the components in TME. The inner circle represents basic pharmacological mechanisms of DNMTi, HDACi and siRNA. In brief, the former two epi-drugs inhibit essential enzymes (DNMT and HDAC) for the maintenance of aberrant epigenetic landscapes (DNA methylation and histone deacetylation), while the latter one directly binds to the target mRNA and blocks protein translation. Those effects eventually alter the tumor-favoring expression profiles in TME, which include but not limit to: 1) normalizing CAFs and anti-angiogenesis, 2) promoting antigen presenting capacity of DCs, 3) reversing the aberrant metastasis-promoting characteristics of NK cells and enhancing their antigen-dependent cell-mediated cytotoxicity, 4) blocking PD-L1, PD-1 and CTAL-4 to exploit the full anti-tumor potency of effector CD8 + T cells, 5) facilitating M2-to-M1 reprogramming process in TAMs, and 6) suppressing the recruitment of Tregs and MDSCs in TMEs while activating their inflammatory characteristics
Epi-drugs normalize CAF phenotype in TME
CAFs in TME often exhibit pro-tumorigenic characteristics due to tumor-induced phenotype reprogramming [293]. Such a process is highly dynamic and plastic that various anti-tumor drugs could easily affect it. Compared with cancer cells, the genomic landscape of CAFs is majorly reprogrammed by epigenetic modification rather than nucleotide mutation, thereby demonstrating higher responsiveness to epi-drugs [8]. Here, we summarized the existing epi-drugs that target CAFs in TME.
Many epi-drugs can directly normalize CAFs by reversing the tumor-induced epigenetic alterations. Among them, DNMTis (including 5-azacitidine, decitabine and eugenol) demonstrated robust therapeutic potential in remodeling the DNA methylation landscape of CAF-associated genes to promote CAF normalization. In 2012, Bian et al. first reported that decitabine could significantly decrease hypermethylation of the phosphatase and tensin homolog (PTEN) promoter in hepatic stellate cells (HSCs) and thus reversed fibrosis-promoting phenotypes by rescuing PTEN expression [254]. Since HSC-mediated fibrosis plays a crucial role in tumor progression and drug resistance of HCC, decitabine is a promising agent to enhance the efficacy of systematic treatment for advanced HCC. Similarly, Al-Kharashi et al. observed that decitabine and eugenol downregulated DNMT1 and DNMT3A through E2F1-mediated pathway [255], which blocked VEGF-A and IL-8 secretion and reversed the pro-angiogenic phenotype of CAFs in breast cancer [257]. Besides, Xiao et al. discovered that decitabine could rescue PDAC-induced methylation of a cytokine signaling suppressor termed suppressor of cytokine signaling 1 (SOCS1) in CAFs, thereby decreasing the secretion of insulin-like growth factor-1 (IGF-1) through JAK1/STAT3 pathway [256]. Intriguingly, combining 5-AZA with ruxolitinib (a JAK1/STAT3 inhibitor) could synergistically abrogate JAK1/STAT3 pathway in CAFs and potentiate CAF normalization in LC and HNSCC. Mechanistically, 5-AZA reversed the hypermethylation state of src homology 2 domain-containing protein tyrosine phosphatase 1 (SHP-1) promoter and increased the expression of endogenous JAK1/STAT3 inhibitor SHP-1, while ruxolitinib suppressed JAK1-induced phosphorylation of STAT3 [133]. Of note, a very recent study reported that DNMT3A could downregulate grainyhead like transcription factor 2 (GRHL2) and cadherin 1 (CDH1) while upregulated zinc finger E-box binding homeobox 1 (ZEB1) and vimentin (VIM) in PC3 cells, promoting CAF-induced epithelial-mesenchymal transition (EMT) and resulting in worse prognosis [294]. Since EMT is a hallmark of castration resistance, cancer stem cells generation, chemoresistance and worse prognosis for prostate cancer, targeting DNMT3A may be of great therapeutic value in improving the prognosis of advanced and metastatic prostate cancer.
Some DNMTis and HDACis could desensitize fibroblasts to CAF-promoting signaling such as TGF-β, thus blocking CAF maturation in TME. For example, a first-in-class HDACi termed CUDC-907 can suppress TGF-β1-induced CAF proliferation as a dual inhibitor of HDACs and PI3K/AKT pathway [142]. Intriguingly, Tang et al. discovered a positive feedback loop that facilitated TGF-β autocrine in breast cancer, where TGF-β1 could activate DNMT3B and thus suppressed the expression of miR-200a, miR-200b, miR-200c and miR-141, leading to enhanced sensitivity of CAFs to TGF-β1 [137]. Exposure of CAFs to decitabine significantly blocked TGF-β1 autocrine in CAFs and thereby normalized CAFs in breast cancer.
SiRNA-based drugs could precisely silence a specific CAF-driving gene and suppress an indicated pro-tumorigenic function. For instance, silencing IL11 and IL15 in CAFs by siRNAs inactivated STAT3 signaling and repressed CAF-induced angiogenesis in breast cancer [295]. In addition, the silence of siRNA-mediated yes1 associated transcriptional regulator (YAP1) in CAFs reduced the generation of collagen fibers and inhibited angiogenesis [296]. Of note, emerging evidence with regard to CAF heterogeneity indicated that certain subgroups of CAFs may serve as a double-edge sword rather than merely enhances tumor progression [293, 297]. In this context, precisely blocking their pro-tumor function by siRNA seems to be a better choice in CAF-targeting therapy.
Epi-drugs suppress neovascularization in TME
Tumor neovascularization is critical for sustained tumor growth and metastasis. Those immature vessel-like structures lead to hyperpermeability, poor perfusion, and hypoxia that further accelerates tumor progression and immune evasion. As mentioned above, DNMTis could refrain the secretion of multiple pro-angiogenetic cytokines such as VEGF-A and IL-8 [257]. However, Hegde et al. found that DNMT1 could inhibit IL-6-induced VEGFR2 upregulation in endothelial cells in breast cancer, suggesting that DNMTi may have a side effect on promoting angiogenesis [298]. For HDACis, a class I HDAC inhibitor termed entinostat could epigenetically activate anti‑angiogenic genes including serpin family F member 1 (SERPINF1) and thrombospondin 2(THBS2) and reduced the formation of vasculogenic mimicry (VM) in triple negative breast cancer, thereby inhibiting tumor metastasis and improving the overall survival of the murine breast cancer models [258].
Notably, siRNA-based drugs are essentially efficient in repressing angiogenesis. A phase II trial demonstrated that ALN-VSP02, an LNP-formulated siRNA drug targeting cadherin 16 (CDH16) and VEGF in neoplastic cells, could dramatically decrease microvessel density and vascular leakage in TME and worked well in repressing liver metastatic sites of EC [259]. Similarly, Tang et al. used the cationic liposomes to deliver the cascaded enzymes lactate oxidase/catalase and siVEGF, which both depleted lactate accumulation and silenced VEGF expression and exerted essential anti-angiogenetic effect for triple negative breast cancer [260]. Moreover, dual silencing of VEGFA in cancer cells and VEGFR1 in endothelial cells significantly suppressed VM formation in breast cancer, suggesting that codelivery of multiple siRNAs is a promising strategy to improve the anti-angiogenetic effects [261]. Intriguingly, a very recent study reported that silencing voltage-dependent anion-selective channel protein 1 (VDAC1) in neoplastic cells could not only inhibit tumor-mediated angiogenesis, but has an overall impact on a battery of TME-associated genes involved in extracellular matrix construction, and intercellular interaction in LC, highlighting the therapeutic potential of siVDAC1 in stromal-abundant solid tumors [262].
Endothelial cells in TME can also acquire tumor-promoting characteristics by epigenetic reprogramming. In this context, siRNA-based drugs demonstrated great efficacy in reversing the aberrant phenotype of endothelial cells. For example, microvessel leakiness is a major cause for drug delivery failure and chemotherapeutic resistance. Leung et al. discovered that silencing LIM domain containing preferred translocation partner (LPP) decreased microvessel leakiness and improved paclitaxel delivery to neoplastic cells, which dramatically improved chemotherapeutic efficacy of paclitaxel in ovarian cancer [265]. Notably, endothelial cells could facilitate tumor metastasis by expressing tumor adhesion ligands. An siRNA drug called Atu027 silenced the expression of a pro-metastatic ligand termed protein kinase N3 (PKN3) in endothelial cells and induced dramatic regression of distant metastasis of pancreatic cancer in phase I and II clinical trials [263, 264]. A very recent study found that the senescence associated secretory phenotype (SASP) of endothelial cells could promote tumor metastasis. SASP inhibition by silencing CXCL11 could significantly attenuate the migration and spheroid invasion of MDA-MB-231 cells in vivo [266].
Taken together, siRNA-based drugs displayed great advantages in suppressing angiogenesis and pro-tumorigenic phenotype of endothelial cells. Of note, synergistic therapy combining siRNAs with other sorts of epi-drugs may serve as a promising strategy to improve the clinical outcomes. However, there is still a long way to go in the development of siRNA-based drugs because most of the carrier particles fail to combine low cytotoxicity and high delivering efficacy simultaneously. Developing bionic nanoparticles with cell-specific ligands would contribute to overcoming the dilemma in clinical practice.
Epi-drugs repress the immunosuppressive components in TME
Immunosuppressive cell populations are widely distributed in TME, including MDSCs, Tregs and regulatory B cells (Bregs). Inborn with the capability to repress anti-tumor immunity, those immunosuppressive components dramatically impair the clinical efficacy of immunotherapy. One way to eliminate the infiltrated MDSCs in TME is apoptosis induced by epi-drugs. For example, decitabine decreased MDSC accumulation by disrupting DNA methylation of RIP1-dependent necroptotic genes and facilitating MDSC elimination in TME [85]. An HDACi called SAHA could induce apoptosis of Gr1+ MDSCs in breast cancer by increasing endogenous ROS in MDSCs [283]. Another HDACi called CG-745 increased IL-2 and IFN-γ, which could suppress M2 macrophages and MDSCs content [284]. Besides, epigenetic inhibition of immunosuppressive biomarkers can serve as another strategy to remodel the immunosuppressive characteristics of MDSCs. For instance, an HDACi called GNE-781 inhibited CBP/EP300-BRD and transformed MDSCs to an inflammatory phenotype by inactivating STAT pathway and repressing Arg1 and iNOS [286]. Intriguingly, a triple combination of 5-AZA, HDACi and α-PD-1 increased the infiltration of CD45+ immune cells, NK cells and active CD8+ T cells, while reducing tumor burden and extending survival through type I IFN signaling, while triple combination provided the best anti-tumor performance in the murine models [287]. However, Sahakian et al. discovered that, in contrast to their wild-type counterparts, tumor-bearing HDAC11-knockout mice displayed increased IL-10 expression and a more suppressive MDSC population [159]. In addition, another HDACi called trichostatin-A was also found to exert dual effects on tumor immunity. On one hand, trichostatin-A modulated the suppressive activity of infiltrating macrophages and inhibited the recruitment of MDSCs. On the other hand, trichostatin-A also upregulated PD-L1 expression on malignant cells, thereby limiting the beneficial therapeutic effects [285].
Unlike the infiltrated MDSCs migrating from other sites, most Treg cells in TME derive in situ due to tumor-mediated intentional reprogramming. In this context, epi-drugs could remarkably contribute to inhibiting Treg maturation and acquirement of immunosuppressive biomarkers [299]. For example, entinostat could reduce STAT3 acetylation by inhibiting HDAC1/3, leading to corollary repression of Foxp3 in Treg cells in renal and prostate cancer [278]. In addition, an EZH2-targeting HDACi called CPI-1205 drove the acquisition of pro-inflammatory functions of Treg cells in the TME, with a coordinate promotion in the recruitment of CD8+ and CD4+ effector T cells [123, 216, 279]. Similarly, another EZH2 inhibitor named Dznep depleted LMP1-HONE1 antigen-induced Tregs in vitro and led to promoting TAA-specific antitumor response in nasopharyngeal cancer [280]. Of note, a novel HDACi PLX51107 (inactivating the histone acetylated protein BRD4) was found to induce EZH2 deficiency in Treg cells. Surprisingly, PLX51107-treated Treg cells in vitro demonstrated a similar gene landscape to Foxp3-deficient Treg cells that lost the immunosuppressive function in melanoma [281, 282]. Compared with HDACis, DNMTis appeared to exert dual function in inhibiting Treg cell derivation in TME. A DNA demethylase ten-eleven translocation (TET) enzyme was found to increase the stability of Foxp3 expression in TGF-β-induced Treg cells through Tet2/Tet3 signaling, suggesting the possible application of DNMTis in repressing Treg cells [300]. However, a very recent study discovered that decitabine could increase Treg infiltration during HBV infection, probably by promoting Treg differentiation from naïve CD4+ T cells [301]. In additional, siRNA-based drugs could precisely block the Treg-activating ligands in TME. Knocking down the expression of membrane-associated phosphatidylinositol transfer protein 3 (PITPNM3) significantly reduced CCL18 recognition in intertumoral Tregs, which subsequently inhibited naive CD4+ T cell recruitment and enhanced immunotherapeutic efficacy of anti-PD-1 antibodies [302].
Epi-drugs facilitate M2-to-M1 repolarization of TAMs
As the pivotal orchestrator in TME, the functional status of TAMs could largely determine the activity of anti-tumor immune response [303]. In this context, M2-to-M1 reprogramming emerges as a robust way to reverse the immunosuppressive landscape of TME due to high plasticity of TAM phenotype in TME. Several epi-drugs have been reported to inhibit the M2-to-M1 repolarization in TME. Shi et al. reported that 5-AZA could epigenetically increase IL-6 expression and p65 phosphorylation, thus stimulating the activation of macrophages toward an M1-like phenotype and subsequent T cell recruitment in TME [273]. Intriguingly, siRNA drugs encapsuled in functionalized nanoparticles (NPs) have attracted great attention in M2-to-M1 repolarization. Compared with nude siRNAs, NP-mediated siRNA delivery can remarkably prevent premature drug degradation, reduce side effects and improve drug absorption. Moreover, multifunctional NPs can be designed to affect the immune-activating pathways and metabolic pattern in TME, which further contributes to reprogram M2-like TAMs. For example, Qian et al. developed M2-like TAM dual-targeting NPs modified with α-peptide and M2 macrophage binding peptide. Loading anti-colony stimulating factor-1 receptor (CS1FR) siRNA into these NPs, they observed significant decrease of IL-10 and TGF-β production and subsequent M2-TAM depletion in melanoma [274]. Another lipid nanoparticle composed of a novel pH-sensitive cationic lipid CL4H6 (CL4H6-LNPs) was developed to deliver siSTAT3, which significantly increased the infiltration of M1 TAMs and reversed TAM-induced angiogenesis in TME [277].
In addition, some smart nanodrugs, which are endowed with specific responsiveness to physicochemical property of TME, exhibit higher specificity and precision in controlling siRNA release. For instance, Song et al. created polyethylene glycol and mannose doubly modified trimethyl chitosan modified with citraconic anhydride grafted poly-based nanoparticles (PEG = MT/PC NPs) with intelligent pH-responsiveness to deliver siVEGF and siPIGF (siRNA for placental growth factor). This strategy achieved efficient gene silencing, promoted M2-to-M1 reprogramming in TAMs, and exerted robust suppression of breast cancer proliferation and lung metastasis [275]. Similarly, Xiao et al. designed a smart micellar nanodrug with M2-targeting peptides hidden in the pH-sheddable PEG corona so that the contents would only be released in the acidic TME. These intelligent NPs could co-deliver siIKKβ and STAT6 inhibitor AS1517499 to reprogram M2-like TAMs, without affecting the normal tissues and causing adverse effects [276]. Taken together, siRNA-based nanodrugs could effectively and specifically target TAMs in TME with high biocompatibility and multifunctional integration, thereby being the most promising strategy for M2-to-M1 repolarization in clinical application.
Epi-drugs reactivate DCs and NK cells in TME
DC vaccines are the newly emerging technique of tumor immunotherapy that boost antigen presentation in TME in order to exploit the full potential of the adaptive immune response to eliminate tumor cells [304]. Unfortunately, most DC vaccines failed to achieve satisfying clinical outcomes due to tumor-induced DC inactivation by epigenetic modification. Frikeche et al. observed that 5-AZA exposure significantly increased the expression of DC-promoting ligands CD40 and CD86 on mature DCs, and suppressed IL-10 and IL-27 secretion that impaired CD8+ T cytotoxicity [269]. Another study revealed that H3K79 demethylation by DOT1Li could reduce forkhead box M1 (FOXM1) expression and increase IL-12 production, thus promoting DCs maturation in breast cancer [271]. Intriguingly, Fragale et al.found that combinatory use of 5-AZA, romidepsin (an HDACi) and IFNα2 (ARI) remodeled the IFN signature and endowed DCs with a marked migratory capability in vivo. Those DCs actively participated in T cell cross-priming in tumor draining lymph nodes [270]. However, Liu et al. proposed an opposite perspective that DNMTi may impair the responsiveness of DCs to TLR3 or TLR4 signaling by increasing the expression level of TLR inhibitor SOCS1, abolishing polyinosinic-polycytidylic acid or LPS-induced DC maturation in TME [305]. Of note, siRNA pretreatment could dramatically potentiate the immunogenicity of DC vaccines, probably due to combinatory role where siRNAs both serve as the immunologic adjuvant and inhibit tumor-induced immunosuppressive phenotype of DCs. Pretreating DC vaccines with siIDO in vitro silenced expression of indoleamine 2,3-DiOxygenase (IDO, an immunosuppressive enzyme) and achieved better tumor-eliminating effects in the murine breast cancer models [272]. Since siRNA pretreatment in vitro could avoid the complex signaling interference in TME, it may serve as a robust strategy to unleash the anti-tumor potential of DC vaccines.
Similar to DCs, the anti-tumor activity of NK cells is often restricted in TME due to tumor-mediated epigenetic modification, whereas some epi-drugs could reactivate NK cells by suppressing NK-inhibiting gene expression. Chan et al. found that decitabine and 5-AZA could rescue NK cells from a metastasis-favoring phenotype in breast cancer by hypomethylation-induced repression of NK-inhibiting genes including T cell immunoreceptor with Ig and ITIM domains (TIGIT) and killer cell lectin like receptor G1 (KLRG1) [267]. In contrast, Kdm5a (an H3K4me3 demethylase) could resisted repressive chromatin configuration at the promoter of NK-activating cytokine suppressor SOCS1 and further impaired NK cell activity, whereas Kdm5a inhibitor re-silenced SOCS1 and rescued the NK-mediated immune response in TME [306]. In addition, siRNA-mediated silencing of the key intrinsic inhibitory NK cell molecules, including SHP-1, Cbl-b, and c-Cbl, could unleash NK cell activity to eliminate tumor cells, providing a new possibility of NK-targeting siRNA therapy [268]. Nevertheless, some epi-drugs like DNMTis and HDACi may have a negative impact on NK activation. 5-AZA and butyric acid (an HDACi) could increase the expression of NK-inactivating ligands called Siglec-7 on NK cells, thus inhibiting NK maturation in TME [307]. Besides, DNMTis could inhibit LPS-induced release of IL-1β, thereby impairing the production of NK-derived proinflammatory factors [308]. Further investigation should be conducted to evaluate the NK-activating effects by epi-drugs in clinical trials.
In summary, epi-drugs exhibit great potency in reinforcing the immunogenicity of DC vaccines and reactivating NK cells in TME, while siRNA-pretreated DC vaccines provide a novel insight into DC-based tumor immunotherapy. Of note, some DNMTis and HDACis appear to have dual effects on reactivating DC and NK cells, but the underlying mechanisms remain unknown. In this context, more efforts should be made to figure out the reason for such dual effects in order to avoid their immunosuppressive effects in clinical practice.
Combining epi-drugs with immune checkpoint inhibitors
The CD8+ T effector cells play a vital role in selective eradicating cancer cells. Nevertheless, overexpression of immune checkpoints on tumor cells, such as PD-L1 and CTLA-4, impairs the tumor-killing potency of CD8+ T cells in TME. In this context, immune checkpoint inhibitors (ICIs) are developed to exploit their full potency of tumor eradication and have demonstrated inspiring clinical efficacy in advanced melanoma and several hematological malignancies [309]. But when applied to treat cancers with immunodeficient TME, ICIs often failed to induce a durable anti-tumor response due to epigenetically induced CD8+ T anergy. Recent clinical trials have revealed that ICIs' therapeutic efficacy could be significantly increased by combining epi-drugs and ICIs in a synergistic therapy. According to one theory, epi-drugs increase the exposure of tumor-associated antigens and upregulate the expression of immune-activating genes in CD8+ T cells, which would synergistically wake up the disabled CD8+ T cells and encourage tumor cleavage in TME [310].
DNMTis could serve as a beneficial partner to improve the clinical efficacy of ICIs. On the one hand, DNMTis could upregulate MHC I and II molecules expression in cancer cells, which increased tumor immunogenicity and stimulated the anti-tumor response in TME. On the other hand, DNMTis could also suppress the infiltration of immunosuppressive components in TME [311]. In 2019, a two-arm phase II study enrolling 86 patients with refractory classic Hodgkin lymphoma demonstrated that patients who received decitabine plus camrelizumab achieved 71% complete remission compared with 32% in camrelizumab monotherapy group. Meanwhile, decitabine significantly reversed resistance to PD-1 inhibitors in patients with refractory classic Hodgkin lymphoma [312]. Recently, a novel DNMTi called CM-272 was found to improve the efficacy of PD-1 inhibitors in advanced bladder cancer by suppressing the expression of G9a and inducing immunogenic cell death [313]. Intriguingly, Srour et al. discovered that protein arginine methyltransferase 7 (PRMT7)-deficient melanoma demonstrated lower level of DNA methylation in the endogenous retroviral elements (ERVs), which enhanced gene expression regarding antigen process, the IFN pathways and chemokine secretion in CD8+T cells. Combining PRMT7 inhibitors with ICI therapy induced a strong anti-tumor T cell immunity in TME and restrained tumor growth in vivo [314].
“HDACis + ICIs” has been the most extensively studied combinatory strategy. Similar to DNMTis, HDACis upregulate the expression of TAP-1 and TAP-2 which participate in the intracellular transportation of tumor-associated antigen, thus enhancing the MHC-I presentation. In addition, HDACis could increase the secretion of IFN-α/β, and reverse T-cell exhaustion in TME. Furthermore, HDACis were found to decrease Foxp3 expression and impair suppressive function of Tregs [315]. In 2019, a phase I/Ib trial testing the combination of an anti-PD-1 antibody pembrolizumab and a pan-HDAC inhibitor vorinostat in 33 advanced NSCLC patients achieved 63% disease control rate and demonstrated preliminary antitumor activity in ICI-naïve patients [316]. Another phase Ib/II trial in 2021 also proved enhanced therapeutic efficacy of pembrolizumab in NSCLC patients with the assistance of the class I specific HDACi entinostat [317]. Similarly, the anti-PD-1 ICI nivolumab and the anti-PD-L1 ICI atezolizumab were also tested in combination with entinostat in several ongoing clinical trials, respectively (NCT02697630, NCT01928576, NCT02453620, NCT02708680) [318]. At present, an open-label phase I study is being conducted to explore the combinatory efficacy of HDAC6-specific inhibitor rocilinostat (ACY-241) with several anti-PD-1 and anti-CTLA4 ICIs in melanoma patients (NCT02935790) [319].
Except for clinical trials, a dual inhibitor for PI3K and HDAC termed BEBT-908 was developed to facilitate tumor ferroptosis by hyperacetylating p53 and upregulating the endogenous expression of MHC-I in tumor cells [320]. BEBT-908 significantly potentiated the effects of anti-PD1 ICIs and suppressed tumor growth in vivo. Qin et al. combined a specific inhibitor for histone lysine-specific demethylase (LSD1) with anti-PD1 ICIs, which significantly suppressed progression and pulmonary metastasis of breast tumor [321]. Similarly, Que et al. tested the combination of chidamide with an anti-PD-1 ICI toripalimab in metastatic sarcoma, which significantly promoted tumor regression and improved overall survival in the murine model [322]. Of note, Yang et al. developed a selective HDAC8 inhibitor which potentiated eradication of established hepatomas by anti-PD-L1 therapy, and the HSCC murine models receiving the combination therapy remained tumor-free for more than 15 months [323]. Another selective HDACi termed PLX51107 could also decrease the PD1 expression on CD8+ T cells, and enhanced therapeutic effects of ICIs in the murine melanoma models [282]. Interestingly, Tay et al. reported that HDAC3 could inhibit CD8+ T cell activation at the early stage, but was later required for persisting the activated status of CD8+ T cells, revealing the dual role of HDAC in regulating CD8+ T cell function [324]. However, it still remains unclear whether HDACis would impair the maturation of CD8+ T cells in TME.
Significantly, siRNA-based drugs demonstrated considerable efficiency in blocking the expression of immune checkpoints, suggesting potential as a novel immunotherapy strategy. For example, polyethylene glycol-chitosan-alginate NPs carrying anti-CTLA-4 siRNA could concomitantly block A2AR and CTLA-4 and facilitated CD8+ T-mediated tumor eradication in TME [288]. Another dual-responsive mPEG-PLA-Phis-ss-PEI polyplexes carrying anti-PD-L1 siRNA and resveratrol (a glycolysis inhibitor) promote CD8+ and CD4+ T cells infiltration by enhancing IFN-γ secretion and reprogramming the metabolic pattern in TME [289]. Moreover, FX@HP nanocomplex composed of fluorinated polymerized CXCR4 antagonism (FX) and paclitaxel-loaded human serum albumin (HP) could increase calreticulin on orthotopic lung tumor cells, which could work synergistically with the encapsuled anti-PD-L1 siRNA to facilitate T cell infiltration [290]. Based on the homing and penetrating peptides of breast tumor, a peptide assembling nano-delivery system was developed to deliver siPD-L1 and indoleamine 2,3-dioxygenase inhibitor in order to activate cytotoxic CD8+ T cells, and ultimately enhanced ICI-induced apoptosis of breast cancer cells [291]. A very recent study reported a polymeric nanoconjugate with innate immunogenicity, consisting of siPD-L1-based polyplexes, PEGylated hyaluronic acid as the CD44-targeting moiety, and ovalbumin as a model foreign antigen. Using such an immunostimulatory NP to carry siPD-L1 can remarkably potentiate DCs and tumor-reactive T cells [292].To draw a brief conclusion, DNMTis and HDACis could affect the expression landscape of immunoregulatory genes and target different immune cell population at the same time (Table 3). Rather than exerting catch-all effects, siRNA-based drugs are more precise and efficient in silencing the expression of immune checkpoints without excessive adverse effects. Notably, since tumor-induced epigenetic remodeling of immune cells plays a vital part in immunotherapeutic resistance, combining epi-drugs with ICIs could serve as a robust weapon to resist tolerance and improve clinical efficacy, representing as a promising trend in clinical immunotherapy.
Enhance the immune response via indirect mechanisms
Despite remodeling of the expression profile of immunoregulatory genes, some epigenetic drugs can indirectly affect tumor immunity via other mechanisms. One way is to induce type I interferon (IFN-I) response in TME. IFN-I response refers to a classic pro-inflammatory response that mobilizes the innate and adaptive immune cells to fight against infections and tumors. In details, various immune cells can produce the IFN-I superfamily ligands that specifically bind to a heterodimeric receptor comprised of IFNAR1 and IFNAR2, activate JAK/STAT pathway and ultimately initiate the transcription of IFN-inducible genes [325]. In TME, IFN-I response plays a crucial role in modulating cell growth, cell differentiation and tumor immune surveillance [326]. Moreover, recent studies have highlighted the potent anti-metastatic properties of IFN-I response in multiple sorts of solid tumors [325]. Intriguingly, epi-drugs such as DNMTis and siRNAs could initiate an unintended IFN-I response in TME through various mechanisms. Chiappinelli et al. reported that DNMTis induced cytosolic sensing of double-stranded RNA and caused IFN-I response in ovarian cancer [327]. In details, DNMTis reversed hypermethylated ERVs in neoplastic cells, which further activated dsRNA sensors TLR3 and melanoma differentiation-associated protein 5 (MDA5) and stimulated IFN-I response in TME. Furthermore, overexpression of ERVs in tumors significantly associates with durable clinical response of ICIs in melanoma patients. Another DNMTi named zebularine specifically sensitized the cGAS-STING pathway in response to DNA stimulation and upregulated interferon-stimulated genes, promoting infiltration of CD8+ T cells and NK cells in TME [328]. For most siRNAs, they often induce an unintended IFN-I response in which the exogenic siRNAs activate the dsRNA-dependent protein kinase termed protein kinase R (PKR) and upregulate IFN-β secretion [329]. Besides, siRNAs could be recognized by TLR8 in immune cells, triggering a TLR-mediated inflammatory response in TME [329, 330]. Nevertheless, since siRNAs are recognized as a foreign antigen in IFN-I response, whether the IFN-I response would attenuate the therapeutic effects of siRNA-based drugs still requires further investigation.
Despite the huge mutation burden carried by tumor cells, few tumors exhibit strong immunogenicity due to the impaired antigen processing system, such as silencing of MHC-I related genes, inactivated antigen-processing cells and decreased expression of costimulatory receptors [331]. Loss of tumor immunogenicity represents one of the core mechanisms in the initiation of immune evasion and immunotherapeutic resistance, posing a great challenge to improve tumor immunotherapy in clinical practice [332]. In this context, some epi-drugs were identified to improve tumor immunogenicity by increasing the expression of MHC-I and other costimulatory receptors on tumor cells. Pharmacological DNMT and HDAC inhibition reversed the repressive chromatin states of HLA-I genes, thereby increasing HLA-I expression on prostate cancer cells and making it more visible to immune surveillance [333]. In addition, HDACis such as Trichostatin A and VPA increased expression of antigen-processing-associated genes (TAP, LMP, tapasin genes and MHC I) on melanoma cells and other malignancies [334, 335]. Of note, a dual inhibitor of HDACs and DNMTs called CM-272 increased therapeutic efficacy of TCR-mediated DC vaccination, which further improved overall survival of B16-OVA tumor-bearing mice [205]. Similarly, decitabine and two HDACis (valproic acid and suberoylanilide hydroxamic acid) coordinately increased tumor antigen expression in malignant pleural mesothelioma and facilitated the therapeutic efficiency of ICIs [336].
Intriguingly, HDACis could enhance NK cytotoxicity by increasing expression of ligands required for the antibody-dependent cellular cytotoxicity (ADCC) on tumor cells. For example, CUDC-907 (a dual HDAC and PI3K inhibitor) caused dramatic regression of chemo-resistant multiple myeloma cell lines by upregulating natural killer group 2D (NKG2D) ligands and enhancing the ADCC effects in multiple myeloma cells [337]. A pan-HDAC inhibitor called panobinostat increased the expression of genes associated with cell adhesion, which guaranteed the process of NK-tumor conjugation and contributed to the increased tumor cytolysis [338]. Cho et al. monitored the effects of six selective HDACis on the expression of NKG2D and MHC-I molecules in lung cancer cells, and discovered that HDAC1 and HDAC2 might be the key orchestrators of NKG2D expression in lung cancer [339]. Mechanistically, HDACis stretch the chromatin structure of the promoter region and increased the accessibility for interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) gene. Consequently, the increased levels of IFIT1 augmented the IFIT1-mediated IRF1, STAT4, and STING pathways and promoted NK cell-mediated IFN-γ production and cytotoxicity [340].
Metabolism pattern of TME components has a profound impact on the activity and functional landscape of immune cells. In details, tumor-derived metabolites can involve in the immunoregulatory signaling crosstalk and remodel the functional state of immune cells [341]. Besides, metabolic competition in the tumor ecosystem limits nutrient availability of immune cells, leading to microenvironmental acidosis and the subsequent impairment of immune cell function [342]. Some epi-drugs are capable of tumor metabolic reprogramming to stimulate the anti-tumor immunity. For example, tumor-derived cytokines could disrupt methionine metabolism in CD8+ T cells, causing methylation at H3K79me2 and impairing T cell immunity [88]. In contrast, blocking tumor SLC43A2 restored H3K79me2 in T cells, thereby boosting their innate potency of tumor eradication. In addition, m6A demethylation mediated by FTO elevated the transcription factors such as C/EBPβ, JunB and c-Jun, in neoplastic cells, which allowed the rewiring of glycolytic metabolism. Indeed, FTO inhibitor Dac51 impaired the glycolytic activity of neoplastic cells and indirectly restored the activity of CD8+ T cells [343].
Outlook
Despite the fact that hundreds of studies have recently concentrated on the role of epigenetic regulation in a variety of tumors along with their microenvironment, revealing the profound influences on tumor initiation, metabolism, angiogenesis, immunosuppression and prognosis, leading to the development of numerous epi-drugs targeting some specific key molecules, further confirming the unique and distinct role of these epi-therapeutic modalities compared with traditional chemotherapy and radiotherapy, however, many questions are waiting to be resolved.
Recent years, increasing studies have uncovered that some ordinary genes regulating normal physiological activities or some non-coding genes silencing in normal tissues may possess potential epigenetic regulatory functions or may have abnormal epigenetic regulatory effects under malignant situations. Except for CLOCK, generally relevant to circadian rhythms but exhibited as the histone acetyltransferase in glioblastoma as mentioned before [127], Alvisi., et al. [344] lately manifested that in intrahepatic cholangiocarcinoma, the high enrichment of a novel transcription factor in tumor-infiltrating Tregs, mesenchyme homeobox 1 (MEOX1), was closely related to EZH2, further reprogram circulating Tregs. Although relevant research is temporarily limited right now, the conventional field of epigenetics may be greatly impacted by increased research on those potential genes, and the tumor landscape may benefit greatly from this innovation.
In addition to the potential epigenetic targets, countless limitations in the traditional field of epigenetic regulation still need further breakthrough. Taking ncRNAs as an example, growing attention has paid to circRNAs, a newly-discovered covalently closed ncRNAs [345] who have been shown to involved in the initiation and regulation of various cancers [346,347,348]. Serve as a competing endogenous RNA, circRNAs have been explored to regulate splicing or transcription, bind or sequester proteins, especially regarded as the miRNA sponge [349,350,351]. As an illustration, circRNA NOP2/Sun RNA methyltransferase 2(circNSUN2), prominently elevated in the cytoplasm of colorectal carcinoma (CRC) cells under m6A modification, stabilizes the structure of high mobility group AT-hook 2 (HMGA2) mRNA via interacting with an RNA-binding protein called Insulin-Like Growth Factor 2 mRNA-Binding Protein 2 (IGF2BP2), ultimately enhancing the aggressive of CRC cells [352]. Meanwhile, by sponging miR-181a-5p, circNSUN2 increased the expression of Rho-associated coiled-coil-containing protein kinase 2 (ROCK2), a key molecule in cancer growth, subsequently improve the proliferation and migration of CRC cells [353] (Fig. 4). As a result, continuous exploration of such traditional epigenetic regulatory areas will not only exceedingly increase the continuous in-depth understanding of epigenetics, also optimize the development and efficacy of cancer-target drugs.

CircRNA (CircNSUN2) enhances the malignant biological behavior of CRC via two mechanisms. Catalyzed by METTLE3 and recognized by YTHDC1, circNSUN2 is exported from the nuclear to the cytoplasm in an m6A methylation-dependent manner. On the one hand, elevated cytoplasmic circNSUN2 prominently promotes the stability of HMGA2 mRNA via interaction with IGF2BP2, ultimately resulting in the aggressive nature of CRC cells. On the other hand, by sponging miR-181a-5p, circNSUN2 represses the miRNA blocking of ROCK translation, upregulates the expression of ROCK and leads to the increased proliferation and migration of CRC
Although targeted epigenetic therapy is fruitful, epi-drugs are not broadly applicated to diverse tumors, with their unsatisfactory efficacy partly due to the low bioavailability, poor stability and relatively unbearable toxicity, as well as irregularities in dosing time and dose, owning plentiful challenges in clinic applications. For example, despite the emerging m6A-associated targets of therapeutic value have been identified in different types of infiltrated immune cells, and METTL3 has been widely verified to deeply impact tumor biology and TME harmony as the predominant catalytic subunit of m6A writer complex [117, 173, 174, 200, 201], only several small molecules targeting m6A-associated enzymes were evaluated in some preclinical trials [125, 186]. Taking together, there remaining huge blanks in the relevant area, thus developing drugs to rescue the activity or to mimic the biological function of those m6A enzymes may provide a novel insight into tumor immunotherapy.
From another perspective, all of those above aspirations should be exceedingly assisted by miscellaneous high-throughput, highly sensitive and domain-wide epigenetic research techniques, such as DNA methylation mapping of cells by genome-wide/precision DNA methylation and hydroxymethylation sequencing, capturing genome-wide interactions and high-level structures of genetic elements using various sensitive spatial genomics techniques, and improving the precision to the single-cell level to analyze the heterogeneity of epi-modifications in different cell subpopulations under complex TME, etc. Further enhancement of these research techniques and analytical methods will be of great help in elucidating TME as a key regulatory component of the tumorigenic process.
In summary, more and more evidence prove that epigenetic events play roles in tumor progression, maintenance, metastasis and prognosis, also take controls in TME remodel and regulation via complicated and dynamic mechanisms, while the potential epi-drugs along with its combination with traditional immunotherapy has rapidly grown into the prospective pattern for cancer treatment. Going forward, there are great expectations provide deeper insight into the extensive exploration and further refinement of the epigenetic processes, supplying boot opportunities for improved and newly explored therapeutic interventions in cancer.
Availability of data and materials
All data used to support the findings of this study are included within the article.
References
Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19:776–800. https://doi.org/10.1038/s41573-020-0077-5.
Dos Santos ES, Wagner VP, Cabral Ramos J, Lambert DW, Castilho RM, Paes Leme AF. Epigenetic modulation of the tumor microenvironment in head and neck cancer: Challenges and opportunities. Crit Rev Oncol Hematol. 2021;164:103397. https://doi.org/10.1016/j.critrevonc.2021.103397.
Hui L, Chen Y. Tumor microenvironment: Sanctuary of the devil. Cancer Letters. 2015;368:7–13. https://doi.org/10.1016/j.canlet.2015.07.039.
Dai E, Zhu Z, Wahed S, Qu Z, Storkus WJ, Guo ZS. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol Cancer. 2021;20:171. https://doi.org/10.1186/s12943-021-01464-x.
Mamrot J, Balachandran S, Steele EJ, Lindley RA. Molecular model linking Th2 polarized M2 tumour-associated macrophages with deaminase-mediated cancer progression mutation signatures. Scand J Immunol. 2019;89:e12760. https://doi.org/10.1111/sji.12760.
Wang D, Yang L, Yue D, Cao L, Li L, Wang D, et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019;452:244–53. https://doi.org/10.1016/j.canlet.2019.03.040.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131. https://doi.org/10.1186/s12943-021-01428-1.
Garcia-Gomez A, Rodríguez-Ubreva J, Ballestar E. Epigenetic interplay between immune, stromal and cancer cells in the tumor microenvironment. Clin Immunol. 2018;196:64–71. https://doi.org/10.1016/j.clim.2018.02.013.
Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669–82. https://doi.org/10.1038/nri3902.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50. https://doi.org/10.1038/s41591-018-0014-x.
Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–8. https://doi.org/10.1016/j.cell.2007.02.006.
Dawson Mark A, Kouzarides T. Cancer Epigenetics: From Mechanism to Therapy Cell. 2012;150:12–27. https://doi.org/10.1016/j.cell.2012.06.013.
Lodewijk I, Nunes SP, Henrique R, Jerónimo C, Dueñas M, Paramio JM. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin Epigenetics. 2021;13:63. https://doi.org/10.1186/s13148-021-01046-0.
Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, et al. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–92. https://doi.org/10.1126/science.1083558.
Jun HJ, Woolfenden S, Coven S, Lane K, Bronson R, Housman D, et al. Epigenetic regulation of c-ROS receptor tyrosine kinase expression in malignant gliomas. Cancer Res. 2009;69:2180–4. https://doi.org/10.1158/0008-5472.Can-08-3351.
Cao J, Yan Q. Cancer Epigenetics, Tumor Immunity, and Immunotherapy. Trends Cancer. 2020;6:580–92. https://doi.org/10.1016/j.trecan.2020.02.003.
Sylvestre M, Tarte K, Roulois D. Epigenetic mechanisms driving tumor supportive microenvironment differentiation and function: a role in cancer therapy? Epigenomics. 2020;12:157–69. https://doi.org/10.2217/epi-2019-0165.
Perrotti D, Silvestri G, Stramucci L, Yu J, Trotta R. Cellular and Molecular Networks in Chronic Myeloid Leukemia: The Leukemic Stem, Progenitor and Stromal Cell Interplay. Curr Drug Targets. 2017;18:377–88. https://doi.org/10.2174/1389450117666160615074120.
Marks DL, Olson RL, Fernandez-Zapico ME. Epigenetic control of the tumor microenvironment. Epigenomics. 2016;8:1671–87. https://doi.org/10.2217/epi-2016-0110.
Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37:1012–27. https://doi.org/10.1016/j.tig.2021.05.002.
Hon GC, Song CX, Du T, Jin F, Selvaraj S, Lee AY, et al. 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol Cell. 2014;56:286–97. https://doi.org/10.1016/j.molcel.2014.08.026.
Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. https://doi.org/10.1038/nrg.2017.80.
Chen Z, Zhang Y. Role of Mammalian DNA Methyltransferases in Development. Annu Rev Biochem. 2020;89:135–58. https://doi.org/10.1146/annurev-biochem-103019-102815.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74. https://doi.org/10.1038/nri2506.
Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16 Spec No 1:R50–59. https://doi.org/10.1093/hmg/ddm018.
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–57. https://doi.org/10.1016/s0092-8674(00)81656-6.
Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908–12. https://doi.org/10.1038/nature06397.
Schmitz RJ, Lewis ZA, Goll MG. DNA Methylation: Shared and Divergent Features across Eukaryotes. Trends Genet. 2019;35:818–27. https://doi.org/10.1016/j.tig.2019.07.007.
Schübeler D. Function and information content of DNA methylation. Nature. 2015;517:321–6. https://doi.org/10.1038/nature14192.
Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–5. https://doi.org/10.1126/science.1170116.
Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18:517–34. https://doi.org/10.1038/nrg.2017.33.
Yu X, Cen L, Chen YA, Markowitz J, Shaw TI, Tsai KY, et al. Tumor Expression Quantitative Trait Methylation Screening Reveals Distinct CpG Panels for Deconvolving Cancer Immune Signatures. Cancer Res. 2022;82:1724–35. https://doi.org/10.1158/0008-5472.Can-21-3113.
Hu Z, Xue C, Zheng J, Lu X, Li J, Dong H, et al. Hyper-Methylated Hub Genes of T-Cell Receptor Signaling Predict a Poor Clinical Outcome in Lung Adenocarcinoma. J Oncol. 2022;2022:5426887. https://doi.org/10.1155/2022/5426887.
Liu XP, Ju L, Chen C, Liu T, Li S, Wang X. DNA Methylation-Based Panel Predicts Survival of Patients With Clear Cell Renal Cell Carcinoma and Its Correlations With Genomic Metrics and Tumor Immune Cell Infiltration. Front Cell Dev Biol. 2020;8:572628. https://doi.org/10.3389/fcell.2020.572628.
Feng Z, Liu Z, Peng K, Wu W. A Prognostic Model Based on Nine DNA Methylation-Driven Genes Predicts Overall Survival for Colorectal Cancer. Front Genet. 2021;12:779383. https://doi.org/10.3389/fgene.2021.779383.
Xiao M, Liang X, Yan Z, Chen J, Zhu Y, Xie Y, et al. A DNA-Methylation-Driven Genes Based Prognostic Signature Reveals Immune Microenvironment in Pancreatic Cancer. Front Immunol. 2022;13:803962. https://doi.org/10.3389/fimmu.2022.803962.
Qin S, Liu G, Jin H, Chen X, He J, Xiao J, et al. The Dysregulation of SOX Family Correlates with DNA Methylation and Immune Microenvironment Characteristics to Predict Prognosis in Hepatocellular Carcinoma. Dis Markers. 2022;2022:2676114. https://doi.org/10.1155/2022/2676114.
Muraro E, Vaccher E, Furlan C, Fratta E, Fanetti G, Fae DA, et al. Predictive Value of CD8 Expression and FoxP3 Methylation in Nasopharyngeal Carcinoma Patients Treated with Chemoradiotherapy in a Non-endemic Area. Pathol Oncol Res. 2020;26:2459–67. https://doi.org/10.1007/s12253-020-00859-3.
Motorin Y, Helm M. RNA nucleotide methylation. Wiley Interdiscip Rev RNA. 2011;2:611–31. https://doi.org/10.1002/wrna.79.
Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2022. https://doi.org/10.1038/s41576-022-00534-0.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5. https://doi.org/10.1038/nchembio.1432.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46. https://doi.org/10.1016/j.cell.2012.05.003.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. https://doi.org/10.1016/j.molcel.2012.10.015.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20. https://doi.org/10.1038/nature12730.
He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Mol Cancer. 2019;18:176. https://doi.org/10.1186/s12943-019-1109-9.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88. https://doi.org/10.1186/s12943-020-01204-7.
Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15:703–8. https://doi.org/10.1038/nrm3890.
Bowman GD, Poirier MG. Post-translational modifications of histones that influence nucleosome dynamics. Chem Rev. 2015;115:2274–95. https://doi.org/10.1021/cr500350x.
Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. 2013;38:621–39. https://doi.org/10.1016/j.tibs.2013.09.004.
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. https://doi.org/10.1038/nrg2905.
Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–8. https://doi.org/10.1038/ng1966.
Guo P, Chen W, Li H, Li M, Li L. The Histone Acetylation Modifications of Breast Cancer and their Therapeutic Implications. Pathol Oncol Res. 2018;24:807–13. https://doi.org/10.1007/s12253-018-0433-5.
Calcagno DQ, Wisnieski F, Mota E, Maia de Sousa SB, Costa da Silva JM, Leal MF, et al. Role of histone acetylation in gastric cancer: implications of dietetic compounds and clinical perspectives. Epigenomics. 2019;11:349–362. https://doi.org/10.2217/epi-2018-0081.
Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. https://doi.org/10.1016/j.canlet.2008.08.016.
Zhao Z, Shilatifard A. Epigenetic modifications of histones in cancer. Genome Biol. 2019;20:245. https://doi.org/10.1186/s13059-019-1870-5.
Dimitrova E, Turberfield AH, Klose RJ. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015;16:1620–1639. https://doi.org/10.15252/embr.201541113.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. https://doi.org/10.1016/j.cell.2007.02.005.
Grønbaek K, Hother C, Jones PA. Epigenetic changes in cancer. APMIS. 2007;115:1039–59. https://doi.org/10.1111/j.1600-0463.2007.apm_636.xml.x.
Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–8. https://doi.org/10.1038/nature12751.
Lawrence M, Daujat S, Schneider R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016;32:42–56. https://doi.org/10.1016/j.tig.2015.10.007.
Li Z, Xu L, Tang N, Xu Y, Ye X, Shen S, et al. The polycomb group protein EZH2 inhibits lung cancer cell growth by repressing the transcription factor Nrf2. FEBS Lett. 2014;588:3000–7. https://doi.org/10.1016/j.febslet.2014.05.057.
Wang C, Shu L, Zhang C, Li W, Wu R, Guo Y, et al. Histone Methyltransferase Setd7 Regulates Nrf2 Signaling Pathway by Phenethyl Isothiocyanate and Ursolic Acid in Human Prostate Cancer Cells. Mol Nutr Food Res. 2018;62:e1700840. https://doi.org/10.1002/mnfr.201700840.
Yu B, Yu X, Xiong J, Ma M. Methylation Modification, Alternative Splicing, and Noncoding RNA Play a Role in Cancer Metastasis through Epigenetic Regulation. Biomed Res Int. 2021;2021:4061525. https://doi.org/10.1155/2021/4061525.
Sullenger BA, Nair S. From the RNA world to the clinic. Science. 2016;352:1417–20. https://doi.org/10.1126/science.aad8709.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. https://doi.org/10.1038/nrg1379.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. https://doi.org/10.1038/nrc1997.
Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. https://doi.org/10.1038/nrc1840.
Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007;8:R214. https://doi.org/10.1186/gb-2007-8-10-r214.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. https://doi.org/10.1038/nature03702.
Day TA, Shirai K, O’Brien PE, Matheus MG, Godwin K, Sood AJ, et al. Inhibition of mTOR Signaling and Clinical Activity of Rapamycin in Head and Neck Cancer in a Window of Opportunity Trial. Clin Cancer Res. 2019;25:1156–64. https://doi.org/10.1158/1078-0432.Ccr-18-2024.
Dourado MR, Guerra ENS, Salo T, Lambert DW, Coletta RD. Prognostic value of the immunohistochemical detection of cancer-associated fibroblasts in oral cancer: A systematic review and meta-analysis. J Oral Pathol Med. 2018;47:443–53. https://doi.org/10.1111/jop.12623.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22. https://doi.org/10.1038/nrd.2016.246.
Zhou Y, Sun W, Qin Z, Guo S, Kang Y, Zeng S, et al. LncRNA regulation: New frontiers in epigenetic solutions to drug chemoresistance. Biochem Pharmacol. 2021;189:114228. https://doi.org/10.1016/j.bcp.2020.114228.
Hahne JC, Valeri N. Non-Coding RNAs and Resistance to Anticancer Drugs in Gastrointestinal Tumors. Front Oncol. 2018;8:226. https://doi.org/10.3389/fonc.2018.00226.
Feng W, Su Z, Yin Q, Zong W, Shen X, Ju S. ncRNAs associated with drug resistance and the therapy of digestive system neoplasms. J Cell Physiol. 2019;234:19143–57. https://doi.org/10.1002/jcp.28551.
Wang Y, Zhang D, Wu K, Zhao Q, Nie Y, Fan D. Long noncoding RNA MRUL promotes ABCB1 expression in multidrug-resistant gastric cancer cell sublines. Mol Cell Biol. 2014;34:3182–93. https://doi.org/10.1128/mcb.01580-13.
Wu X, Zheng Y, Han B, Dong X. Long noncoding RNA BLACAT1 modulates ABCB1 to promote oxaliplatin resistance of gastric cancer via sponging miR-361. Biomed Pharmacother. 2018;99:832–8. https://doi.org/10.1016/j.biopha.2018.01.130.
Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–25. https://doi.org/10.1038/s41568-019-0144-6.
Senthebane DA, Jonker T, Rowe A, Thomford NE, Munro D, Dandara C, et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int J Mol Sci. 2018;19. https://doi.org/10.3390/ijms19102861.
Senthebane DA, Rowe A, Thomford NE, Shipanga H, Munro D, Mazeedi M, et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int J Mol Sci. 2017;18. https://doi.org/10.3390/ijms18071586.
Ghasemi S. Cancer’s epigenetic drugs: where are they in the cancer medicines? Pharmacogenomics J. 2020;20:367–79. https://doi.org/10.1038/s41397-019-0138-5.
DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep. 2019;27:502-513.e505. https://doi.org/10.1016/j.celrep.2019.03.037.
Bam M, Chintala S, Fetcko K, Williamsen BC, Siraj S, Liu S, et al. Genome wide DNA methylation landscape reveals glioblastoma's influence on epigenetic changes in tumor infiltrating CD4+ T cells. Oncotarget. 2021;12:967–981. https://doi.org/10.18632/oncotarget.27955.
Ai L, Mu S, Sun C, Fan F, Yan H, Qin Y, et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. 2019;18:88. https://doi.org/10.1186/s12943-019-1011-5.
Smith AD, Lu C, Payne D, Paschall AV, Klement JD, Redd PS, et al. Autocrine IL6-Mediated Activation of the STAT3-DNMT Axis Silences the TNFα-RIP1 Necroptosis Pathway to Sustain Survival and Accumulation of Myeloid-Derived Suppressor Cells. Cancer Res. 2020;80:3145–56. https://doi.org/10.1158/0008-5472.Can-19-3670.
Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The Fate of Th17 Cells is Shaped by Epigenetic Modifications and Remodeled by the Tumor Microenvironment. Int J Mol Sci. 2020;21. https://doi.org/10.3390/ijms21051673.
Herremans KM, Szymkiewicz DD, Riner AN, Bohan RP, Tushoski GW, Davidson AM, et al. The interleukin-1 axis and the tumor immune microenvironment in pancreatic ductal adenocarcinoma. Neoplasia. 2022; 28:100789. https://doi.org/10.1016/j.neo.2022.100789.
Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature. 2020;585:277–82. https://doi.org/10.1038/s41586-020-2682-1.
Pal R, Rakshit S, Shanmugam G, Paul N, Bhattacharya D, Chatterjee A, et al. Involvement of Xeroderma Pigmentosum Complementation Group G (XPG) in epigenetic regulation of T-Helper (T(H)) cell differentiation during breast cancer. Immunobiol. 2022;227:152259. https://doi.org/10.1016/j.imbio.2022.152259.
Fang J, Hu M, Sun Y, Zhou S, Li H. Expression Profile Analysis of m6A RNA Methylation Regulators Indicates They Are Immune Signature Associated and Can Predict Survival in Kidney Renal Cell Carcinoma. DNA Cell Biol. 2020. https://doi.org/10.1089/dna.2020.5767.
Wang F, Wang F, Zhang S, Xu X. MicroRNA-325 inhibits the proliferation and induces the apoptosis of T cell acute lymphoblastic leukemia cells in a BAG2-dependent manner. Exp Ther Med. 2021;21:631. https://doi.org/10.3892/etm.2021.10063.
Berglund-Brown I, Nissen E, Koestler DC, Butler RA, Eliot MN, Padbury JF, et al. A core of differentially methylated CpG loci in gMDSCs isolated from neonatal and adult sources. Clin Epigenetics. 2022;14:27. https://doi.org/10.1186/s13148-022-01247-1.
Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. 2017;129:1791–801. https://doi.org/10.1182/blood-2016-07-730614.
Ibrahim ML, Klement JD, Lu C, Redd PS, Xiao W, Yang D, et al. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep. 2018;25:3036-3046.e3036. https://doi.org/10.1016/j.celrep.2018.11.050.
Dejaegher J, Solie L, Hunin Z, Sciot R, Capper D, Siewert C, et al. DNA methylation based glioblastoma subclassification is related to tumoral T-cell infiltration and patient survival. Neuro Oncol. 2021;23:240–50. https://doi.org/10.1093/neuonc/noaa247.
Zhu Y, Zhao Y, Zou L, Zhang D, Aki D, Liu YC. The E3 ligase VHL promotes follicular helper T cell differentiation via glycolytic-epigenetic control. J Exp Med. 2019;216:1664–81. https://doi.org/10.1084/jem.20190337.
Halperin C, Hey J, Weichenhan D, Stein Y, Mayer S, Lutsik P, et al. Global DNA Methylation Analysis of Cancer-Associated Fibroblasts Reveals Extensive Epigenetic Rewiring Linked with RUNX1 Upregulation in Breast Cancer Stroma. Cancer Res. 2022:Of1-of14. https://doi.org/10.1158/0008-5472.Can-22-0209.
Pantelaiou-Prokaki G, Mieczkowska I, Schmidt GE, Fritzsche S, Prokakis E, Gallwas J, et al. HDAC8 suppresses the epithelial phenotype and promotes EMT in chemotherapy-treated basal-like breast cancer. Clin Epigenetics. 2022;14:7. https://doi.org/10.1186/s13148-022-01228-4.
Xu L, Deng Q, Pan Y, Peng M, Wang X, Song L, et al. Cancer-associated fibroblasts enhance the migration ability of ovarian cancer cells by increasing EZH2 expression. Int J Mol Med. 2014;33:91–6. https://doi.org/10.3892/ijmm.2013.1549.
Yan J, Ye G, Shao Y. High expression of the ferroptosis-associated MGST1 gene in relation to poor outcome and maladjusted immune cell infiltration in uterine corpus endometrial carcinoma. J Clin Lab Anal. 2022;36:e24317. https://doi.org/10.1002/jcla.24317.
Yang SC, Wang WY, Zhou JJ, Wu L, Zhang MJ, Yang QC, et al. Inhibition of DNMT1 potentiates antitumor immunity in oral squamous cell carcinoma. Int Immunopharmacol. 2022;111:109113. https://doi.org/10.1016/j.intimp.2022.109113.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)A Demethylase ALKBH5 Regulates PD-L1 Expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021;81:4778–93. https://doi.org/10.1158/0008-5472.Can-21-0468.
Zhao H, Liu X, Yu L, Lin S, Zhang C, Xu H, et al. Comprehensive landscape of epigenetic-dysregulated lncRNAs reveals a profound role of enhancers in carcinogenesis in BC subtypes. Mol Ther Nucleic Acids. 2021;23:667–81. https://doi.org/10.1016/j.omtn.2020.12.024.
Su SF, Ho H, Li JH, Wu MF, Wang HC, Yeh HY, et al. DNA methylome and transcriptome landscapes of cancer-associated fibroblasts reveal a smoking-associated malignancy index. J Clin Invest. 2021; 131. https://doi.org/10.1172/jci139552.
Feng H, Yu Z, Tian Y, Lee YY, Li MS, Go MY, et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J Hepatol. 2015;62:1100–11. https://doi.org/10.1016/j.jhep.2014.11.040.
Zhou J, Liu M, Sun H, Feng Y, Xu L, Chan AWH, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67:931–44. https://doi.org/10.1136/gutjnl-2017-314032.
Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-Small Cell Lung Cancer. Cancer Discov. 2017;7:852–67. https://doi.org/10.1158/2159-8290.Cd-16-1020.
Wu Y, Sang M, Liu F, Zhang J, Li W, Li Z, et al. Epigenetic modulation combined with PD-1/PD-L1 blockade enhances immunotherapy based on MAGE-A11 antigen-specific CD8+T cells against esophageal carcinoma. Carcinogenesis. 2020;41:894–903. https://doi.org/10.1093/carcin/bgaa057.
Kim TD, Lee SU, Yun S, Sun HN, Lee SH, Kim JW, et al. Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood. 2011;118:5476–86. https://doi.org/10.1182/blood-2011-04-347526.
Yun S, Lee SU, Kim JM, Lee HJ, Song HY, Kim YK, et al. Integrated mRNA-microRNA profiling of human NK cell differentiation identifies MiR-583 as a negative regulator of IL2Rγ expression. PLoS One. 2014;9:e108913. https://doi.org/10.1371/journal.pone.0108913.
Chen H, Pan Y, Zhou Q, Liang C, Wong CC, Zhou Y, et al. METTL3 Inhibits Antitumor Immunity by Targeting m(6)A-BHLHE41-CXCL1/CXCR2 Axis to Promote Colorectal Cancer. Gastroenterology. 2022;163:891–907. https://doi.org/10.1053/j.gastro.2022.06.024.
Labib Salem M, Zidan AA, Ezz El-Din El-Naggar R, Attia Saad M, El-Shanshory M, Bakry U, et al. Myeloid-derived suppressor cells and regulatory T cells share common immunoregulatory pathways-related microRNAs that are dysregulated by acute lymphoblastic leukemia and chemotherapy. Hum Immunol. 2021;82:36–45. https://doi.org/10.1016/j.humimm.2020.10.009.
Tseng WY, Huang YS, Clanchy F, McNamee K, Perocheau D, Ogbechi J, et al. TNF receptor 2 signaling prevents DNA methylation at the Foxp3 promoter and prevents pathogenic conversion of regulatory T cells. Proc Natl Acad Sci U S A. 2019;116:21666–72. https://doi.org/10.1073/pnas.1909687116.
Smiline Girija AS. Protean role of epigenetic mechanisms and their impact in regulating the Tregs in TME. Cancer Gene Ther. 2022;29:661–4. https://doi.org/10.1038/s41417-021-00371-z.
Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity. 2018;49:178-193.e177. https://doi.org/10.1016/j.immuni.2018.06.006.
Hart M, Walch-Rückheim B, Friedmann KS, Rheinheimer S, Tänzer T, Glombitza B, et al. miR-34a: a new player in the regulation of T cell function by modulation of NF-κB signaling. Cell Death Dis. 2019;10:46. https://doi.org/10.1038/s41419-018-1295-1.
Tong J, Cao G, Zhang T, Sefik E, Amezcua Vesely MC, Broughton JP, et al. m(6)A mRNA methylation sustains Treg suppressive functions. Cell Res. 2018;28:253–6. https://doi.org/10.1038/cr.2018.7.
Sun S, Yu F, Xu D, Zheng H, Li M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim Biophys Acta Rev Cancer. 2022;1877:188700. https://doi.org/10.1016/j.bbcan.2022.188700.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. Embo J. 2020;39:e104514. https://doi.org/10.15252/embj.2020104514.
Xu H, Cheung IY, Guo HF, Cheung NK. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7–H3: potential implications for immune based therapy of human solid tumors. Cancer Res. 2009;69:6275–81. https://doi.org/10.1158/0008-5472.Can-08-4517.
Reddy D, Bhattacharya S, Shah S, Rashid M, Gupta S. DNA methylation mediated downregulation of histone H3 variant H3.3 affects cell proliferation contributing to the development of HCC. Biochim Biophys Acta Mol Basis Dis. 2022;1868:166284.https://doi.org/10.1016/j.bbadis.2021.166284.
Götze S, Schumacher EC, Kordes C, Häussinger D. Epigenetic Changes during Hepatic Stellate Cell Activation. PLoS One. 2015;10:e0128745. https://doi.org/10.1371/journal.pone.0128745.
Wang S, Chai P, Jia R, Jia R. Novel insights on m(6)A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. 2018;17:101. https://doi.org/10.1186/s12943-018-0847-4.
Qin S, Liu G, Jin H, Chen X, He J, Xiao J, et al. The comprehensive expression and functional analysis of m6A modification "readers" in hepatocellular carcinoma. Aging (Albany NY). 2022;14:6269–6298.https://doi.org/10.18632/aging.204217.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159–70. https://doi.org/10.1073/pnas.1918986117.
Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–53. https://doi.org/10.1038/nature15520.
Chen P, Hsu WH, Chang A, Tan Z, Lan Z, Zhou A, et al. Circadian Regulator CLOCK Recruits Immune-Suppressive Microglia into the GBM Tumor Microenvironment. Cancer Discov. 2020;10:371–81. https://doi.org/10.1158/2159-8290.Cd-19-0400.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–7. https://doi.org/10.1073/pnas.1320318110.
Cioffi M, Trabulo SM, Vallespinos M, Raj D, Kheir TB, Lin ML, et al. The miR-25–93–106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget. 2017;8:21609–21625.https://doi.org/10.18632/oncotarget.15450.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. https://doi.org/10.1038/nrc1877.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659.https://doi.org/10.1056/nejm198612253152606.
Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18:99–115. https://doi.org/10.1038/s41573-018-0004-1.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204. https://doi.org/10.1038/ncomms10204.
Vizoso M, Puig M, Carmona FJ, Maqueda M, Velásquez A, Gómez A, et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis. 2015;36:1453–63. https://doi.org/10.1093/carcin/bgv146.
Al-Kharashi LA, Al-Mohanna FH, Tulbah A, Aboussekhra A. The DNA methyl-transferase protein DNMT1 enhances tumor-promoting properties of breast stromal fibroblasts. Oncotarget. 2018;9:2329–2343.https://doi.org/10.18632/oncotarget.23411.
Wu W, Wang X, Yu X, Lan HY. Smad3 Signatures in Renal Inflammation and Fibrosis. Int J Biol Sci. 2022;18:2795–806. https://doi.org/10.7150/ijbs.71595.
Tang X, Tu G, Yang G, Wang X, Kang L, Yang L, et al. Autocrine TGF-β1/miR-200s/miR-221/DNMT3B regulatory loop maintains CAF status to fuel breast cancer cell proliferation. Cancer Lett. 2019;452:79–89. https://doi.org/10.1016/j.canlet.2019.02.044.
Kuninty PR, Bojmar L, Tjomsland V, Larsson M, Storm G, Östman A, et al. MicroRNA-199a and -214 as potential therapeutic targets in pancreatic stellate cells in pancreatic tumor. Oncotarget. 2016;7:16396–16408.https://doi.org/10.18632/oncotarget.7651.
Yang J, Lu Y, Lin YY, Zheng ZY, Fang JH, He S, et al. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016;383:18–27. https://doi.org/10.1016/j.canlet.2016.09.012.
Melling GE, Flannery SE, Abidin SA, Clemmens H, Prajapati P, Hinsley EE, et al. A miRNA-145/TGF-β1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis. 2018;39:798–807. https://doi.org/10.1093/carcin/bgy032.
Li Y, Li X, Deng M, Ye C, Peng Y, Lu Y. Cancer-Associated Fibroblasts Hinder Lung Squamous Cell Carcinoma Oxidative Stress-Induced Apoptosis via METTL3 Mediated m(6)A Methylation of COL10A1. Oxid Med Cell Longev. 2022;2022:4320809. https://doi.org/10.1155/2022/4320809.
Zhang W, Zhang Y, Tu T, Schmull S, Han Y, Wang W, et al. Dual inhibition of HDAC and tyrosine kinase signaling pathways with CUDC-907 attenuates TGFβ1 induced lung and tumor fibrosis. Cell Death Dis. 2020;11:765. https://doi.org/10.1038/s41419-020-02916-w.
Tassinari M, Gandellini P. Noncoding RNAs in the Interplay between Tumor Cells and Cancer-Associated Fibroblasts: Signals to Catch and Targets to Hit. Cancers (Basel). 2021;13.https://doi.org/10.3390/cancers13040709.
Kunita A, Morita S, Irisa TU, Goto A, Niki T, Takai D, et al. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Sci Rep. 2018;8:8838. https://doi.org/10.1038/s41598-018-27128-3.
Chen S, Chen X, Shan T, Ma J, Lin W, Li W, et al. MiR-21-mediated Metabolic Alteration of Cancer-associated Fibroblasts and Its Effect on Pancreatic Cancer Cell Behavior. Int J Biol Sci. 2018;14:100–10. https://doi.org/10.7150/ijbs.22555.
Bullock MD, Pickard KM, Nielsen BS, Sayan AE, Jenei V, Mellone M, et al. Pleiotropic actions of miR-21 highlight the critical role of deregulated stromal microRNAs during colorectal cancer progression. Cell Death Dis. 2013;4:e684.https://doi.org/10.1038/cddis.2013.213.
Zhang L, Yao J, Li W, Zhang C. Micro-RNA-21 Regulates Cancer-Associated Fibroblast-Mediated Drug Resistance in Pancreatic Cancer. Oncol Res. 2018;26:827–35. https://doi.org/10.3727/096504017x14934840662335.
Givel AM, Kieffer Y, Scholer-Dahirel A, Sirven P, Cardon M, Pelon F, et al. miR200-regulated CXCL12β promotes fibroblast heterogeneity and immunosuppression in ovarian cancers. Nat Commun. 2018;9:1056. https://doi.org/10.1038/s41467-018-03348-z.
Chow MT, Möller A, Smyth MJ. Inflammation and immune surveillance in cancer. Semin Cancer Biol. 2012;22:23–32. https://doi.org/10.1016/j.semcancer.2011.12.004.
Law AMK, Valdes-Mora F, Gallego-Ortega D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells. 2020;9.https://doi.org/10.3390/cells9030561.
Rodríguez-Ubreva J, Català-Moll F, Obermajer N, Álvarez-Errico D, Ramirez RN, Company C, et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017;21:154–67. https://doi.org/10.1016/j.celrep.2017.09.018.
Wu L, Du H, Li Y, Qu P, Yan C. Signal transducer and activator of transcription 3 (Stat3C) promotes myeloid-derived suppressor cell expansion and immune suppression during lung tumorigenesis. Am J Pathol. 2011;179:2131–41. https://doi.org/10.1016/j.ajpath.2011.06.028.
Trikha P, Carson WE. Signaling pathways involved in MDSC regulation. Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 2014;1846:55–65.https://doi.org/10.1016/j.bbcan.2014.04.003.
Mace TA, Bloomston M, Lesinski GB. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunol. 2013;2:e24891.https://doi.org/10.4161/onci.24891.
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res. 2009;69:2506–13. https://doi.org/10.1158/0008-5472.Can-08-4323.
Wu CT, Hsieh CC, Lin CC, Chen WC, Hong JH, Chen MF. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J Mol Med (Berl). 2012;90:1343–55. https://doi.org/10.1007/s00109-012-0916-x.
Shang W, Gao Y, Tang Z, Zhang Y, Yang R. The Pseudogene Olfr29-ps1 Promotes the Suppressive Function and Differentiation of Monocytic MDSCs. Cancer Immunol Res. 2019;7:813–27. https://doi.org/10.1158/2326-6066.Cir-18-0443.
Sasidharan Nair V, Saleh R, Toor SM, Taha RZ, Ahmed AA, Kurer MA, et al. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin Epigenetics. 2020;12:13. https://doi.org/10.1186/s13148-020-0808-9.
Sahakian E, Powers JJ, Chen J, Deng SL, Cheng F, Distler A, et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol. 2015;63:579–85. https://doi.org/10.1016/j.molimm.2014.08.002.
Cheng F, Lienlaf M, Perez-Villarroel P, Wang HW, Lee C, Woan K, et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014;60:44–53. https://doi.org/10.1016/j.molimm.2014.02.019.
Su Y, Qiu Y, Qiu Z, Qu P. MicroRNA networks regulate the differentiation, expansion and suppression function of myeloid-derived suppressor cells in tumor microenvironment. J Cancer. 2019;10:4350–6. https://doi.org/10.7150/jca.35205.
Guo X, Qiu W, Wang J, Liu Q, Qian M, Wang S, et al. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int J Cancer. 2019;144:3111–26. https://doi.org/10.1002/ijc.32052.
Guo X, Qiu W, Liu Q, Qian M, Wang S, Zhang Z, et al. Immunosuppressive effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten Pathways. Oncogene. 2018;37:4239–59. https://doi.org/10.1038/s41388-018-0261-9.
Huber V, Vallacchi V, Fleming V, Hu X, Cova A, Dugo M, et al. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J Clin Invest. 2018;128:5505–16. https://doi.org/10.1172/jci98060.
Tao Z, Xu S, Ruan H, Wang T, Song W, Qian L, et al. MiR-195/-16 Family Enhances Radiotherapy via T Cell Activation in the Tumor Microenvironment by Blocking the PD-L1 Immune Checkpoint. Cell Physiol Biochem. 2018;48:801–14. https://doi.org/10.1159/000491909.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904. https://doi.org/10.1038/nrd.2018.169.
Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, et al. Type-2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol. 2014;307:C25-38. https://doi.org/10.1152/ajpcell.00084.2014.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. https://doi.org/10.1016/j.immuni.2014.06.008.
Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci. 2021;22.https://doi.org/10.3390/ijms22136995.
Arlauckas SP, Garren SB, Garris CS, Kohler RH, Oh J, Pittet MJ, et al. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics. 2018;8:5842–54. https://doi.org/10.7150/thno.26888.
Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014;28:565–74. https://doi.org/10.1210/me.2013-1293.
Travers M, Brown SM, Dunworth M, Holbert CE, Wiehagen KR, Bachman KE, et al. DFMO and 5-Azacytidine Increase M1 Macrophages in the Tumor Microenvironment of Murine Ovarian Cancer. Cancer Res. 2019;79:3445–54. https://doi.org/10.1158/0008-5472.Can-18-4018.
Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m(6)A as a positive regulator of macrophage activation. Sci Adv. 2021;7.https://doi.org/10.1126/sciadv.abd4742.
Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394. https://doi.org/10.1038/s41467-021-21514-8.
Su B, Han H, Gong Y, Li X, Ji C, Yao J, et al. Let-7d inhibits intratumoral macrophage M2 polarization and subsequent tumor angiogenesis by targeting IL-13 and IL-10. Cancer Immunol Immunother. 2021;70:1619–34. https://doi.org/10.1007/s00262-020-02791-6.
Fei L, Ren X, Yu H, Zhan Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front Immunol. 2021;12:771210.https://doi.org/10.3389/fimmu.2021.771210.
Hou P, Kapoor A, Zhang Q, Li J, Wu CJ, Li J, et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020;10:1058–77. https://doi.org/10.1158/2159-8290.Cd-19-0597.
Zheng Y, Wang Z, Wei S, Liu Z, Chen G. Epigenetic silencing of chemokine CCL2 represses macrophage infiltration to potentiate tumor development in small cell lung cancer. Cancer Lett. 2021;499:148–63. https://doi.org/10.1016/j.canlet.2020.11.034.
Xia L, Zhu X, Zhang L, Xu Y, Chen G, Luo J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol Appl Biochem. 2020;67:1011–9. https://doi.org/10.1002/bab.1875.
Wang YF, Yu L, Hu ZL, Fang YF, Shen YY, Song MF, et al. Regulation of CCL2 by EZH2 affects tumor-associated macrophages polarization and infiltration in breast cancer. Cell Death Dis. 2022;13:748. https://doi.org/10.1038/s41419-022-05169-x.
Zhao J, Li H, Zhao S, Wang E, Zhu J, Feng D, et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol Cancer. 2021;20:46. https://doi.org/10.1186/s12943-021-01343-5.
Xun J, Du L, Gao R, Shen L, Wang D, Kang L, et al. Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B. Theranostics. 2021;11:6847–59. https://doi.org/10.7150/thno.51864.
Gerloff D, Lützkendorf J, Moritz RKC, Wersig T, Mäder K, Müller LP, et al. Melanoma-Derived Exosomal miR-125b-5p Educates Tumor Associated Macrophages (TAMs) by Targeting Lysosomal Acid Lipase A (LIPA). Cancers (Basel). 2020;12.https://doi.org/10.3390/cancers12020464.
Guo J, Duan Z, Zhang C, Wang W, He H, Liu Y, et al. Mouse 4T1 Breast Cancer Cell-Derived Exosomes Induce Proinflammatory Cytokine Production in Macrophages via miR-183. J Immunol. 2020;205:2916–25. https://doi.org/10.4049/jimmunol.1901104.
Li X, Tang M. Exosomes released from M2 macrophages transfer miR-221-3p contributed to EOC progression through targeting CDKN1B. Cancer Med. 2020;9:5976–88. https://doi.org/10.1002/cam4.3252.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell. 2020;38:79-96.e11. https://doi.org/10.1016/j.ccell.2020.04.017.
Lei J, Chen P, Zhang F, Zhang N, Zhu J, Wang X, et al. M2 macrophages-derived exosomal microRNA-501-3p promotes the progression of lung cancer via targeting WD repeat domain 82. Cancer Cell Int. 2021;21:91. https://doi.org/10.1186/s12935-021-01783-5.
Ma YS, Wu TM, Ling CC, Yu F, Zhang J, Cao PS, et al. M2 macrophage-derived exosomal microRNA-155-5p promotes the immune escape of colon cancer by downregulating ZC3H12B. Mol Ther Oncolytics. 2021;20:484–98. https://doi.org/10.1016/j.omto.2021.02.005.
Li X, Chen Z, Ni Y, Bian C, Huang J, Chen L, et al. Tumor-associated macrophages secret exosomal miR-155 and miR-196a-5p to promote metastasis of non-small-cell lung cancer. Transl Lung Cancer Res. 2021;10:1338–1354.https://doi.org/10.21037/tlcr-20-1255.
Yuan Y, Wang Z, Chen M, Jing Y, Shu W, Xie Z, et al. Macrophage-Derived Exosomal miR-31-5p Promotes Oral Squamous Cell Carcinoma Tumourigenesis Through the Large Tumor Suppressor 2-Mediated Hippo Signalling Pathway. J Biomed Nanotechnol. 2021;17:822–37. https://doi.org/10.1166/jbn.2021.3066.
Zhang Z, Xu J, Chen Z, Wang H, Xue H, Yang C, et al. Transfer of MicroRNA via Macrophage-Derived Extracellular Vesicles Promotes Proneural-to-Mesenchymal Transition in Glioma Stem Cells. Cancer Immunol Res. 2020;8:966–81. https://doi.org/10.1158/2326-6066.Cir-19-0759.
Guan H, Peng R, Fang F, Mao L, Chen Z, Yang S, et al. Tumor-associated macrophages promote prostate cancer progression via exosome-mediated miR-95 transfer. J Cell Physiol. 2020;235:9729–42. https://doi.org/10.1002/jcp.29784.
Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012;21:402–17. https://doi.org/10.1016/j.ccr.2012.01.008.
Nishita M, Park SY, Nishio T, Kamizaki K, Wang Z, Tamada K, et al. Ror2 signaling regulates Golgi structure and transport through IFT20 for tumor invasiveness. Sci Rep. 2017;7:1. https://doi.org/10.1038/s41598-016-0028-x.
Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016;30:324–36. https://doi.org/10.1016/j.ccell.2016.06.003.
Gupta YH, Khanom A, Acton SE. Control of Dendritic Cell Function Within the Tumour Microenvironment. Front Immunol. 2022;13:733800.https://doi.org/10.3389/fimmu.2022.733800.
Gordy JT, Luo K, Kapoor A, Kim ES, Ayeh SK, Karakousis PC, et al. Treatment with an immature dendritic cell-targeting vaccine supplemented with IFN-α and an inhibitor of DNA methylation markedly enhances survival in a murine melanoma model. Cancer Immunol Immunother. 2020;69:569–80. https://doi.org/10.1007/s00262-019-02471-0.
Simonović N, Witalisz-Siepracka A, Meissl K, Lassnig C, Reichart U, Kolbe T, et al. NK Cells Require Cell-Extrinsic and -Intrinsic TYK2 for Full Functionality in Tumor Surveillance and Antibacterial Immunity. J Immunol. 2019;202:1724–34. https://doi.org/10.4049/jimmunol.1701649.
Shi Q, Shen L, Gan J, He L, Lin J, Guo S, et al. Integrative analysis identifies DNMTs against immune-infiltrating neutrophils and dendritic cells in colorectal cancer. Epigenetics. 2019;14:392–404. https://doi.org/10.1080/15592294.2019.1588684.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat Commun. 2019;10:1898. https://doi.org/10.1038/s41467-019-09903-6.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4. https://doi.org/10.1038/s41586-019-0916-x.
Liu J, Zhang X, Chen K, Cheng Y, Liu S, Xia M, et al. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity. 2019;50:600-615.e615. https://doi.org/10.1016/j.immuni.2019.01.021.
Shinde P, Fernandes S, Melinkeri S, Kale V, Limaye L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci Rep. 2018;8:5705. https://doi.org/10.1038/s41598-018-23943-w.
Guenther C, Faisal I, Fusciello M, Sokolova M, Harjunpää H, Ilander M, et al. β2-Integrin Adhesion Regulates Dendritic Cell Epigenetic and Transcriptional Landscapes to Restrict Dendritic Cell Maturation and Tumor Rejection. Cancer Immunol Res. 2021;9:1354–69. https://doi.org/10.1158/2326-6066.Cir-21-0094.
De Beck L, Awad RM, Basso V, Casares N, De Ridder K, De Vlaeminck Y, et al. Inhibiting Histone and DNA Methylation Improves Cancer Vaccination in an Experimental Model of Melanoma. Front Immunol. 2022;13:799636 https://doi.org/10.3389/fimmu.2022.799636.
Chen J, Wang S, Jia S, Ding G, Jiang G, Cao L. Integrated Analysis of Long Non-Coding RNA and mRNA Expression Profile in Pancreatic Cancer Derived Exosomes Treated Dendritic Cells by Microarray Analysis. J Cancer. 2018;9:21–31. https://doi.org/10.7150/jca.21749.
Liu D, Heij LR, Czigany Z, Dahl E, Lang SA, Ulmer TF, et al. The role of tumor-infiltrating lymphocytes in cholangiocarcinoma. J Exp Clin Cancer Res. 2022;41:127. https://doi.org/10.1186/s13046-022-02340-2.
Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18:842–59. https://doi.org/10.1038/s41423-020-00565-9.
Du Y, Wei Y. Therapeutic Potential of Natural Killer Cells in Gastric Cancer. Front Immunol. 2018;9:3095. https://doi.org/10.3389/fimmu.2018.03095.
Cui F, Qu D, Sun R, Nan K. Circulating CD16+CD56+ nature killer cells indicate the prognosis of colorectal cancer after initial chemotherapy. Med Oncol. 2019;36:84. https://doi.org/10.1007/s12032-019-1307-8.
Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, et al. Phenotypic and functional separation of memory and effector human CD8+ T cells. J Exp Med. 1997;186:1407–18. https://doi.org/10.1084/jem.186.9.1407.
Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun. 2018;95:77–99. https://doi.org/10.1016/j.jaut.2018.08.007.
Safaei S, Mohme M, Niesen J, Schüller U, Bockmayr M. DIMEimmune: Robust estimation of infiltrating lymphocytes in CNS tumors from DNA methylation profiles. Oncoimmunology. 2021;10:1932365. https://doi.org/10.1080/2162402x.2021.1932365.
Janson PC, Marits P, Thörn M, Ohlsson R, Winqvist O. CpG methylation of the IFNG gene as a mechanism to induce immunosuppression [correction of immunosupression] in tumor-infiltrating lymphocytes. J Immunol. 2008;181:2878–86. https://doi.org/10.4049/jimmunol.181.4.2878.
Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 Epigenetically Silences Th1-Type Chemokines to Suppress Effector T-Cell Trafficking in Colon Cancer. Cancer Res. 2016;76:275–82. https://doi.org/10.1158/0008-5472.Can-15-1938.
Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018;23:3262–74. https://doi.org/10.1016/j.celrep.2018.05.050.
Li Y, Wang J, Yin J, Liu X, Yu M, Li T, et al. Chromatin state dynamics during NK cell activation. Oncotarget. 2017;8:41854–41865 https://doi.org/10.18632/oncotarget.16688.
Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 2018;115:E3509-e3518. https://doi.org/10.1073/pnas.1802691115.
Cribbs A, Hookway ES, Wells G, Lindow M, Obad S, Oerum H, et al. Inhibition of histone H3K27 demethylases selectively modulates inflammatory phenotypes of natural killer cells. J Biol Chem. 2018;293:2422–37. https://doi.org/10.1074/jbc.RA117.000698.
Ramakrishnan S, Granger V, Rak M, Hu Q, Attwood K, Aquila L, et al. Inhibition of EZH2 induces NK cell-mediated differentiation and death in muscle-invasive bladder cancer. Cell Death Differ. 2019;26:2100–14. https://doi.org/10.1038/s41418-019-0278-9.
Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 Axis Controls Treg Infiltration in the Tumor Microenvironment by Inducing CCR4 Expression in Breast Cancer. Front Immunol. 2022;13:740588.https://doi.org/10.3389/fimmu.2022.740588.
MaruYama T, Kobayashi S, Nakatsukasa H, Moritoki Y, Taguchi D, Sunagawa Y, et al. The Curcumin Analog GO-Y030 Controls the Generation and Stability of Regulatory T Cells. Front Immunol. 2021;12:687669 https://doi.org/10.3389/fimmu.2021.687669.
Vignard V, Labbé M, Marec N, André-Grégoire G, Jouand N, Fonteneau JF, et al. MicroRNAs in Tumor Exosomes Drive Immune Escape in Melanoma. Cancer Immunol Res. 2020;8:255–67. https://doi.org/10.1158/2326-6066.Cir-19-0522.
Zhou C, Zhang Y, Yan R, Huang L, Mellor AL, Yang Y, et al. Exosome-derived miR-142-5p remodels lymphatic vessels and induces IDO to promote immune privilege in the tumour microenvironment. Cell Death Differ. 2021;28:715–29. https://doi.org/10.1038/s41418-020-00618-6.
Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol Res. 2018;6:1578–92. https://doi.org/10.1158/2326-6066.Cir-17-0479.
Jin F, Du Z, Tang Y, Wang L, Yang Y. Impact of microRNA-29b on natural killer cells in T-cell acute lymphoblastic leukemia. Oncol Lett. 2019;18:2394–403. https://doi.org/10.3892/ol.2019.10559.
Shen J, Pan J, Du C, Si W, Yao M, Xu L, et al. Silencing NKG2D ligand-targeting miRNAs enhances natural killer cell-mediated cytotoxicity in breast cancer. Cell Death Dis. 2017; 8:e2740.https://doi.org/10.1038/cddis.2017.158.
Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125:5591–6. https://doi.org/10.1242/jcs.116392.
Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B-side of Cancer Immunity: The Underrated Tune. Cells. 2019;8.https://doi.org/10.3390/cells8050449.
Kim SS, Shen S, Miyauchi S, Sanders PD, Franiak-Pietryga I, Mell L, et al. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin Cancer Res. 2020;26:3345–59. https://doi.org/10.1158/1078-0432.Ccr-19-3211.
Gong L, Kwong DLW, Dai W, Wu P, Li S, Yan Q, et al. Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma. Nature Commun. 2021;12(1540).https://doi.org/10.1038/s41467-021-21795-z.
Alberghini F, Petrocelli V, Rahmat M, Casola S. An epigenetic view of B-cell disorders. Immunol Cell Biol. 2015;93:253–260.https://doi.org/10.1038/icb.2014.116.
Bao Y, Cao X. Epigenetic Control of B Cell Development and B-Cell-Related Immune Disorders. Clin Rev Allergy Immunol. 2016;50:301–11. https://doi.org/10.1007/s12016-015-8494-7.
Pavlasova G, Mraz M. The regulation and function of CD20: an “enigma” of B-cell biology and targeted therapy. Haematologica. 2020;105:1494–506. https://doi.org/10.3324/haematol.2019.243543.
Tomita A, Hiraga J, Kiyoi H, Ninomiya M, Sugimoto T, Ito M, et al. Epigenetic regulation of CD20 protein expression in a novel B-cell lymphoma cell line, RRBL1, established from a patient treated repeatedly with rituximab-containing chemotherapy. Int J Hematol. 2007;86:49–57. https://doi.org/10.1532/ijh97.07028.
Hiraga J, Tomita A, Sugimoto T, Shimada K, Ito M, Nakamura S, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009;113:4885–93. https://doi.org/10.1182/blood-2008-08-175208.
Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. https://doi.org/10.1038/35102167.
Toh TB, Lim JJ, Chow EK. Epigenetics in cancer stem cells. Mol Cancer. 2017;16:29. https://doi.org/10.1186/s12943-017-0596-9.
Jin L, Vu TT, Datta PK. Abstract 1709: STRAP mediates the stemness of human colorectal cancer cells by epigenetic regulation of Notch pathway. Can Res. 2016;76:1709–1709. https://doi.org/10.1158/1538-7445.Am2016-1709.
Hajkova P, Ancelin K, Waldmann T, Lacoste N, Lange UC, Cesari F, et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–81. https://doi.org/10.1038/nature06714.
Liu S, Cheng K, Zhang H, Kong R, Wang S, Mao C, et al. Methylation Status of the Nanog Promoter Determines the Switch between Cancer Cells and Cancer Stem Cells. Adv Sci (Weinh). 2020;7:1903035. https://doi.org/10.1002/advs.201903035.
Suelves M, Carrió E, Núñez-Álvarez Y, Peinado MA. DNA methylation dynamics in cellular commitment and differentiation. Brief Funct Genomics. 2016;15:443–53. https://doi.org/10.1093/bfgp/elw017.
Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, et al. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69:3570–8. https://doi.org/10.1158/0008-5472.Can-08-2807.
Jiang X, Tan J, Li J, Kivimäe S, Yang X, Zhuang L, et al. DACT3 is an epigenetic regulator of Wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications. Cancer Cell. 2008;13:529–41. https://doi.org/10.1016/j.ccr.2008.04.019.
Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–42. https://doi.org/10.1038/ncb2013.
Ghoshal P, Nganga AJ, Moran-Giuati J, Szafranek A, Johnson TR, Bigelow AJ, et al. Loss of the SMRT/NCoR2 corepressor correlates with JAG2 overexpression in multiple myeloma. Cancer Res. 2009;69:4380–7. https://doi.org/10.1158/0008-5472.Can-08-3467.
Xie Q, Wang S, Zhao Y, Zhang Z, Qin C, Yang X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget. 2017; 8:22003–22013.https://doi.org/10.18632/oncotarget.15781.
Wang X, Meng Q, Qiao W, Ma R, Ju W, Hu J, et al. miR-181b/Notch2 overcome chemoresistance by regulating cancer stem cell-like properties in NSCLC. Stem Cell Res Ther. 2018;9:327. https://doi.org/10.1186/s13287-018-1072-1.
Xu L, Zhang J, Sun J, Hou K, Yang C, Guo Y, et al. Epigenetic regulation of cancer stem cells: Shedding light on the refractory/relapsed cancers. Biochem Pharmacol. 2022; 202:115110.https://doi.org/10.1016/j.bcp.2022.115110.
Khan AQ, Ahmed EI, Elareer NR, Junejo K, Steinhoff M, Uddin S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells. 2019;8.https://doi.org/10.3390/cells8080840.
French R, Pauklin S. Epigenetic regulation of cancer stem cell formation and maintenance. Int J Cancer. 2021;148:2884–97. https://doi.org/10.1002/ijc.33398.
Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019;14:1164–76. https://doi.org/10.1080/15592294.2019.1640546.
Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173–84. https://doi.org/10.1038/nrg2006.
Bian EB, Huang C, Ma TT, Tao H, Zhang H, Cheng C, et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol. 2012;264:13–22. https://doi.org/10.1016/j.taap.2012.06.022.
Al-Kharashi LA, Bakheet T, AlHarbi WA, Al-Moghrabi N, Aboussekhra A. Eugenol modulates genomic methylation and inactivates breast cancer-associated fibroblasts through E2F1-dependent downregulation of DNMT1/DNMT3A. Mol Carcinog. 2021;60:784–95. https://doi.org/10.1002/mc.23344.
Xiao Q, Zhou D, Rucki AA, Williams J, Zhou J, Mo G, et al. Cancer-Associated Fibroblasts in Pancreatic Cancer Are Reprogrammed by Tumor-Induced Alterations in Genomic DNA Methylation. Cancer Res. 2016;76:5395–404. https://doi.org/10.1158/0008-5472.Can-15-3264.
Al-Kharashi LA, Tulbah A, Arafah M, Eldali AM, Al-Tweigeri T, Aboussekhra A. High DNMT1 Expression in Stromal Fibroblasts Promotes Angiogenesis and Unfavorable Outcome in Locally Advanced Breast Cancer Patients. Front Oncol. 2022;12:877219.https://doi.org/10.3389/fonc.2022.877219.
Maiti A, Qi Q, Peng X, Yan L, Takabe K, Hait NC. Class I histone deacetylase inhibitor suppresses vasculogenic mimicry by enhancing the expression of tumor suppressor and anti-angiogenesis genes in aggressive human TNBC cells. Int J Oncol. 2019;55:116–30. https://doi.org/10.3892/ijo.2019.4796.
Tabernero J, Shapiro GI, LoRusso PM, Cervantes A, Schwartz GK, Weiss GJ, et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013;3:406–17. https://doi.org/10.1158/2159-8290.Cd-12-0429.
Tang Y, Jia C, Wang Y, Wan W, Li H, Huang G, et al. Lactate Consumption via Cascaded Enzymes Combined VEGF siRNA for Synergistic Anti-Proliferation and Anti-Angiogenesis Therapy of Tumors. Adv Healthc Mater. 2021;10:e2100799.https://doi.org/10.1002/adhm.202100799.
Egorova AA, Shtykalova SV, Maretina MA, Sokolov DI, Selkov SA, Baranov VS, et al. Synergistic Anti-Angiogenic Effects Using Peptide-Based Combinatorial Delivery of siRNAs Targeting VEGFA, VEGFR1, and Endoglin Genes. Pharmaceutics. 2019;11.https://doi.org/10.3390/pharmaceutics11060261.
Zerbib E, Arif T, Shteinfer-Kuzmine A, Chalifa-Caspi V, Shoshan-Barmatz V. VDAC1 Silencing in Cancer Cells Leads to Metabolic Reprogramming That Modulates Tumor Microenvironment. Cancers (Basel). 2021;13.https://doi.org/10.3390/cancers13112850.
Schultheis B, Strumberg D, Santel A, Vank C, Gebhardt F, Keil O, et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J Clin Oncol. 2014;32:4141–8. https://doi.org/10.1200/jco.2013.55.0376.
Schultheis B, Strumberg D, Kuhlmann J, Wolf M, Link K, Seufferlein T, et al. Safety, Efficacy and Pharcacokinetics of Targeted Therapy with The Liposomal RNA Interference Therapeutic Atu027 Combined with Gemcitabine in Patients with Pancreatic Adenocarcinoma. A Randomized Phase Ib/IIa Study. Cancers (Basel). 2020;12.https://doi.org/10.3390/cancers12113130.
Leung CS, Yeung TL, Yip KP, Wong KK, Ho SY, Mangala LS, et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J Clin Invest. 2018;128:589–606. https://doi.org/10.1172/jci95200.
Hwang HJ, Lee YR, Kang D, Lee HC, Seo HR, Ryu JK, et al. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020;490:100–10. https://doi.org/10.1016/j.canlet.2020.06.019.
Chan IS, Knútsdóttir H, Ramakrishnan G, Padmanaban V, Warrier M, Ramirez JC, et al. Cancer cells educate natural killer cells to a metastasis-promoting cell state. J Cell Biol. 2020;219.https://doi.org/10.1083/jcb.202001134.
Biber G, Sabag B, Raiff A, Ben-Shmuel A, Puthenveetil A, Benichou JIC, et al. Modulation of intrinsic inhibitory checkpoints using nano-carriers to unleash NK cell activity. EMBO Mol Med. 2022;14:e14073.https://doi.org/10.15252/emmm.202114073.
Frikeche J, Clavert A, Delaunay J, Brissot E, Grégoire M, Gaugler B, et al. Impact of the hypomethylating agent 5-azacytidine on dendritic cells function. Exp Hematol. 2011;39:1056–63. https://doi.org/10.1016/j.exphem.2011.08.004.
Fragale A, Romagnoli G, Licursi V, Buoncervello M, Del Vecchio G, Giuliani C, et al. Antitumor Effects of Epidrug/IFNα Combination Driven by Modulated Gene Signatures in Both Colorectal Cancer and Dendritic Cells. Cancer Immunol Res. 2017;5:604–16. https://doi.org/10.1158/2326-6066.Cir-17-0080.
Zhou Z, Chen H, Xie R, Wang H, Li S, Xu Q, et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol Oncol. 2019;13:873–93. https://doi.org/10.1002/1878-0261.12443.
Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer. 2013;132:967–77. https://doi.org/10.1002/ijc.27710.
Shi R, Zhao K, Wang T, Yuan J, Zhang D, Xiang W, et al. 5-aza-2’-deoxycytidine potentiates anti-tumor immunity in colorectal peritoneal metastasis by modulating ABC A9-mediated cholesterol accumulation in macrophages. Theranostics. 2022;12:875–90. https://doi.org/10.7150/thno.66420.
Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L, et al. Molecular-Targeted Immunotherapeutic Strategy for Melanoma via Dual-Targeting Nanoparticles Delivering Small Interfering RNA to Tumor-Associated Macrophages. ACS Nano. 2017;11:9536–49. https://doi.org/10.1021/acsnano.7b05465.
Song Y, Tang C, Yin C. Combination antitumor immunotherapy with VEGF and PIGF siRNA via systemic delivery of multi-functionalized nanoparticles to tumor-associated macrophages and breast cancer cells. Biomaterials. 2018;185:117–32. https://doi.org/10.1016/j.biomaterials.2018.09.017.
Xiao H, Guo Y, Li B, Li X, Wang Y, Han S, et al. M2-Like Tumor-Associated Macrophage-Targeted Codelivery of STAT6 Inhibitor and IKKβ siRNA Induces M2-to-M1 Repolarization for Cancer Immunotherapy with Low Immune Side Effects. ACS Cent Sci. 2020;6:1208–22. https://doi.org/10.1021/acscentsci.9b01235.
Shobaki N, Sato Y, Suzuki Y, Okabe N, Harashima H. Manipulating the function of tumor-associated macrophages by siRNA-loaded lipid nanoparticles for cancer immunotherapy. J Control Release. 2020;325:235–48. https://doi.org/10.1016/j.jconrel.2020.07.001.
Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, et al. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012;7:e30815. https://doi.org/10.1371/journal.pone.0030815.
Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest. 2018;128:3813–8. https://doi.org/10.1172/jci99760.
Sun W, Chen L, Tang J, Zhang C, Wen Y, Wen W. Targeting EZH2 depletes LMP1-induced activated regulatory T cells enhancing antitumor immunity in nasopharyngeal carcinoma. J Cancer Res Ther. 2020;16:309–19. https://doi.org/10.4103/jcrt.JCRT_986_19.
DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–38. https://doi.org/10.1016/j.immuni.2015.01.007.
Nikbakht N, Tiago M, Erkes DA, Chervoneva I, Aplin AE. BET Inhibition Modifies Melanoma Infiltrating T Cells and Enhances Response to PD-L1 Blockade. J Invest Dermatol. 2019;139:1612–5. https://doi.org/10.1016/j.jid.2018.12.024.
Wang HF, Ning F, Liu ZC, Wu L, Li ZQ, Qi YF, et al. Histone deacetylase inhibitors deplete myeloid-derived suppressor cells induced by 4T1 mammary tumors in vivo and in vitro. Cancer Immunol Immunother. 2017;66:355–66. https://doi.org/10.1007/s00262-016-1935-1.
Kim YD, Park SM, Ha HC, Lee AR, Won H, Cha H, et al. HDAC Inhibitor, CG-745, Enhances the Anti-Cancer Effect of Anti-PD-1 Immune Checkpoint Inhibitor by Modulation of the Immune Microenvironment. J Cancer. 2020;11:4059–72. https://doi.org/10.7150/jca.44622.
Li X, Su X, Liu R, Pan Y, Fang J, Cao L, et al. HDAC inhibition potentiates anti-tumor activity of macrophages and enhances anti-PD-L1-mediated tumor suppression. Oncogene. 2021;40:1836–50. https://doi.org/10.1038/s41388-020-01636-x.
de Almeida Nagata DE, Chiang EY, Jhunjhunwala S, Caplazi P, Arumugam V, Modrusan Z, et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019;27:269-281.e264. https://doi.org/10.1016/j.celrep.2019.03.008.
Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A. 2017;114:E10981-e10990. https://doi.org/10.1073/pnas.1712514114.
Ghasemi-Chaleshtari M, Kiaie SH, Irandoust M, Karami H, Nabi Afjadi M, Ghani S, et al. Concomitant blockade of A2AR and CTLA-4 by siRNA-loaded polyethylene glycol-chitosan-alginate nanoparticles synergistically enhances antitumor T-cell responses. J Cell Physiol. 2020;235:10068–80. https://doi.org/10.1002/jcp.29822.
Jia L, Gao Y, Zhou T, Zhao XL, Hu HY, Chen DW, et al. Enhanced response to PD-L1 silencing by modulation of TME via balancing glucose metabolism and robust co-delivery of siRNA/Resveratrol with dual-responsive polyplexes. Biomaterials. 2021;271:120711.https://doi.org/10.1016/j.biomaterials.2021.120711.
Li Z, Wang Y, Shen Y, Qian C, Oupicky D, Sun M. Targeting pulmonary tumor microenvironment with CXCR4-inhibiting nanocomplex to enhance anti-PD-L1 immunotherapy. Sci Adv. 2020;6:eaaz9240.https://doi.org/10.1126/sciadv.aaz9240.
Li G, Gao Y, Gong C, Han Z, Qiang L, Tai Z, et al. Dual-Blockade Immune Checkpoint for Breast Cancer Treatment Based on a Tumor-Penetrating Peptide Assembling Nanoparticle. ACS Appl Mater Interfaces. 2019;11:39513–24. https://doi.org/10.1021/acsami.9b13354.
Kim S, Heo R, Song SH, Song KH, Shin JM, Oh SJ, et al. PD-L1 siRNA-hyaluronic acid conjugate for dual-targeted cancer immunotherapy. J Control Release. 2022;346:226–39. https://doi.org/10.1016/j.jconrel.2022.04.023.
Biffi G, Tuveson DA. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev. 2021;101:147–76. https://doi.org/10.1152/physrev.00048.2019.
Pistore C, Giannoni E, Colangelo T, Rizzo F, Magnani E, Muccillo L, et al. DNA methylation variations are required for epithelial-to-mesenchymal transition induced by cancer-associated fibroblasts in prostate cancer cells. Oncogene. 2017;36:5551–66. https://doi.org/10.1038/onc.2017.159.
Du YE, Tu G, Yang G, Li G, Yang D, Lang L, et al. MiR-205/YAP1 in Activated Fibroblasts of Breast Tumor Promotes VEGF-independent Angiogenesis through STAT3 Signaling. Theranostics. 2017;7:3972–88. https://doi.org/10.7150/thno.18990.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46. https://doi.org/10.1038/ncb2756.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47. https://doi.org/10.1016/j.ccr.2014.04.021.
Hegde M, Guruprasad KP, Ramachandra L, Satyamoorthy K, Joshi MB. Interleukin-6-mediated epigenetic control of the VEGFR2 gene induces disorganized angiogenesis in human breast tumors. J Biol Chem. 2020;295:12086–98. https://doi.org/10.1074/jbc.RA120.012590.
Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol. 2019;10:3100. https://doi.org/10.3389/fimmu.2019.03100.
Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, et al. Control of Foxp3 stability through modulation of TET activity. J Exp Med. 2016;213:377–97. https://doi.org/10.1084/jem.20151438.
Fang Y, Yuan XD, Liu HH, Xiang L, Chen LM, Fan YC, et al. 5-Aza-2’-deoxycytidine may enhance the frequency of T regulatory cells from CD4(+) naïve T cells isolated from the peripheral blood of patients with chronic HBV infection. Expert Rev Clin Immunol. 2021;17:177–85. https://doi.org/10.1080/1744666x.2020.1866987.
Su S, Liao J, Liu J, Huang D, He C, Chen F, et al. Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 2017;27:461–82. https://doi.org/10.1038/cr.2017.34.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084.https://doi.org/10.3389/fimmu.2020.583084.
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95. https://doi.org/10.1038/cr.2016.157.
Liu L, Yi H, Wang C, He H, Li P, Pan H, et al. Integrated Nanovaccine with MicroRNA-148a Inhibition Reprograms Tumor-Associated Dendritic Cells by Modulating miR-148a/DNMT1/SOCS1 Axis. J Immunol. 2016;197:1231–41. https://doi.org/10.4049/jimmunol.1600182.
Zhao D, Zhang Q, Liu Y, Li X, Zhao K, Ding Y, et al. H3K4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating with p50 to Suppress SOCS1. Cell Rep. 2020;30:2460. https://doi.org/10.1016/j.celrep.2020.01.104.
Huang HT, Su SC, Chiou TJ, Lin YH, Shih YC, Wu YX, et al. DNA methylation-mediated Siglec-7 regulation in natural killer cells via two 5’ promoter CpG sites. Immunology. 2020;160:38–51. https://doi.org/10.1111/imm.13179.
Muhammad JS, Jayakumar MN, Elemam NM, Venkatachalam T, Raju TK, Hamoudi RA, et al. Gasdermin D Hypermethylation Inhibits Pyroptosis And LPS-Induced IL-1β Release From NK92 Cells. Immunotargets Ther. 2019;8:29–41. https://doi.org/10.2147/itt.S219867.
Ostroumov D, Fekete-Drimusz N, Saborowski M, Kühnel F, Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci. 2018;75:689–713. https://doi.org/10.1007/s00018-017-2686-7.
Terranova-Barberio M, Thomas S, Munster PN. Epigenetic modifiers in immunotherapy: a focus on checkpoint inhibitors. Immunotherapy. 2016;8:705–19. https://doi.org/10.2217/imt-2016-0014.
Hu C, Liu X, Zeng Y, Liu J, Wu F. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Epigenetics. 2021;13:166. https://doi.org/10.1186/s13148-021-01154-x.
Nie J, Wang C, Liu Y, Yang Q, Mei Q, Dong L, et al. Addition of Low-Dose Decitabine to Anti-PD-1 Antibody Camrelizumab in Relapsed/Refractory Classical Hodgkin Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2019;37:1479–89. https://doi.org/10.1200/jco.18.02151.
Segovia C, San José-Enériz E, Munera-Maravilla E, Martínez-Fernández M, Garate L, Miranda E, et al. Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat Med. 2019;25:1073–81. https://doi.org/10.1038/s41591-019-0499-y.
Srour N, Villarreal OD, Hardikar S, Yu Z, Preston S, Miller WH, Jr., et al. PRMT7 ablation stimulates anti-tumor immunity and sensitizes melanoma to immune checkpoint blockade. Cell Reports. 2022;38:110582.https://doi.org/10.1016/j.celrep.2022.110582.
Borcoman E, Kamal M, Marret G, Dupain C, Castel-Ajgal Z, Le Tourneau C. HDAC Inhibition to Prime Immune Checkpoint Inhibitors. Cancers. 2021;14. https://doi.org/10.3390/cancers14010066.
Gray JE, Saltos A, Tanvetyanon T, Haura EB, Creelan B, Antonia SJ, et al. Phase I/Ib Study of Pembrolizumab Plus Vorinostat in Advanced/Metastatic Non-Small Cell Lung Cancer. Clin Cancer Res. 2019;25:6623–32. https://doi.org/10.1158/1078-0432.Ccr-19-1305.
Hellmann MD, Jänne PA, Opyrchal M, Hafez N, Raez LE, Gabrilovich DI, et al. Entinostat plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2021;27:1019–28. https://doi.org/10.1158/1078-0432.Ccr-20-3305.
Jespersen H, Olofsson Bagge R, Ullenhag G, Carneiro A, Helgadottir H, Ljuslinder I, et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study): protocol for a multicenter phase II open label study. BMC Cancer. 2019;19:415. https://doi.org/10.1186/s12885-019-5623-3.
Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci Rep. 2019;9:6136. https://doi.org/10.1038/s41598-019-42237-3.
Fan F, Liu P, Bao R, Chen J, Zhou M, Mo Z, et al. A Dual PI3K/HDAC Inhibitor Induces Immunogenic Ferroptosis to Potentiate Cancer Immune Checkpoint Therapy. Can Res. 2021;81:6233–45. https://doi.org/10.1158/0008-5472.Can-21-1547.
Qin Y, Vasilatos SN, Chen L, Wu H, Cao Z, Fu Y, et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene. 2019;38:390–405. https://doi.org/10.1038/s41388-018-0451-5.
Que Y, Zhang XL, Liu ZX, Zhao JJ, Pan QZ, Wen XZ, et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunotherapy Cancer. 2021;9.https://doi.org/10.1136/jitc-2020-001696.
Yang W, Feng Y, Zhou J, Cheung OK, Cao J, Wang J, et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci Transl Med. 2021;13.https://doi.org/10.1126/scitranslmed.aaz6804.
Tay RE, Olawoyin O, Cejas P, Xie Y, Meyer CA, Ito Y, et al. Hdac3 is an epigenetic inhibitor of the cytotoxicity program in CD8 T cells. J Exp Med. 2020;217.https://doi.org/10.1084/jem.20191453.
Negishi H, Taniguchi T, Yanai H. The Interferon (IFN) Class of Cytokines and the IFN Regulatory Factor (IRF) Transcription Factor Family. Cold Spring Harb Perspect Biol. 2018;10.https://doi.org/10.1101/cshperspect.a028423.
Lazear HM, Schoggins JW, Diamond MS. Shared and Distinct Functions of Type I and Type III Interferons. Immunity. 2019;50:907–23. https://doi.org/10.1016/j.immuni.2019.03.025.
Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2017;169:361. https://doi.org/10.1016/j.cell.2017.03.036.
Lai J, Fu Y, Tian S, Huang S, Luo X, Lin L, et al. Zebularine elevates STING expression and enhances cGAMP cancer immunotherapy in mice. Mol Ther. 2021;29:1758–71. https://doi.org/10.1016/j.ymthe.2021.02.005.
Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nat Cell Biol. 2003;5:834–9. https://doi.org/10.1038/ncb1038.
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A. 2012;109:E2110-2116. https://doi.org/10.1073/pnas.1209414109.
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128. https://doi.org/10.1186/s12943-019-1055-6.
Booth L, Roberts JL, Kirkwood J, Poklepovic A, Dent P. Unconventional Approaches to Modulating the Immunogenicity of Tumor Cells. Adv Cancer Res. 2018;137:1–15. https://doi.org/10.1016/bs.acr.2017.11.004.
Rodems TS, Heninger E, Stahlfeld CN, Gilsdorf CS, Carlson KN, Kircher MR, et al. Reversible epigenetic alterations regulate class I HLA loss in prostate cancer. Commun Biol. 2022;5:897. https://doi.org/10.1038/s42003-022-03843-6.
Khan AN, Gregorie CJ, Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008;57:647–54. https://doi.org/10.1007/s00262-007-0402-4.
Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, et al. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008;68:9601–7. https://doi.org/10.1158/0008-5472.Can-07-5270.
Leclercq S, Gueugnon F, Boutin B, Guillot F, Blanquart C, Rogel A, et al. A 5-aza-2’-deoxycytidine/valproate combination induces cytotoxic T-cell response against mesothelioma. Eur Respir J. 2011;38:1105–16. https://doi.org/10.1183/09031936.00081310.
Hirano M, Imai Y, Kaito Y, Murayama T, Sato K, Ishida T, et al. Small-molecule HDAC and Akt inhibitors suppress tumor growth and enhance immunotherapy in multiple myeloma. J Exp Clin Cancer Res. 2021;40:110. https://doi.org/10.1186/s13046-021-01909-7.
Afolabi LO, Bi J, Li X, Adeshakin AO, Adeshakin FO, Wu H, et al. Synergistic Tumor Cytolysis by NK Cells in Combination With a Pan-HDAC Inhibitor, Panobinostat. Front Immunol. 2021;12:701671.https://doi.org/10.3389/fimmu.2021.701671.
Cho H, Son WC, Lee YS, Youn EJ, Kang CD, Park YS, et al. Differential Effects of Histone Deacetylases on the Expression of NKG2D Ligands and NK Cell-Mediated Anticancer Immunity in Lung Cancer Cells. Molecules. 2021;26.https://doi.org/10.3390/molecules26133952.
Idso JM, Lao S, Schloemer NJ, Knipstein J, Burns R, Thakar MS, et al. Entinostat augments NK cell functions via epigenetic upregulation of IFIT1-STING-STAT4 pathway. Oncotarget. 2020;11:1799–1815. https://doi.org/10.18632/oncotarget.27546.
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. 2021;20:28. https://doi.org/10.1186/s12943-021-01316-8.
Afonso J, Santos LL, Longatto-Filho A, Baltazar F. Competitive glucose metabolism as a target to boost bladder cancer immunotherapy. Nat Rev Urol. 2020;17:77–106. https://doi.org/10.1038/s41585-019-0263-6.
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021;33:1221-1233.e1211. https://doi.org/10.1016/j.cmet.2021.04.001.
Alvisi G, Termanini A, Soldani C, Portale F, Carriero R, Pilipow K, et al. Multimodal single-cell profiling of intrahepatic cholangiocarcinoma defines hyperactivated Tregs as a potential therapeutic target. J Hepatol. 2022;77:1359–72. https://doi.org/10.1016/j.jhep.2022.05.043.
Sufianov A, Begliarzade S, Beilerli A, Liang Y, Ilyasova T, Beylerli O. Circular RNAs as biomarkers for lung cancer. Noncoding RNA Res. 2023;8:83–8. https://doi.org/10.1016/j.ncrna.2022.11.002.
Chen Y, Ling Z, Cai X, Xu Y, Lv Z, Man D, et al. Activation of YAP1 by N6-Methyladenosine-Modified circCPSF6 Drives Malignancy in Hepatocellular Carcinoma. Cancer Res. 2022;82:599–614. https://doi.org/10.1158/0008-5472.Can-21-1628.
Hu C, Xia R, Zhang X, Li T, Ye Y, Li G, et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol Cancer. 2022;21:24. https://doi.org/10.1186/s12943-022-01501-3.
An M, Zheng H, Huang J, Lin Y, Luo Y, Kong Y, et al. Aberrant Nuclear Export of circNCOR1 Underlies SMAD7-Mediated Lymph Node Metastasis of Bladder Cancer. Cancer Res. 2022;82:2239–53. https://doi.org/10.1158/0008-5472.Can-21-4349.
Tang X, Ren H, Guo M, Qian J, Yang Y, Gu C. Review on circular RNAs and new insights into their roles in cancer. Comput Struct Biotechnol J. 2021;19:910–28. https://doi.org/10.1016/j.csbj.2021.01.018.
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, et al. Translation of CircRNAs. Mol Cell. 2017;66:9-21.e27. https://doi.org/10.1016/j.molcel.2017.02.021.
Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–11. https://doi.org/10.1038/nrm.2015.32.
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695. https://doi.org/10.1038/s41467-019-12651-2.
Chi J, Liu S, Wu Z, Shi Y, Shi C, Zhang T, et al. circNSUN2 promotes the malignant biological behavior of colorectal cancer cells via the miR‑181a‑5p/ROCK2 axis. Oncol Rep. 2021;46.https://doi.org/10.3892/or.2021.8093.
Acknowledgements
We are very grateful to BioRender (https://www.biorender.com) and the creators (Mirko Scrivano and Charlotte Butterworth) for providing some of the images and elements in our figures.
Disclosure of relationships and activities
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (NO. 81972193), and the Scientific Research Foundation for Recruited Talents, West China Hospital of Stomatology Sichuan University (NO.QDIF2019-1).
Author information
Authors and Affiliations
Contributions
Zhuojun Xie and Zirui Zhou contributed equally to this work. Shiwen Zhang and Bin Shao designed the study and reviewed the manuscript. Zhuojun Xie, Zirui Zhou and Shuxian Yang wrote the manuscript. Zhuojun Xie and Zirui Zhou reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xie, Z., Zhou, Z., Yang, S. et al. Epigenetic regulation and therapeutic targets in the tumor microenvironment. Mol Biomed 4, 17 (2023). https://doi.org/10.1186/s43556-023-00126-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43556-023-00126-2