
Abstract
Background
Ewing sarcoma is a malignant round cell neoplasm and categorized as part of Ewing family tumor (EFT). Only 1-4% of all Ewing sarcomas are primarily located in the head and neck region and the isolated primary sinonasal sarcoma represents a rare entity. This case was reported due to its rarity and dilemma in diagnosis of recurrent post-treatment.
Case presentation
We report a rare case of isolated left maxillary sinus Ewing sarcoma, presented with frequent unprovoked epistaxis, nasal blockage, and rhinorrhea from the left nostril for more than 2 years. Histopathological and immunohistochemical studies performed on the tissue taken from the left nasal mass confirmed Ewing sarcoma. He responded well to the 9 cycles of chemotherapy, evidenced by regression of mass from repeated CECT. However, the post-treatment scan showed multifocal nodular mucosal thickening involving the medial, anterior, and lateral walls of the maxillary sinus with corresponding bony resorption of the medial wall, raising a suspicion of residual disease. As such, he underwent endoscopic medial maxillectomy, which intraoperative findings revealed mild thickening of normal mucosa within the left maxillary sinus and histopathological examination showed no evidence of malignant cell. He showed no signs of recurrence during subsequent follow-up.
Conclusion
As a conclusion, in a case of suspicious residual or recurrent disease detected post-chemotherapy, MRI is an option to differentiate between mucosal thickening and residual tumor. However, the role of surgical resection is equally important in assisting the tissue diagnosis and ensures a better local control, perhaps can increase the survival rate.
Similar content being viewed by others


Background
Ewing sarcoma is a malignant round cell neoplasm originating from both skeletal and extraskeletal structures [1, 2]. In children, osteosarcoma is the most common malignant bone tumor followed by Ewing sarcoma [2]. In general, skeletal forms are more common. Long bones of the extremities are the most common areas for skeletal forms while soft tissue of lower extremities, paravertebral tissues, chest wall, and retroperitoneum are common areas for the extraskeletal form [1]. Primary sinonasal location is rare. Only 1–4% of all Ewing sarcomas are primarily located in the head and neck region [3,4,5]. The term Ewing sarcoma is a part of Ewing family tumor (EFT) used in soft tissue neoplasms which originally described as primitive neuroectodermal tumors (PNET) [5] Multiple differential diagnoses of round cells neoplasms and specific tumors for the site makes diagnosis challenging [2]. We report a case of extraskeletal Ewing sarcoma of the maxillary sinus in a 26-year-old male who presented with epistaxis, nasal obstruction, and rhinorrhea, mimicking symptoms of chronic rhinosinusitis with nasal polyposis.
Case presentation
A 26-year-old Malay male presented to the otorhinolaryngology outpatient department with history of frequent mild to moderate unprovoked epistaxis from the left nostril for more than 2 years. His other complaints were left nasal blockage and rhinorrhea, with no other symptoms from ear, throat, and neck. Apart from that, he was a nonsmoker with no significant family history of malignancy.
Clinical examination showed no external nasal deformity and normal skin with no nasal or facial swelling. However, there was an obvious polyp-like-mass covered with crusting which filled up the entire left nostril and the cold spatula test showed no misting. Further examination with nasoendoscopy revealed the polypoidal mass was arising from middle meatus and extended to posterior choana. The mass was inflamed and friable. The eye, neck, and ear examinations were unremarkable.
A biopsy was taken from the nasal mass in the office following careful topical decongestion. No immediate complications or excessive bleeding occurred post-procedure. Histopathological examination on the tissue taken from the left nasal mass showed uniform small round blue cells with fine nuclear chromatin and vacuolated cytoplasm arranged in sheet with occasional mitoses. Given the various differential diagnoses of small round blue cell tumors in the sinonasal tract, immunohistochemical test carried out. Further analysis showed the small blue cells to be immunoreactive toward vimentin, CD99 and FLI-1 and negative for CK, S-100 and desmin. These collective results clinched the diagnosis of Ewing sarcoma (Fig. 1).

All pictures taken under magnification ×40 using microscope Olympus BX43. a The tumor consists of uniform small round blue arranged in sheets. b Tumor cells positive for FLI-1. c Tumor cells positive for CD99
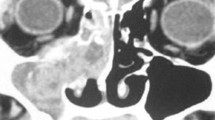
An initial contrast-enhanced computed tomography (CECT) scan from brain till pelvis was performed to stage the disease. The CECT revealed an isolated lesion of the left maxillary sinus with no regional, distant metastasis or corresponding skeletal, or extraskeletal lesions (Fig. 2).

CECT scan showed a heterogeneous mass arising from the left maxillary sinus extending to the left nasal cavity through the widened secondary ostium (arrow)
Patient was treated by oncology team with chemotherapy (vincristine, doxorubicin, cyclophosphamide and ifosfamide, etoposide). Following 9 cycles of chemotherapy, repeated nasoendoscopy examination of the left nasal cavity showed complete resolution of the polypoidal nasal mass. This finding was supported by a repeated CECT imaging, which revealed a significant regression of left sinonasal mass. However, multiple nodular mucosal thickening was seen at anterior, lateral, and medial wall of the left maxillary sinus with evidence of bony resorption on the medial wall of the maxillary sinus, raising suspicion of residual disease (Fig. 3).

Repeated CECT scan post-chemotherapy showed multiple nodular opacification (arrow) at medial wall (a) and anterior and lateral wall (b) of left maxillary sinus
Following that, a diagnosis of residual Ewing sarcoma of the left maxillary sinus was made. He was then underwent endoscopic medial maxillectomy. However intraoperatively, normal mucosa was encountered within the left maxillary sinus. Correspondingly, the histopathological examination showed no evidence of malignant cells. Up to date, regular monitoring for this patient showed neither any signs of residual nor recurrence of the disease at 12 months post completion of chemotherapy and surgery.
Discussion
Ewing sarcoma was first described by James Ewing in 1921 [4]. However, the first case of an Ewing sarcoma arising in soft tissue/extraskeletal was reported by Angervall and Enzinger in 1975, 50 years later owing to its rarity [6]. Extraskeletal Ewing sarcoma is a rare, rapidly growing, round cell malignant tumor that can develop in any soft tissues at any locations [1, 6, 7].
They are more common in white populations, with a slight predominance in male [8]. Peak incidence in the second decade with nearly 80% of patients are younger than 20 years old [7]. About a three quarter of Ewing sarcomas arise in bone [8]. Primary sinonasal Ewing sarcoma, subgroup of head and neck is rare. The exact percentage is unknown as most studies have not focused specifically on the sinonasal region [2]. About a quarter of patients have detectable metastases at diagnosis with lung as the commonest site [8].
Tumors arising in the nasal cavities and paranasal sinuses can present with nonspecific symptoms such as nasal obstruction, rhinorrhea, and epistaxis making the diagnosis of Ewing sarcoma difficult. Therefore, patients usually present with tumors that are locally advanced or even metastatic at the time of diagnosis [2, 9]. In this case, although the patient presented with symptoms mimicking chronic rhinosinusitis, the friable unilateral nasal mass found on nasoendoscopy raised a suspicion of a malignant condition, making biopsy mandatory even at the first visit. Our initial differential diagnosis included inverted papilloma or sinonasal carcinoma which were more commoner for the unilateral sinonasal mass [7].
Based on clinical and radiological examination alone, it is difficult to differentiate Ewing sarcoma from similar tumors arising from the sinonasal tract; hence, it requires a histopathological examination, immunohistochemistry, and a cytogenetic analysis to make the diagnosis [7]. There are various differential diagnosis of small round blue cell tumors in the sinonasal tract, specifically rhabdomyosarcoma, lymphoma, poorly differentiated carcinomas, melanoma, olfactory neuroblastoma, and Ewings family tumors (EFT) [7]. On immunohistochemical examination, the neoplastic cells strongly and diffusely express membranous CD99 in nearly all cases. Nuclear FLI-1 is seen in large percentage of cases. Myogenic and hematolymphoid markers typically absent [10]. In our case, the immunohistochemistry test showed the small blue cells are immunoreactive for vimentin, CD99 and FLI-1, and negative for CK, S-100, and desmin. In Ewing family tumor (EFT), the most common translocation diagnostic for Ewing sarcoma is the t(11,22)(q24;q12) translocation, accounting more than 85% of cases. Other diagnostic translocations involving the Ewing sarcoma locus on chromosome 22, including t(21,22)(q22;q12) and t(7,22)(p22;q12) [6, 10]. However, in our case, we did not performed the cytogenetic test in view of costs related issues. Therefore, the diagnosis of Ewing sarcoma was based on the histological features and its confirmatory immunohistochemical staining.
Option of treatment for Ewing sarcoma can be broadly divided into surgical resection, chemotherapy, and radiotherapy. The effective treatment plan includes combined surgical excision and modern chemotherapy or radiotherapy [5, 7, 11]. Combined treatment has increased the overall 5-year survival rate 20 to 70% [4, 11]. Chemotherapy reduces the tumor size and clear micrometastasis. Responses to chemotherapy were categories to complete response, partial response, stable response, and progressive disease depend on tumor size changes or any new lesion noted [11]. Surgical resection is recommended after chemotherapy eradicate micrometastatic disease and facilitate effective local control measures with wide negative margins unless otherwise contraindicated [4, 6]. Imaging evaluation required in monitoring the disease. Computed tomography is used to monitor bone destruction and mass propagation fields, while MRI is an ideal method for the evaluation of soft tissue involvement around the primary lesion [12].
In this case, a suspicious residual lesion was detected in the left maxillary sinus with evidence of bony resorption in the medial wall of maxillary sinus showed in repeated CECT. Subsequent left endoscopic medial maxillectomy was performed; however, a normal mucosa within the left maxillary sinus was found intraoperatively with no evidence of malignancy cell reported from the histopathological examination. There was a similar reported case of residual lesion by Nasser F et al. He has reported a residual soft tissue density in CT scan after completion of chemoradiotherapy, which MRI showed it was represent underlying fibrosis rather than residual tumor. The patient was reported to have no recurrence in 2 years upon completion treatment [4]. On the other hand, there was a reported case of recurrence post initial surgical excision and chemotherapy which required a second surgery for local control [12].
Early diagnosis followed by wide resection, chemotherapy, and radiotherapy can leave the patient disease-free for a long time. However, there has been debate about the need to use radiotherapy to guarantee local control in patients who seem to have achieved complete remission with surgery and chemotherapy [9]. Many authors agree that radiotherapy is not required when surgical resection had provided with wide clear margins [9]. If histological examination of a radically resected tumor reveals more than 10% of viable tumor cells, chemoradiotherapy can be administered postoperatively for control of local recurrence in view of strict anatomic constraints with a high rate of unresectability in tumors of the head and neck [13]. Furthermore, the chemoradiotherapy regimen is the exclusive treatment for inoperable localized tumors or in case of refusal of surgery [2, 13]. In this case, the patient has undergone chemotherapy and followed by surgical resection as a diagnostic and therapeutic method in view present of suspicious lesion query of residual in repeated CECT scan.
The patient’s age, stage, anatomic location, and size of the tumor are important prognostic factors. Patients younger than 15 years old without metastasis have a better outcome with 55% 5-year survival rate compared to 22% with metastasis [11].
Conclusion
Ewing sarcoma involves a wide histological spectrum that makes significant diagnostic challenges, which need expert immunohistochemistry and cytogenetics examination. Primary head and neck Ewing sarcoma is rare, and sinonasal location is even rarer. It is a multidisciplinary management for Ewing sarcoma, with an effective treatment plan including chemotherapy and surgery or chemoradiotherapy is needed to guarantee local recurrence and healing with a better survival rate. In a case of suspicious residual or recurrent disease detected post neoadjuvant chemotherapy, MRI is an option to differentiate the lesion. However, the rule of surgical resection is equally important in assisting the tissue diagnosis and ensures a better local control, perhaps can increase the survival rate.
Availability of data and materials
N/A
Abbreviations
- CECT:
-
Contrast-enhanced computed tomography
- EFT:
-
Ewing family tumor
- MRI:
-
Magnetic resonance imaging
- PNET:
-
Primitive neuroectodermal tumors
References
Kothari DC, Kumar M. Extraskeletal primary Ewing’s sarcoma of nasopharyngeal region - rare tumour a case report. 2015;2(2):143 –8.
Souheil J, Skander K, Sawssen D, Sana M, Delia Y, Khalil M et al (2016) Ewing sarcomas of the sino-nasal tract and maxillary bone. Egypt J Ear, Nose, Throat Allied Sci 17(3):147–153
Shah S, Huh K-H, Yi W-J, Heo M-S, Lee S-S, Choi S-C (2014) Primitive neuroectodermal tumor of the maxillary sinus in an elderly male: a case report and literature review. Imaging Sci Dent. 44(4):307
Nasser F, Al Khalil M, Al Homsi O, Bin ME (2015) Ewing ’s sarcoma of the maxillary sinus. Egypt J Ear, Nose, Throat Allied Sci. 16(2):177–180
Kawabata M, Yoshifuku K, Sagara Y, Kurono Y (2008) Ewing’s sarcoma/primitive neuroectodermal tumour occurring in the maxillary sinus. Rhinology 46(1):75–78
Iwamoto Y (2007) Diagnosis and treatment of Ewing’s sarcoma. Jpn. J. Clin. Oncol. 37(2):79–89
Satish D, Nanadakumar R, Balasubramanya AM, Mathew N (2018) A rare case of Ewing’s sarcoma in the sinonasal tract. Int J Otorhinolaryngol Head Neck Surg. 4(1):304–307
Balamuth NJ, Womer RB (2010) Ewing’s sarcoma. Lancet Oncol. 11(2):184–192
Gradoni P, Giordano D, Oretti G, Fantoni M, Barone A, La Cava S et al (2011) Clinical outcomes of rhabdomyosarcoma and Ewing’s sarcoma of the head and neck in children. Auris Nasus Larynx 38(4):480–486
K. Unni FMCDMF (2004) Pathology and genetics of tumours of soft tissue and bone. Published 2002, 1st edition, ISBN 92 832 24132. Surg. Oncol. 13(1):297
Yeshvanth S, Bhandary S, Shetty J, Makannavar J, Ninan K, Lakshinarayana KP (2012) Rare case of extraskeletal Ewings sarcoma of the sinonasal tract. J. Cancer Res. Ther. 8(1):142
Yaprak N, Toru HS, Ozbudak IH, Derin AT. Primary extraskeletal Ewing’s sarcoma of the maxillary sinus. Braz J Otorhinolaryngol. 2019 J;85(4):538-541.
Whaley JT, Indelicato DJ, Morris CG, Hinerman RW, Amdur RJ, Mendenhall WM et al (2010) Ewing tumors of the head and neck. Am J Clin Oncol Cancer Clin Trials. 33(4):321–326
Acknowledgements
We would like to thank all the persons involved in this article directly or indirectly for the contributions.
Funding
None
Author information
Authors and Affiliations
Contributions
IA contributes as a main writer for the case report. AA contributes the idea to check the grammar, sentences, contents, and give expert opinions regarding case and discussion. EH contributes the idea to polish the grammar, input regarding contents, expert opinion regarding disease and discussion. MA contributes the idea to polish the grammar, contents as the whole and expert opinion regarding the discussion. All authors have read and approved the final manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
N/A
Consent for publication
Verbal consent taken from patient.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hamid, I.A., Md Arepen, S.A., Hassan, N.E. et al. A case report of rare isolated Ewing sarcoma of left maxillary sinus. Egypt J Otolaryngol 36, 6 (2020). https://doi.org/10.1186/s43163-020-00008-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43163-020-00008-2