
Abstract
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by insulin resistance (IR) and hyperglycemia. The development of inflammatory disorders in T2DM triggers the activation of different growth factors as a compensatory mechanism to reduce IR and adipose tissue dysfunction in T2DM. Fibroblast growth factor 21 (FGF21) which is involved in the regulation of glucose homeostasis is attractive to be a novel therapeutic target in the management of T2DM. FGF21 has poor pharmacokinetic profile as it rapidly degraded; therefore, FGF21 analogs which are more stable can be used in T2DM patients. However, FGF21 analogs are tested pre-clinically but not approved in clinical settings. Therefore, searching for anti-diabetic agents who enhance FGF21 expression is mandatory. It has been shown that metformin which used as a first-line in the management of T2DM can positively affect the expression of FGF21, though the underlying mechanisms for metformin-induced FGF21 expression are not fully elucidated. Therefore, this review from published studies aimed to find how metformin improves insulin sensitivity through FGF21-dependent pathway in T2DM. In conclusion, metformin improves FGF21 signaling in T2DM, and this could be a novel mechanism for metformin in the amelioration of glucose homeostasis and metabolic disorders in T2DM patients.
Similar content being viewed by others


Introduction
Type 2 diabetes mellitus (T2DM) is a chronic metabolic disorder characterized by insulin resistance (IR) and hyperglycemia [1]. T2DM is linked with inflammatory disorders and end-organ injury due to hyperglycemia-induced oxidative stress and the release of pro-inflammatory cytokines [2]. IR and relative insulin deficiency due to pancreatic β cell dysfunction is the major feature of T2DM [3]. Activation of inflammatory disorders in T2DM occurs due to immune cell deregulation and infiltration of immune cells into adipose tissue that advances the expression of pro-inflammatory cytokines with the development of systemic inflammation [3]. Extended low-grade inflammation in T2DM by hyperglycemia and adipose tissue activation increases the development of IR and associated complications [4]. Inflammatory disorders participate in the progression of IR, T2DM, and systemic complications [5]. It has been revealed that hypoglycemia and hyperglycemia as well as glucose variability activate oxidative stress which enhances inflammatory disorders [6]. Furthermore, environmental and genetic factors such as stress, diet, and smoking are affianced with the activation of chronic inflammation in T2DM [7]. These inflammatory changes trigger the activation of different growth factors as a compensatory mechanism to reduce IR and adipose tissue dysfunction in T2DM [8]. One of the most important growth factors is fibroblast growth factor 21 (FGF21) which is involved in the regulation of glucose homeostasis by increasing insulin sensitivity [9]. Of note, the insulin-sensitizing drug metformin which is used as a first-line in the management of T2DM can positively affect the expression of FGF21 [10]. Therefore, this review of published studies aimed to find how metformin improves insulin sensitivity through the FGF21-dependent pathway in T2DM.
Fibroblast growth factor 21
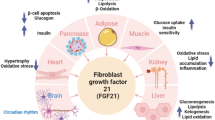
FGF21 is a peptide hormone released from the liver as a member of different hormones called hepatocytes [11]. FGF21 is extremely expressed in the liver, pancreas, and adipose tissues [12]. Skeletal muscles and other tissues also produce FGF21 via a phosphoinositide 3 kinase (PI3K)-mediated pathway [13]. Expression of FGF21 differs by diverse pathophysiological conditions, fasting, and exercise which increases FGF21 expression in the liver and muscles correspondingly [14]. In addition, satiety and cold exposure augment FGF21 expression in the pancreas and adipose tissue correspondingly [15]. Different cellular signaling affects FGF21 expression like liver X receptor (LXR) which inhibits FGF21 expression [16]. FGF21 expression is also induced by thyroid hormones and fructose [17]. A chronic low-protein diet promotes FGF21 expression which improves the metabolic profile [18]. Hepatic FGF21 expression is induced by peroxisome proliferator activator receptor alpha (PPAR-α), and adipose tissue FGF21 expression is induced by PPAR gamma (PPAR-α) [19]. However, mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS2) and sirtuin-1 (SIRT1) specifically induce FGF21 expression [20]. FGF21 binds four types of FGF receptors 1–4. Interaction of FGF21 with its receptor is improved by β-Klotho which is a transmembrane protein that acts as a co-receptor for FGF21 [15]. FGF21 augments glucose uptake and gluconeogenesis [21]. FGF21 has many beneficial effects on different body systems (Fig. 1).

Endogenous FGF21 expression and interaction in metabolic organs. The liver, white and brown adipose tissue, skeletal muscle, the pancreas, and the heart are among the metabolic organs that express and secrete FGF21 in response to diverse stimuli [13, 15, 18, 20]. The FGFR1/KLB complex in the brain and white adipose tissue can be targeted by systemic FGF21, which is mostly produced by the liver. CRF, corticotropin-releasing factor; SNS, sympathetic nervous system; FGF21, fibroblast growth factor-21; WAT, white adipose tissue
Pharmacology of metformin
Metformin is an insulin-sensitizing agent reduces IR [22, 23]. Metformin is 3-(diaminomethylidene)-1,1-dimethylguanidine [24] (Fig. 2).

Chemical structure of metformin
Metformin is an orally active drug, absorbed from the small intestine via plasma membrane monoamine transporter (PMAT) expressed in the enterocytes [25]. Organic cation transporter 2 (OCT2) which is expressed on the brush border of enterocytes is concerned with the uptake of metformin [26]. Hepatic uptake of metformin is mostly by OCT1 and less by OCT3. The uptake of metformin by the renal epithelial cell is mediated by OCT2, and its excretion by the kidney is through multidrug and toxin extrusion 1 (MATE1) [27]. Metformin is not metabolized and is excreted unchanged by the kidney. Metformin half-life is 5 h, highly distributed, and its plasma steady state ranged 54–4133 [28]. Metformin has an exclusive pharmacodynamic effect (Fig. 3); metformin is a positive charge molecule, extremely accumulated in the mitochondria because of the negative charge of the mitochondrial membrane [29].

Metformin’s pharmacodynamics pathway. Cells have been stylized to show how metformin works. Metformin appears to elevate insulin sensitivity and AMPK levels, which improves glucose transport [30, 31]. AMP, adenosine monophosphate; AMPK, adenosine monophosphate-activated protein kinase; mGPD, mitochondrial glycerophosphate dehydrogenase; OCT1, organic cation transporter 1; NAD, nicotinamide adenine dinucleotide; NADH, H for hydrogen; FADH, flavin adenine dinucleotide
Metformin inhibits ATP production through the inhibition of mitochondrial complex I leading to an increase AMP: ATP with increasing levels of adenosine monophosphate protein kinase (AMPK) [32]. AMPK inhibits gluconeogenesis and fat synthesis, decreases hepatic fat storage, and improves insulin sensitivity and anaerobic glucose metabolism in the enterocytes [30]. Metformin promotes glucose utilization by gut microbiota with the activation release of glucagon-like peptide 1 (GLP-1) from L cells in the intestine [33]. Furthermore, metformin improves peripheral glucose utilization by increasing the expression of glucose transporter type 4 (GLUT4) with subsequent improvement of insulin sensitivity [31]. Moreover, metformin has pleiotropic properties like anti-inflammatory and oxidant effects thereby reducing the risk of diabetic complications [34].
Prolonged use of metformin is linked with the development of various adverse effects counting gastrointestinal disorders like diarrhea, nausea, vomiting, abdominal pain, and loss of appetite [35]. Nevertheless, prolonged use of metformin is linked with the development of weight loss, B12, and folate deficiency with a risk of peripheral neuropathy and cognitive impairment [36]. A rare but serious adverse effect related to metformin use is lactic acidosis which developed due to a reduction in the use of lactate by inhibited gluconeogenesis process [37]. Metformin toxicity due to overdosage leads to hypoglycemia and lactic acidosis; it is treated by hemodialysis [38].
Moreover, metformin had low interaction with other drugs as it was not metabolized and excreted unchanged by the kidney [39]. Aspirin and anti-diabetic agents increase the risk of hypoglycemia when used with metformin [40], though some drugs like cimetidine, topiramate, ranolazine, and cephalexin increase the risk for the development of lactic acidosis by competing with metformin renal excretions [39].
Effects of metformin on FGF21 in T2DM
FGF21 in T2DM
It has been reported that FGF21 plays a critical role in the regulation of glucose metabolism and can be used as a monotherapy in the management of T2DM [41]. FGF21 exerts a beneficial effect on T2DM and obesity by reducing blood glucose and restoring the function of adipose tissue respectively [42]. Of note, FGF21 expression is increased in T2DM patients as a compensatory mechanism to counteract inflammatory disorders and associated IR [43]. An experimental study illustrated that hepatic expression of FGF21 mRNA was increased in mice with a high-fat diet [44]. Obesity in children increases circulating FGF21 levels due to the development of FGF21, and weight loss decreases FGF21 levels [45]. It has been shown that FGF21 level is increased during fasting; however, this reaction is impaired in mice with experimental diabetes [46]. FGF21 analog LY2405319 was confirmed to improve blood glucose in streptozotocin-induced diabetes in mice through modulation metabolism of brown adipose tissue (BAT) [46]. Furthermore, there is a significant change in postprandial FGF21 level in diabetes according to preclinical and clinical findings [47, 48]. FGF21 level is higher in T2DM patients compared to controls due to the development of FGF21 resistance [47]. Chavez et al. [49] observed that circulating FGF21 level is increased in patients with impaired glucose tolerance and T2DM that correlated with IR. A population-based prospective study in China observed that higher FGF21 level in prediabetes subjects was a predictor for the development of T2DM within 5.4 years [50].
Under normal physiological conditions, glucose stimulates while insulin inhibits FGF21 secretion [51]. However, FGF21 secretion is mainly driven by blood glucose independent of insulin or glucagon-like peptide 1 (GLP-1) secretion in normal healthy subjects [51]. In addition, many studies reported that insulin did not affect FGF21 secretion [52, 53]. However, super-physiological insulin level enhances FGF21 secretion [54]. In addition, glucagon enhances FGF21 secretion independent of insulin level [55]. Similarly, glucagon promotes hepatic expression of FGF21 [56]. In T2DM, glucagon and insulin levels are augmented and implicated in the development of diabetic complications [57]. Therefore, increasing glucagon and insulin levels in T2DM together with FGF21 resistance may explain a higher level of FGF21 in T2DM patients.
Of note, FGF21 has a poor pharmacokinetic profile; it rapidly degraded in vitro and in vivo, so it has a short half-life [46]. Therefore, FGF21 analogs could be novel therapeutic agents for the management of T2DM and metabolic disorders [58]. Therefore, FGF21 analogs that are more stable can be used in T2DM patients. It has been shown that in two administrations of FGF21 analogs, mFGF21 was more effective than insulin glargine and GLP-1 receptor agonist liraglutide in the reduction of glycated hemoglobin level, improvement of insulin sensitivity, and lipid profile [59]. The half-life of mFGF21 is 20 times longer than FGF21, so it induces rapid and persistent reduction of blood glucose independent of insulin secretion without risk of hypoglycemia [58]. In addition, mFGF21 increases the expression of glucokinase (GK) and GLUT-1 leading to more reduction of blood glucose [59]. Additionally, FGF21 analogs AKR-001 improve insulin sensitivity and reduce metabolic complications in FGF21 analogs [60]. A previous clinical trial on the use of FGF21 variant LY2405319 (LY) compared to placebo in obese patients with T2DM revealed that LY use for 4 weeks improves dyslipidemia and insulin sensitivity with significant reduction of atherogenic risk [61]. These findings indicated that FGF21 analogs are highly effective in the management of T2DM.
The underlying mechanism for the reduction of blood glucose is related to the inhibition of G6Pase and activation of GK and GLUT-1 as illustrated in Fig. 4.

The glucose-lowering effect of FGF21. FGF21, fibroblast growth factor-21; ATP, adenosine triphosphate; ADP, adenosine diphosphate
Metformin and FGF21
It has been reported from preclinical and clinical findings that metformin increases the expression of FGF21 [62,63,64]. In vitro and in vivo studies showed that metformin promotes the expression of FGF21 through AMPK dependent pathway [62]. In addition, metformin increases FGF21 expression through the induction of expression of activating transcription factor 4 (ATF4) [62]. Metformin therapy for 6 months in T2DM patients increased FGF21 circulating level through ATF4 [62]. ATF4 in addition to its neurological is also involved in the regulation of lipid and glucose metabolism through modulation of insulin secretion and sensitivity [65]. It has been shown that metformin has protected against lipopolysaccharide (LPS)-induced inflammation by increasing the expression of FGF21 in rats [66]. Furthermore, metformin improves blood glucose and increases the expression of hepatic FGF21 via AMPK pathway [63]. Therefore, FGF21 seems to be a potential mediator for the action of metformin in the regulation of glucose homeostasis and metabolic adaptive response [67].
Moreover, metformin regulates metabolic balance through the induction expression of FGF21 in adipocytes and the liver [64]. Increasing expression of FGF21 by metformin not only regulates blood glucose but also contributes in the regulation of autoimmune response and atherogenic risk [68, 69]. Metformin through AMPK/FGF21 improves the differentiation of brown adipose tissue and regulates immune balance in obese mice [68]. In addition, metformin reduces the progression of atherosclerosis by increasing the expression of FGF21 [69].
Of note, β-Klotho which is a co-receptor for FGF21 [15] is also deregulated in T2DM [70]. Notoriously, β-Klotho/FGF21 complex seems to be an attractive target in the management of T2DM [70]. It has been observed that β-Klotho serum is reduced in T2DM [71], and this may explain the development of FGF21 resistance in T2DM patients. A case–control study involving 261 T2DM and 106 healthy controls observed that β-Klotho serum reduced as compared to controls [71]. Notably, β-Klotho is highly downregulated in T2DM patients, and downregulation induces the development of diabetic complications [70]. Therefore, amelioration of β-Klotho expression by anti-diabetic agents may reduce the risk of diabetic complications. Interestingly, metformin promotes β-Klotho expression thereby it acts as anti-aging agent [23]. In addition, metformin prevents diabetic nephropathy by increasing the expression of β-Klotho [72].
It has been shown that diabetic mice had low FGF21 sensitivity due to higher circulating miR34a levels [73]. Consequently, increasing of β-Klotho and its effectors SIRT1 and ERK by metformin was shown to improve FGF21 sensitivity [73], though over-expression of FGF21 in T2DM may lead to FGF21 resistance [74]. Furthermore, IR and high pro-inflammatory cytokines like TNF-α repress the expression of β-Klotho leading to FGF21 resistance in adipocytes that further aggravate inflammatory disorders [75]. Thus, mitigation of IR by metformin can regulate β-Klotho expression and abrogate FGF21 resistance. These observations suggest that metformin via increasing β-Klotho expression can enhance the functional activity of FGF21.
Chau et al. [76] illustrated that FGF21 regulates energy metabolism by increasing the expression of PGC-1α, SIRT1, and AMPK. Inhibition of PGC-1α, SIRT1, and AMPK reduces the effect of FGF21 on gene expression and oxygen consumption by adipocytes with the development of FGF21 resistance [76]. Of note, metformin activates PGC-1α, SIRT1, and AMPK [77, 78]. Metformin attenuates pancreatic β cell apoptosis and prevents IR via the activation of PGC-1α, SIRT1, and AMPK [77]. In addition, metformin prevents gluconeogenesis through increasing the expression of hepatic PGC-1α [78]. Henceforth, metformin through modulation of PGC-1α, SIRT1, and AMPK improves FGF21 signaling and reduces FGF21 resistance.
Moreover, metformin increases the production of GLP-1 from L cells with significant protection of GLP-1-producing cells [79]. Analysis from clinical trials illustrated that metformin increases the release of GLP-1 [80]. GLP-1 acts additively with FGF21 against the development of T2DM in mice [81]. Genetic ablation of glucagon receptor increases FGF21 expression [81]; therefore, increasing GLP-1 by metformin reduces glucagon and improves the release of FGF21. Remarkably, GLP-1 blocks hepatic glucose output through increasing expression of FGF21 [82]. Therefore, glucose homeostasis induced by GLP-1 is mediated by FGF21 signaling. Supporting this notion, the inhibition of FGF21 receptors by antibodies reduced the inhibitory effect of GLP-1 on hepatic glucose output [82]. Thus, metformin improves FGF21 by increasing the expression of GLP-1 in patients with T2DM.
Furthermore, metformin augments the expression of anti-inflammatory growth differentiation factor 15 (GDF15) which reduces body weight and improves insulin sensitivity [24, 83]. GDF15 improves the expression of β-Klotho in experimental acute kidney injury [84]. Fasting promotes the expression of FGF21 which enhances the release of GDF15 which in turn enhance the release of FGF21 [85]. In addition, both of FGF21 and GDF15 are augmented in response to mitochondrial dysfunction [86]. Of note, cold exposure activates sympathetic drive which promotes release of GDF15 though FGF21 [87]. Therefore, metformin through activation of GDF15 improves the expression and release of FGF21. Regarding role of inflammatory disorders in T2DM and their effects on the expression on GDF15 and FGF21, it has been shown that pro-inflammatory cytokines, p53, and angiotensin II promote the expression and release of GDF15 [88, 89]. Besides, proinflammatory cytokines attenuate the metabolic effect of FGF21 leading to FGF21 resistance [90]. Therefore, increasing of GDF15 by metformin can mitigates the inflammatory disorders in T2DM and enhances FGF21 action. In addition, FGF21 is anti-inflammatory by inhibiting the inflammatory signaling pathway NF-κB and increasing expression of anti-inflammatory nuclear factor erythroid 2-related factor 2 (Nrf2) [91]. Therefore, the anti-inflammatory effects of metformin might be mediated by FGF21. In this state, there is a complex interaction between GDF15 and FGF21 during inflammatory reactions in T2DM.
Of note, liver X receptor (LXR) inhibits FGF21 expression [16]. High-cholesterol fed promotes expression of LXR which reduces expression of FGF21 expression [16]. Likewise, LXR agonists TO-901317 reduce expression of FGF21 to protect the liver from cholesterol accumulation and intrahepatic lipolysis [16]. In addition, fasting-induced expression of FGF21 is inhibited by LXR agonists via the activation of histone deacetylase 3 (HDAC3) co-repressor in mice [92]. LXR is involved in lipid and glucose homeostasis, and dysregulation of LXR is involved in the pathogenesis of T2DM. Genetic variation of LXR is implicated in T2DM as confirmed in a clinical study [93]. Dysregulation of LXR in T2DM increases the risk for the development of nonalcoholic fatty liver disease (NAFLD) [94]. Different studies illustrated that metformin is effective against the development NAFLD in T2DM by various molecular mechanisms inclusion repression expression of LXR [95, 96]. It has been shown by many studies that metformin can reduce the expression of LXR [97, 98]. Metformin attenuates the development of NAFLD by downregulating the expression of LXR in mice [97]. Similarly, metformin reduces the hypothalamic pituitary adrenal axis in T2DM through induction phosphorylation of LXR in the pituitary [98]. Therefore, metformin through modulation of LXR can improve FGF21 expression.
Furthermore, hepatic FGF21 expression is induced by PPAR-α [19]; thus, PPAR-α agonists can enhance FGF21 expression [99]. Interestingly, metformin induces expression of GLP-1 independent of AMPK pathway but through activation of PPAR-α in mice [100]. In addition, metformin reduces the risk of atrial fibrillation in T2DM patients through induction expression of PPAR-α which regulates lipid metabolism in the atria [101]. Herein, metformin through induction of PPAR-α improves FGF21 expression.
Taken together, metformin improves FGF21 signaling in T2DM, and this could be a novel mechanism for metformin in the amelioration of glucose homeostasis and metabolic disorders in T2DM patients.
Conclusions
T2DM is a chronic metabolic disorder characterized by IR and hyperglycemia. Commencement of inflammatory disorders in T2DM occurs due to immune cell deregulation and infiltration of immune cells into adipose tissue that advances the expression of pro-inflammatory cytokines with the development of systemic inflammation. These inflammatory changes stimulate the activation of different growth factors as a compensatory mechanism to reduce IR and adipose tissue dysfunction in T2DM. One of the most important growth factors is FGF21 which is concerned with the regulation of glucose homeostasis by increasing insulin sensitivity. Of note, the insulin-sensitizing drug metformin which is used as a first-line in the management of T2DM can positively affect the expression of FGF21. FGF21 plays a critical role in the regulation of glucose metabolism and can be used as a monotherapy in the management of T2DM. FGF21 expression is increased in T2DM patients as a compensatory mechanism to counteract inflammatory disorders and associated IR. Glucagon promotes hepatic expression of FGF21 and in T2DM; glucagon and insulin levels are augmented and implicated in the development of diabetic complications. Therefore, increasing glucagon and insulin levels in T2DM together with FGF21 resistance may explain a higher level of FGF21 in T2DM patients. FGF21 has a poor pharmacokinetic profile; it rapidly degraded, so it has a short half-life. Thus, FGF21 analogs could be novel therapeutic agents for the management of T2DM and metabolic disorders. FGF21 analogs which are more stable can be used in T2DM patients. However, FGF21 analogs are tested preclinically but not approved clinical settings. Therefore, searching for anti-diabetic agents who enhance FGF21 expression is mandatory.
Metformin increases expression of FGF21 through AMPK dependent pathway. In addition, metformin increases FGF21 expression via induction of expression of different signaling pathways including PGC-1α, SIRT1, GDF15, PPAR-α, and GLP-1. In addition, β-Klotho which is a co-receptor for FGF21 is also deregulated in T2DM. Interestingly, metformin promotes β-Klotho expression. Therefore, mitigation of IR by metformin can regulate β-Klotho expression and abrogate FGF21 resistance. Henceforth, metformin improves FGF21 signaling in T2DM, and this could be a novel mechanism for metformin in the amelioration of glucose homeostasis and metabolic disorders in T2DM patients. Clinical trials and large-scale clinical studies are recommended in this regard.
Availability of data and materials
Not applicable.
References
Rasheed HA, Al-Kuraishy HM, Al-Gareeb AI, Hussien NR, Al-Nami MS (2019) Effects of diabetic pharmacotherapy on prolactin hormone in patients with type 2 diabetes mellitus: bane or boon. J Adv Pharm Technol Res 10(4):163
Al-Kuraishy HM, Al-Gareeb AI, Alblihed M, Cruz-Martins N, Batiha GE-S (2021) COVID-19 and risk of acute ischemic stroke and acute lung injury in patients with type ii diabetes mellitus: the anti-inflammatory role of metformin. Front Med 8:110
Al-Kuraishy HM, Al-Gareeb AI, Waheed HJ, Al-Maiahy TJ (2018) Differential effect of metformin and/or glyburide on apelin serum levels in patients with type 2 diabetes mellitus: concepts and clinical practice. J Adv Pharm Technol Res 9(3):80
Al-Nami MS, Al-Kuraishy HM, Al-Gareeb AI, Al-Mamoori F (2019) Metabolic profile and prolactin serum levels in men with type 2 diabetes mellitus: old-new rubric. Int J Crit Illn Inj Sci 9(3):120
Al-Kuraishy HM, Al-Gareeb AI, Alsayegh AA et al (2023) A potential link between visceral obesity and risk of Alzheimer’s disease. Neurochem Res 48:745–766. https://doi.org/10.1007/s11064-022-03817-4
Kovatchev B, Cobelli C (2016) Glucose variability: timing, risk analysis, and relationship to hypoglycemia in diabetes. Diabetes Care 39(4):502–510
Beulens JW, Pinho MG, Abreu TC, den Braver NR, Lam TM, Huss A, Vlaanderen J, Sonnenschein T, Siddiqui NZ, Yuan Z, Kerckhoffs J (2022) Environmental risk factors of type 2 diabetes—an exposome approach. Diabetologia 65(2):263–274
Qiao YC, Chen YL, Pan YH, Ling W, Tian F, Zhang XX, Zhao, HL (2017) Changes of transforming growth factor beta 1 in patients with type 2 diabetes and diabetic nephropathy: A PRISMA-compliant systematic review and meta-analysis. Medicine 96:e6583. https://doi.org/10.1097/MD.0000000000006583
Barb D, Bril F, Kalavalapalli S, Cusi K (2019) Plasma fibroblast growth factor 21 is associated with severity of nonalcoholic steatohepatitis in patients with obesity and type 2 diabetes. J Clin Endocrinol Metab 104(8):3327–3336
Al-Kuraishy HM, Al-Gareeb AI, Saad HM, Batiha GE (2023) Long-term use of metformin and Alzheimer’s disease: beneficial or detrimental effects. Inflammopharmacology 28:1–9
Woo YC, Xu A, Wang Y, Lam KS (2013) Fibroblast growth factor 21 as an emerging metabolic regulator: clinical perspectives. Clin Endocrinol 78(4):489–496
Nygaard EB, Møller CL, Kievit P, Grove KL, Andersen B (2014) Increased fibroblast growth factor 21 expression in high-fat diet-sensitive non-human primates (Macaca mulatta). Int J Obes 38(2):183–191
Sun H, Sherrier M, Li H (2021) Skeletal muscle and bone—emerging targets of fibroblast growth factor-21. Front Physiol 12:625287. https://doi.org/10.3389/fphys.2021.625287
Cuevas-Ramos D, Almeda-Valdes P, Gómez-Pérez FJ, Meza-Arana CE, Cruz-Bautista I, Arellano-Campos O, Navarrete-López M, Aguilar-Salinas CA (2010) Daily physical activity, fasting glucose, uric acid, and body mass index are independent factors associated with serum fibroblast growth factor 21 levels. Eur J Endocrinol 163(3):469–477
Pérez-Martí A, Sandoval V, Marrero PF, Haro D, Relat J (2017) Nutritional regulation of fibroblast growth factor 21: from macronutrients to bioactive dietary compounds. Hormone Mole Biol Clin Invest 30(1):20160034. https://doi.org/10.1515/hmbci-2016-0034
Uebanso T, Taketani Y, Yamamoto H, Amo K, Tanaka S, Arai H, Takei Y, Masuda M, Yamanaka-Okumura H, Takeda E (2012) Liver X receptor negatively regulates fibroblast growth factor 21 in the fatty liver induced by cholesterol-enriched diet. J Nutr Biochem 23(7):785–790
Rodgers M, Heineman B, Dushay J (2019) Increased fructose consumption has sex-specific effects on fibroblast growth factor 21 levels in humans. Obes Sci Pract 5(5):503–510
Larson KR, Chaffin AT, Goodson ML, Fang Y, Ryan KK (2019) Fibroblast growth factor-21 controls dietary protein intake in male mice. Endocrinology 160(5):1069–1080
Yu J, Yu B, Jiang H, Chen D (2012) Conjugated linoleic acid induces hepatic expression of fibroblast growth factor 21 through PPAR-α. Br J Nutr 107(4):461–465
Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, Yang Q, Gao B, Guarente L, Zang M (2014) Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 146(2):539–549
Li K, Li L, Yang M, Liu H, Boden G, Yang G (2012) The effects of fibroblast growth factor-21 knockdown and over-expression on its signaling pathway and glucose–lipid metabolism in vitro. Mol Cell Endocrinol 348(1):21–26
Zhang L, Liu T (2018) Clinical implication of alterations in serum Klotho levels in patients with type 2 diabetes mellitus and its associated complications. J Diabetes Complications 32(10):922–30
Prud’homme GJ, Kurt M, Wang Q. Pathobiology of the klotho antiaging protein and therapeutic considerations. Frontiers in Aging. 2022;3.
Al-kuraishy HM, Al-Gareeb AI, Alexiou A, Papadakis M, Nadwa EH, Albogami SM, Alorabi M, Saad HM, Batiha GE. Metformin and growth differentiation factor 15 (GDF15) in type 2 diabetes mellitus: a hidden treasure. J Diabetes. 2022.
Zhou M, Xia L, Wang J (2007) Metformin transport by a newly cloned proton-stimulated organic cation transporter (plasma membrane monoamine transporter) expressed in human intestine. Drug Metab Dispos 35(10):1956–1962
Chen Y, Li S, Brown C, Cheatham S, Castro RA, Leabman MK, Urban TJ, Chen L, Yee SW, Choi JH, Huang Y (2009) Effect of genetic variation in the organic cation transporter 2, OCT2, on the renal elimination of metformin. Pharmacogenet Genomics 19(7):497
Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH (2009) Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: a preliminary study. Diabetes 58(3):745–749
Triggle CR, Ding H (2017) Metformin is not just an antihyperglycaemic drug but also has protective effects on the vascular endothelium. Acta Physiol 219(1):138–151
Kenechukwu FC, Nnamani DO, Momoh MA, Attama AA (2022) Enhanced circulation longevity and pharmacodynamics of metformin from surface-modified nanostructured lipid carriers based on solidified reverse micellar solutions. Heliyon 8(3):e09100
Rena G, Hardie DG, Pearson ER (2017) The mechanisms of action of metformin. Diabetologia 60(9):1577–1585
Herman R, Kravos NA, Jensterle M, Janež A, Dolžan V (2022) Metformin and insulin resistance: a review of the underlying mechanisms behind changes in GLUT4-mediated glucose transport. Int J Mol Sci 23(3):1264
Ouyang J, Parakhia RA, Ochs RS (2011) Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 286(1):1–1
Bahne E, Hansen M, Brønden A, Sonne DP, Vilsbøll T, Knop FK (2016) Involvement of glucagon-like peptide-1 in the glucose-lowering effect of metformin. Diabetes Obes Metab 18(10):955–961
Markowicz-Piasecka M, Sadkowska A, Huttunen KM, Podsiedlik M, Mikiciuk-Olasik E, Sikora J. An investigation into the pleiotropic activity of metformin. A glimpse of haemostasis. Eur J Pharmacol. 2020;872:172984.
Piskovatska V, Stefanyshyn N, Storey KB, et al (2019) Metformin as a geroprotector: experimental and clinical evidence. Biogerontology 20:33–48. https://doi.org/10.1007/s10522-018-9773-5
Xu L, Huang Z, He X, Wan X, Fang D, Li Y (2013) Adverse effect of metformin therapy on serum vitamin B12 and folate: short-term treatment causes disadvantages? Med Hypotheses 81(2):149–151
Salpeter SR, Greyber E, Pasternak GA, Salpeter EE. Risk of fatal and nonfatal lactic acidosis with metformin use in type 2 diabetes mellitus. Cochrane Database Syst Rev. 2010(4).
Espada L, Dakhovnik A, Chaudhari P, Martirosyan A, Miek L, Poliezhaieva T, Schaub Y, Nair A, Döring N, Rahnis N, Werz O (2020) Loss of metabolic plasticity underlies metformin toxicity in aged Caenorhabditis elegans. Nat Metab 2(11):1316–1331
Stage TB, Brøsen K, Christensen MM (2015) A comprehensive review of drug–drug interactions with metformin. Clin Pharmacokinet 54:811–824
Rennert G, Rennert HS, Gronich N, Pinchev M, Gruber SB. Use of metformin and risk of breast and colorectal cancer. Diabetes Res Clin Pract. 2020;165:108232. https://doi.org/10.1016/j.diabres.2020.108232.
Guo C, Zhao L, Li Y, Deng X, Yuan G (2021) Relationship between FGF21 and drug or nondrug therapy of type 2 diabetes mellitus. J Cell Physiol 236(1):55–67
Zhang J, Weng W, Wang K, Lu X, Cai L, Sun J (2018) The role of FGF21 in type 1 diabetes and its complications. Int J Biol Sci 14(9):1000
Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, Defronzo RA, Tripathy D (2009) Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 32:1542–1546
Hale C, Chen MM, Stanislaus S, Chinookoswong N, Hager T, Wang M et al (2012) Lack of overt FGF21 resistance in two mouse models of obesity and insulin resistance. Endocrinology 153:69–80
Reinehr T, Woelfle J, Wunsch R, Roth CL (2012) Fibroblast growth factor 21 (FGF-21) and its relation to obesity, metabolic syndrome, and nonalcoholic fatty liver in children: a longitudinal analysis. J Clin Endocrinol Metab 97:2143–2150
Kim JH, Bae KH, Choi YK, Go Y, Choe M, Jeon YH et al (2015) Fibroblast growth factor 21 analogue LY2405319 lowers blood glucose in streptozotocin-induced insulin-deficient diabetic mice by restoring brown adipose tissue function. Diabetes Obes Metab 17:161–169
Xiao Y, Xu A, Law LS, Chen C, Li H, Li X et al (2012) Distinct changes in serum fibroblast growth factor 21 levels in different subtypes of diabetes. J Clin Endocrinol Metab 97:E54–E58
Zibar K, Blaslov K, Bulum T, Cuca JK, Smircic-Duvnjak L (2015) Basal and postprandial change in serum fibroblast growth factor-21 concentration in type 1 diabetic mellitus and in healthy controls. Endocrine 48:848–855
Chavez AO, Molina-Carrion M, Abdul-Ghani MA, Folli F, DeFronzo RA, Tripathy D (2009) Circulating fibroblast growth factor-21 is elevated in impaired glucose tolerance and type 2 diabetes and correlates with muscle and hepatic insulin resistance. Diabetes Care 32(8):1542–1546
Chen C, Cheung BM, Tso AW, Wang Y, Law LS, Ong KL, Wat NM, Xu A, Lam KS (2011) High plasma level of fibroblast growth factor 21 is an Independent predictor of type 2 diabetes: a 5.4-year population-based prospective study in Chinese subjects. Diabetes Care. 34(9):2113–5
Solomon TP, Carter S, Haus JM, Karstoft K, von Holstein-Rathlou S, Nielsen MS, Gillum MP (2022) Plasma FGF21 concentrations are regulated by glucose independently of insulin and GLP-1 in lean, healthy humans. PeerJ 19(10):e12755
Samms RJ, Lewis JE, Norton L, Stephens FB, Gaffney CJ, Butterfield T, Smith DP, Cheng CC, Perfield JW, Adams AC, Ebling FJ (2017) FGF21 is an insulin-dependent postprandial hormone in adult humans. J Clin Endocrinol Metab 102(10):3806–3813
Arner P, Pettersson A, Mitchell PJ, Dunbar JD, Kharitonenkov A, Rydén M (2008) FGF21 attenuates lipolysis in human adipocytes–a possible link to improved insulin sensitivity. FEBS Lett 582(12):1725–1730
Mai K, Andres J, Biedasek K, Weicht J, Bobbert T, Sabath M et al (2009) Free fatty acids link metabolism and regulation of the insulin-sensitizing fibroblast growth factor-21. Diabetes 58:1532–1538
Arafat AM, Kaczmarek P, Skrzypski M, Pruszyńska-Oszmalek E, Kołodziejski P, Szczepankiewicz D, Sassek M, Wojciechowicz T, Wiedenmann B, Pfeiffer AF, Nowak KW (2013) Glucagon increases circulating fibroblast growth factor 21 independently of endogenous insulin levels: a novel mechanism of glucagon-stimulated lipolysis? Diabetologia 56:588–597
Cyphert HA, Alonge KM, Ippagunta SM, Hillgartner FB (2014) Glucagon stimulates hepatic FGF21 secretion through a PKA-and EPAC-dependent posttranscriptional mechanism. PLoS ONE 9(4):e94996
Ojha A, Ojha U, Mohammed R, Chandrashekar A, Ojha H (2019) Current perspective on the role of insulin and glucagon in the pathogenesis and treatment of type 2 diabetes mellitus. Clin Pharmacol 9:57–65
Watanabe H, Miyahisa M, Chikamatsu M, Nishida K, Minayoshi Y, Takano M, Ichimizu S, Kobashigawa Y, Morioka H, Maeda H, Maruyama T (2020) Development of a long acting FGF21 analogue-albumin fusion protein and its anti-diabetic effects. J Control Release 10(324):522–531
Ye X, Qi J, Yu D, Wu Y, Zhu S, Li S, Wu Q, Ren G, Li D (2017) Pharmacological efficacy of FGF21 analogue, liraglutide and insulin glargine in treatment of type 2 diabetes. J Diabetes Complications 31(4):726–734
Kaufman A, Abuqayyas L, Denney WS, Tillman EJ, Rolph T (2020) AKR-001, an Fc-FGF21 analog, showed sustained pharmacodynamic effects on insulin sensitivity and lipid metabolism in type 2 diabetes patients. Cell Rep Med 1(4):100057
Gaich G, Chien JY, Fu H, Glass LC, Deeg MA, Holland WL, Kharitonenkov A, Bumol T, Schilske HK, Moller DE (2013) The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab 18(3):333–340
Kim KH, Jeong YT, Kim SH, Jung HS, Park KS, Lee HY, Lee MS (2013) Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem Biophys Res Commun 440:76–81. https://doi.org/10.1016/j.bbrc.2013.09.026
Nygaard EB, Vienberg SG, Ørskov C, Hansen HS, Andersen B (2012) Metformin stimulates FGF21 expression in primary hepatocytes. Exp Diabetes Res 15:2012
Kim EK, Lee SH, Jhun JY, Byun JK, Jeong JH, Lee SY, Kim JK, Choi JY, Cho ML. Metformin prevents fatty liver and improves balance of white/brown adipose in an obesity mouse model by inducing FGF21. Mediators of inflammation. 2016;2016.
Wang C, Guo F (2012) Effects of activating transcription factor 4 deficiency on carbohydrate and lipid metabolism in mammals. IUBMB Life 64(3):226–230
Kar E, Alataş Ö, Şahıntürk V, Öz S (2022) Effects of metformin on lipopolysaccharide induced inflammation by activating fibroblast growth factor 21. Biotech Histochem 97(1):44–52
Kim KH, Lee MS (2015) FGF21 as a mediator of adaptive responses to stress and metabolic benefits of anti-diabetic drugs. J Endocrinol 226(1):R1-16
Kim EK, Lee SH, Lee SY, Kim JK, Jhun JY, Na HS, Kim SY, Choi JY, Yang CW, Park SH, Cho ML (2018) Metformin ameliorates experimental-obesity-associated autoimmune arthritis by inducing FGF21 expression and brown adipocyte differentiation. Exp Mole Med. 50(1):e432
Luo F, Guo Y, Ruan G, Li X (2016) Metformin promotes cholesterol efflux in macrophages by up-regulating FGF21 expression: a novel anti-atherosclerotic mechanism. Lipids Health Dis 15(1):1–2
Hua S, Liu Q, Li J, Fan M, Yan K, Ye D (2021) Beta-klotho in type 2 diabetes mellitus: from pathophysiology to therapeutic strategies. Rev Endocr Metab Disord 22(4):1091–1109
Nie F, Wu D, Du H, Yang X, Yang M, Pang X, Xu Y (2017) Serum klotho protein levels and their correlations with the progression of type 2 diabetes mellitus. J Diabetes Complicat 31:594–98. https://doi.org/10.1016/j.jdiacomp.2016.11.008
Jing Xue et al (2019) Basic research in diabetic nephropathy health care: a study of the renoprotective mechanism of metformin. J Med Syst. 43:1–13
Majeed Y, Upadhyay R, Lakshmanan A, Triggle C, Ding H. Down-regulation of Erk and Sirt1 signaling may lead to reduced Fgf-21 sensitivity in a mouse model of diabetes. InQatar Foundation Annual Research Conference Proceedings 2016 . (Vol. 2016, No. 1, p. HBPP1804). Qatar: HBKU Press.
So WY, Leung PS (2016) Fibroblast growth factor 21 as an emerging therapeutic target for type 2 diabetes mellitus. Med Res Rev 36(4):672–704
Díaz-Delfín J, Hondares E, Iglesias R, Giralt M, Caelles C, Villarroya F (2012) TNF-α represses β-Klotho expression and impairs FGF21 action in adipose cells: involvement of JNK1 in the FGF21 pathway. Endocrinology 153(9):4238–4245
Chau MD, Gao J, Yang Q, Wu Z, Gromada J (2010) Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK–SIRT1–PGC-1α pathway. Proc Natl Acad Sci 107(28):12553–12558
Li Q, Jia S, Xu L, Li B, Chen N (2019) Metformin-induced autophagy and irisin improves INS-1 cell function and survival in high-glucose environment via AMPK/SIRT1/PGC-1α signal pathway. Food Sci Nutr 7(5):1695–1703
Aatsinki SM, Buler M, Salomäki H, Koulu M, Pavek P, Hakkola J (2014) Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br J Pharmacol 171(9):2351–2363
Kappe C, Patrone C, Holst JJ, Zhang Q, Sjöholm Å (2013) Metformin protects against lipoapoptosis and enhances GLP-1 secretion from GLP-1-producing cells. J Gastroenterol 48:322–332
DeFronzo RA, Buse JB, Kim T, Burns C, Skare S, Baron A, Fineman M (2016) Once-daily delayed-release metformin lowers plasma glucose and enhances fasting and postprandial GLP-1 and PYY: results from two randomised trials. Diabetologia 59:1645–1654
Omar BA, Andersen B, Hald J, Raun K, Nishimura E, Ahrén B (2014) Fibroblast growth factor 21 (FGF21) and glucagon-like peptide 1 contribute to diabetes resistance in glucagon receptor–deficient mice. Diabetes 63(1):101–110
Liu J, Yang K, Yang J, Xiao W, Le Y, Yu F, Gu L, Lang S, Tian Q, Jin T, Wei R (2019) Liver-derived fibroblast growth factor 21 mediates effects of glucagon-like peptide-1 in attenuating hepatic glucose output. EBioMedicine 1(41):73–84
Babalghith AO, Al-Kuraishy HM, Al-Gareeb AI, De Waard M, Sabatier JM, Saad HM, Batiha GE (2022) The potential role of growth differentiation factor 15 in COVID-19: a corollary subjective effect or not? Diagnostics 12(9):2051
Valiño-Rivas L, Cuarental L, Ceballos MI, Pintor-Chocano A, Perez-Gomez MV, Sanz AB, Ortiz A, Sanchez-Niño MD (2022) Growth differentiation factor-15 preserves Klotho expression in acute kidney injury and kidney fibrosis. Kidney Int 101(6):1200–1215
Zhang M, Sun W, Qian J, Tang Y (2018) Fasting exacerbates hepatic growth differentiation factor 15 to promote fatty acid β-oxidation and ketogenesis via activating XBP1 signaling in liver. Redox Biol 1(16):87–96
Huddar A, Govindaraj P, Chiplunkar S, Deepha S, Ponmalar JJ, Philip M, Nagappa M, Narayanappa G, Mahadevan A, Sinha S, Taly AB (2021) Serum fibroblast growth factor 21 and growth differentiation factor 15: two sensitive biomarkers in the diagnosis of mitochondrial disorders. Mitochondrion 1(60):170–177
Campderrós L, Moure R, Cairó M, Gavaldà-Navarro A, Quesada-López T, Cereijo R, Giralt M, Villarroya J, Villarroya F (2019) Brown adipocytes secrete GDF15 in response to thermogenic activation. Obesity 27(10):1606–1616
Adela R, Banerjee SK (2015) GDF-15 as a target and biomarker for diabetes and cardiovascular diseases: a translational prospective. J Diabetes Res 2015:1–14
Ikram M, Magdy Beshbishy A, Kifayatullah M et al (2020) Chemotherapeutic potential of Carthamus oxycantha root extract as antidiarrheal and in vitro antibacterial activities. Antibiotics 9(5):226
Slusher AL, Whitehurst M, Zoeller RF, Mock JT, Maharaj M, Huang CJ (2015) Attenuated fibroblast growth factor 21 response to acute aerobic exercise in obese individuals. Nutr Metab Cardiovasc Dis 25(9):839–845
Yu Y, He J, Li S, Song L, Guo X, Yao W, Zou D, Gao X, Liu Y, Bai F, Ren G (2016) Fibroblast growth factor 21 (FGF21) inhibits macrophage-mediated inflammation by activating Nrf2 and suppressing the NF-κB signaling pathway. Int Immunopharmacol 1(38):144–152
Archer A, Venteclef N, Mode A, Pedrelli M, Gabbi C, Clément K, Parini P, Gustafsson JÅ, Korach-André M (2012) Fasting-induced FGF21 is repressed by LXR activation via recruitment of an HDAC3 corepressor complex in mice. Mol Endocrinol 26(12):1980–1990
Dahlman I, Nilsson M, Gu HF, Lecoeur C, Efendic S, Östenson CG, Brismar K, Gustafsson JÅ, Froguel P, Vaxillaire M, Dahlman-Wright K (2009) Functional and genetic analysis in type 2 diabetes of liver X receptor alleles–a cohort study. BMC Med Genet 10:1–3
Targher G, Corey KE, Byrne CD, Roden M (2021) The complex link between NAFLD and type 2 diabetes mellitus—mechanisms and treatments. Nat Rev Gastroenterol Hepatol 18(9):599–612
Zhang D, Ma Y, Liu J, Deng Y, Zhou B, Wen Y, Li M, Wen D, Ying Y, Luo S, Shi C (2021) Metformin alleviates hepatic steatosis and insulin resistance in a mouse model of high-fat diet-induced nonalcoholic fatty liver disease by promoting transcription factor EB-dependent autophagy. Front Pharmacol 23(12):689111
Pinyopornpanish K, Leerapun A, Pinyopornpanish K, Chattipakorn N (2021) Effects of metformin on hepatic steatosis in adults with nonalcoholic fatty liver disease and diabetes: insights from the cellular to patient levels. Gut and liver 15(6):827
Lin MJ, Dai W, Scott MJ, Li R, Zhang YQ, Yang Y, Chen LZ, Huang XS (2017) Metformin improves nonalcoholic fatty liver disease in obese mice via down-regulation of apolipoprotein A5 as part of the AMPK/LXRα signaling pathway. Oncotarget 8(65):108802
Cho K, Chung JY, Cho SK, Shin HW, Jang IJ, Park JW, Yu KS, Cho JY (2015) Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRα/POMC pathway. Sci Rep 5(1):8145
Christodoulides C, Dyson P, Sprecher D, Tsintzas K, Karpe F (2009) Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J Clin Endocrinol Metab 94(9):3594–3601
Maida A, Lamont BJ, Cao X, Drucker DJ (2011) Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-α in mice. Diabetologia 54:339–349
Bai F, Liu Y, Tu T, Li B, Xiao Y, Ma Y, Qin F, Xie J, Zhou S, Liu Q (2019) Metformin regulates lipid metabolism in a canine model of atrial fibrillation through AMPK/PPAR-α/VLCAD pathway. Lipids Health Dis 18(1):1–9
Acknowledgements
The authors extend their appreciation to Al-Mustansiriyah University, the University of Technology, and the University of Misan for their technical support.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology, Hayder M. Al-kuraishy and Ali I. Al-Gareeb; formal analysis, Majid S. Jabir and Salim Albukhaty; investigation and data curation, Salim Albukhaty; validation, Majid S. Jabir; original draft preparation, Hayder M. Al-kuraishy and Ali I. Al-Gareeb; writing—review and editing, Majid S. Jabir and Salim Albukhaty; supervision, Hayder M. Al-kuraishy. All authors gave approval to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-kuraishy, H.M., Al-Gareeb, A.I., Jabir, M.S. et al. Effects of metformin on fibroblast growth factor 21 in patients with type 2 diabetes mellitus: faraway but so close. Egypt J Intern Med 35, 65 (2023). https://doi.org/10.1186/s43162-023-00238-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43162-023-00238-9