
Abstract
Myelofibrosis (MF) is a haematopoietic stem cell tumour caused by the lack of BCR-ABL translocation due to point mutations in Janus kinases (JAKs). In previous years, dealing with MF included several protocols such as traditional drugs that control general symptoms, splenectomy, blood transfusion, and allogeneic haematopoietic stem-cell transplantation (HSCT). Allogeneic HSCT is remaining the only treatment that has the potential to alter MF’s progression. However, clinical trials of JAK inhibitors and non-JAK targeted therapies have been increasingly carried out in earlier years. The most prominent JAK inhibitors for the treatment of MF are ruxolitinib, fedratinib, momelotinib, pacritinib, gandotinib, ilginatinib, itacitinib, and lestaurtinib. On the other hand, the non-JAK targeted therapies that showed strong efficacy and safety are alisertib, imetelstat, pembrolizumab, nivolumab, and sotatercept. In this review, we summarized the recent clinical trials carried out on these drugs to understand their efficacy and safety. Also, we talked briefly about allogeneic HSCT as powerful therapy until the present for patients suffering from MF.
Similar content being viewed by others

Background
Myeloproliferative neoplasms (MPNs) are a group of nine blood disorders defined by the World Health Organization (WHO). MPNs are characterized by blood cells overproduction and bone marrow dysfunction. Primary myelofibrosis (PMF), polycythemia vera (PV), and essential thrombocytopenia (ET) are among the 9 disorders of MPNs. PV and ET in their advanced levels are called PPV-MF (post-PV myelofibrosis) and PET-MF (post-ET myelofibrosis), and both are categorized under the name of secondary marrow fibrosis (SMF). It is now clear that myelofibrosis (MF) includes both PMF and SMF [1].
Pathogenesis
Genetic mutations
MF is a haematopoietic stem cell tumour that leads to bone marrow fibrosis. This process is marked by the lack of BCR-ABL translocation due to point mutations in Janus kinases (JAKs), the most important of which is the somatic mutation in one nucleotide (G to T) at exon 14 of the gene JAK2 which leads to the transformation of valine (V) to phenylalanine (F) at the codon 617 (V617F) [2].
Of note, JAKs are a class of tyrosine kinases (JAK1, JAK2, JAK3, and TYK4) found in mammals. JAK2 is one of the important molecules engaged in signalling pathways in the immune and haematopoietic systems. JAK2 is also essential for the production of red blood cells and the activation of different immune cells [3].
Cytokines
Additionally, significantly elevated levels of circulating pro-inflammatory cytokines are a key biological feature of MF. Cytokines are believed to be a result of the malignant clone as well as a component of the bone marrow microenvironment, encouraging malignant haematopoiesis [4].
Cytokines are thought to have a significant role in the onset, development, and phenotypic manifestation of MF [5]. One-hundred twenty-seven individuals with PMF had their plasma levels of 30 cytokines and chemokines examined, revealing common cytokines. Interleukin (IL)-8 and IL-2R exhibited the highest connection with phenotype and prognosis of MF. Elevated levels of these cytokines were linked to constitutional symptoms, transfusion requirements, leukocytosis, and leukaemia [6]. IL-8 is a pleiotropic pro-inflammatory cytokine that is secreted by a variety of cells and possesses angiogenic, mitogenic, and growth factor action. IL-8 expression has been linked to a poor prognosis in a variety of solid tumours. The pathogenic role of IL-8 in MF is not entirely understood; however, it may be implicated in leukaemic transformation via its growth factor and mitogenic properties [7]. Also, both immune activation and a rise in the number of tumour cells can be reflected by an increase in IL-2R levels [6].
A potent inflammatory cytokine known as lipocalin-2 has been found to preferentially promote the proliferation of MF CD34+ cells. It has a role in double-stranded DNA breaks and the apoptosis of healthy bone marrow cells and increase the proliferation of stromal cells by releasing reactive oxygen species in the blood of MF patients [8].
Additionally, mesenchymal stem cells’ production of the extracellular matrix protein collagen type 1 (COL1A1) is stimulated by lipocalin-2, and this, along with other changes in the bone marrow microenvironment, presumably contributes to the development of the malignant haematopoietic stem cell (HSC) [5].
Osteoblastic lineage cells
On the other hand, the endosteal niche, which is physically defined by its close proximity to trabecular or cortical bone, is made up of osteoblastic lineage cells. Osteoblastic lineage cells, which are produced from multipotent stromal cells, are crucial for the maintenance and support of healthy HSC [9, 10].
Genes that control extracellular matrix, cell adhesion, and inflammatory reactions are expressed more often in the osteoblastic lineage cells and have transforming growth factor-beta 1 (TGF-β1) targets. These cells have increased expression of TGF-β1, which may promote myeloid neoplastic differentiation [9, 10].
Score system of myelofibrosis
GIPSS (genetically inspired prognostic scoring system) and MIPSS70 (mutation-enhanced international prognostic scoring system for transplant-age patients) are two modern examples of MF prognostic algorithms [11].
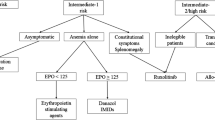
The most recent and up-to-date prognostic method for PMF is called MIPSS70 and covers clinical risk factors, mutations (MIPSS70), and karyotype (MIPSS70+ and MIPSS70+ version 2.0) [11]. In order to be directly applicable to transplant decision-making, MIPSS70, MIPSS70+, and MIPSS70+ version 2.0 were created in patients who were 70 years of age or younger. Three genetic (the absence of CALR type 1/like mutation; the presence of high-molecular-risk mutations, specifically ASXL1, SRSF2, EZH2, IDH1, or IDH2; and the presence of ≥ 2 high-molecular-risk mutations) and six clinical risk factors (haemoglobin < 10 g/dl, leucocytes > 25 × 109/l, platelets < 100 × 109/l, circulating blast ≥ 2%, bone marrow fibrosis grade ≥ 2, and constitutional symptoms) are among the nine variables in the MIPSS70. A total score of 0–1, 2–4, and 5 defined the three-tiered MIPSS70 low-, intermediate-, and high-risk categories. Subsequently, a hazard ratio-weighted score of “2” was given to leucocytes > 25 109/l, platelets 100 109/l, and the presence of 2 high-molecular-risk mutations [11].
GIPSS is based mainly on mutations and karyotype markers. GIPPS provides a less complicated predictive technique that accurately determines candidates for allogeneic stem-cell transplantation (GIPSS high-risk illness) or long-term monitoring with little to no therapeutic intervention (GIPSS low-risk disease). GIPSS risk classes are divided into four levels: low (zero points), intermediate-1 (one point), intermediate-2 (two points), and high (three or more). The median survival rates (5-year survival rate) for each level are 26.4 years (94%), 8.0 years (73%), 4.2 years (40%), and 2 years (14%). The optimal strategy is a step-by-step prognostication technique that begins with GIPSS but also takes into account MIPSS70 to validate the most effective course of treatment for the specific patient [11].
Trend toward novel therapy
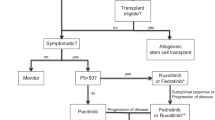
Although allogeneic haematopoietic stem-cell transplantation (HSCT) is remaining the only treatment that has the potential to alter MF's progression, its success rate is not as good as expected. Therefore, the trend toward developing new drugs has become a necessary requirement in the scientific community. To date, there are many JAK and non-JAK targeted therapies have been discovered for the management of MF. Importantly, ruxolitinib and fedratinib are JAK2 inhibitors authorized by the US Food and Drug Administration (FDA) for the management of patients with intermediate and severe cases of MF. They controlled spleen size and decreased disease symptoms, but they did not affect genetic mutations or bone marrow fibrosis [1]. In this review, we will discuss the most prominent clinical trials that achieved the therapeutic targets of MF.
Main text
Treatment
JAK inhibitors and non-JAK targeted drugs, traditional drugs, splenectomy, and blood transfusion are used for patients with intermediate- to high-risk conditions of MF controlling general symptoms. Therefore, these treatments can improve only the quality of life and reduce mortality in MF patients. However, stem-cell transplantation therapy is remaining the most common solution [1].
Splenectomy
Interestingly, splenectomy is an option for MF patients who have extensive splenomegaly, particularly in situations when the condition is resistant to traditional therapy, but it usually leads to a variety of health problems as well as significant morbidity and mortality [12].
Allogeneic haematopoietic stem-cell transplantation (HSCT)
HSCT is the process of transfusion of haematopoietic stem cells into the bloodstream restoring normal hematopoiesis and curing tumours. Haematopoietic stem cells (HSCs) can be collected from different sources such as bone marrow, peripheral blood, and umbilical cord as well as induced pluripotent stem cell (iPSC) technology. Then, HSC can replicate replacing the tumour with normal healthy tissues [13]. Nowadays, allogeneic HSCT is remaining the only treatment that has the potential to alter MF’s progression. Therefore, over the past years, the number of trials performed for the treatment of MF increased significantly, despite the FDA approval of some JAK inhibitors [14].
JAK inhibitors for myelofibrosis
JAK2 V617F and JAK2 exon 12 mutations are important for the tumour progression of several haematological disorders, including MPNs, acute myeloid leukaemia (AML), and chronic myeloid leukaemia (CML) [3]. Therefore, the discovery of JAK2 inhibitors has occupied a large space in recently published scientific papers focused on the treatment of MF. The most common clinical trials are summarized in the timeline shown in Fig. 1 and Table 1.

Timeline history of clinical trials representing JAK inhibitors and non-JAK targeted drugs
Ruxolitinib, a JAK1/2 inhibitor, was authorized by the FDA in 2011 for the treatment of intermediate- or high-risk MF [27]. Until now, ruxolitinib is still the first-line therapy for MF in patients with intermediate or severe conditions.
In phase III clinical trial (COMFORT-I), 309 MF patients (intermediate and high risk) were randomized to receive ruxolitinib or placebo (1:1). At 24 weeks, the primary endpoint was 35% of patients had a reduction in spleen volume, while the secondary endpoints were improvement of survival and decreasing symptoms. Patients who received ruxolitinib showed a decrease in score-evaluating symptoms by 50% than patients who received a placebo. On the other hand, in phase III clinical trial (COMFORT-II), 219 MF patients (intermediate and high risk) were randomized to receive ruxolitinib or the best available therapy (BAT) (2:1). At 48 weeks, 28% of the ruxolitinib group had a 35% reduction in spleen volume while no one in the BAT group showed any reduction [15, 16].
As we discussed, the drug did not affect myelofibrosis or genetic mutations of the JAK2 gene. Moreover, many side effects appeared during treatment such as anaemia and thrombocytopenia.
Therefore, ruxolitinib in combination with another drug is intended to improve its limits against MF. Accordingly, many experiments have been conducted to achieve these points, and all of them are still in the first- or second-phase trial, as follows: PEG-IFNα2 (phase I/II trials), danazol (phase II trials), lenalidomide (phase II trials), thalidomide (phase II trials), pomalidomide (phase I/II trials), panobinostat (phase I trials), pracinostat (phase II trials), sonidegib (phase I trials), vismodegib (phase I trials), buparlisib (phase I trials), enasidenib (phase II trials), navitoclax (phase II trials), pelabresib (phase I/II trials), PU-H71 (phase I trials), 9-ING-41 (phase II trials), pevonedistat (phase I trials), siremadlin (phase I/II trials), crizanlizumab (phase I/II trials), and MBG453(phase I/II trials) [1].
Fedratinib is another molecule that showed a high effect as a JAK2 inhibitor. It also showed inhibition for JAK1 and JAK3 but with a lower effect. In a phase II trial named JAKARTA-2, fedratinib was tested in MF patients who showed resistance or tolerance to ruxolitinib. Then, in a phase III trial named JAKARTA, fedratinib was evaluated in MF patients who had recently been treated with ruxolitinib [17, 18]. In the JAKARTA-2 trial, 25 to 30% of patients achieved the endpoints (reduction in symptoms and spleen volume), while in the JAKARTA trial, 35 to 40% of patients achieved the endpoints. Recently, in August 2019, fedratinib (Inrebic) was authorized by the FDA as a new therapy for adults with intermediate-2 or high-risk MF [28].
Momelotinib is a nonselective JAK1/2 inhibitor. In phase III trial (SIMPLIFY-1), the study showed an improvement in blood transfusion rate. In another phase III trial called SIMPLIFY-2, the study compared the effectiveness and safety of momelotinib to BAT in MF patients who had unsatisfactory responses or adverse effects with ruxolitinib. Momelotinib was found to treat anaemia; however, it can also develop or increase thrombocytopenia [19, 20].
Pacritinib is a selective JAK2 inhibitor. In the phase II trial (PERSIST-1), pacritinib and BAT were randomized for 327 MF patients. After 24 weeks, the pacritinib group achieved significantly the primary and secondary endpoints than the BAT group. Besides, the phase III trial (PERSIST-2) was carried out aiming to understand the effect of pacritinib after exposure of MF patients to ruxolitinib. Also, this study tried to show the effect of pacritinib on adult MF patients as well as patients who have platelet levels below 50,000/μL [21, 22]. Interestingly, in March 2022, the FDA authorized pacritinib under the trading name of Vonjo for the treatment of adults with MF and who have platelet levels below 50,000/μL [29].
Gandotinib effectiveness, toxicity, and pharmacokinetics were studied in a multicentre, phase II study in patients with MF. Fortunately, gandotinib succeeded in treating patients with V617F mutations in JAK2 [23].
Ilginatinib, a JAK2 inhibitor, was found to decrease leukocytosis and splenomegaly but not the JAK2 V617F mutant allele load. In phase I study included 48 MF patients, the spleen size dropped by more than 50% in almost half of the patients who used ilginatinib for 4 weeks and that was greater than the number of patients treated with other JAK2 inhibitors [24].
Itacitinib is a JAK1 inhibitor that is only effective when taken orally. By targeting JAK1, it inhibits the release of cytokines, lowering the risk of bone marrow suppression. In the phase II study, 87 participants with MF received the drug. After 24 weeks of higher-dose therapy, 64.1% of patients had reduced more than 50% of their overall symptoms, while 17% had reduced their spleen volume [25]. Due to drug toxicity and limitations, lestaurtinib has been withdrawn from a phase II clinical study [1].
Non-JAK targeted drugs for myelofibrosis
There are few studies of non-JAK targeted agents for MF. The most common clinical trials are summarized in Fig. 1 and Table 2 (alisertib, Imetelstat, Pembrolizumab, nivolumab, and sotatercept).
Megakaryocyte dysregulation is a primary driving force behind MF [35]. Inhibition of aurora kinase A improves the viability and differentiation of megakaryocytes [36]. Alisertib is an aurora kinase A inhibitor. In a phase I trial including 24 MF patients, alisertib showed a reduction in megakaryocytes, myelofibrosis, spleen size (29% of patients), and overall symptoms (32% of patients) [30].
Telomerase is a ribonuclear protein enzyme selectively activated in cancer cells compared to somatic cells. Imetelstat is a powerful competitive inhibitor of telomerase activity because it specifically targets the RNA template of human telomerase [37].
This phase II research of two imetelstat dosages, 9.4 mg/kg once every 3 weeks, showed clinical advantages in response rate, with a good safety profile for this JAKi R/R group with poor risk. The use of biomarkers and measurements of bone marrow fibrosis revealed that the malignant clone had a selective impact [31].
Pembrolizumab is an IgG4/kappa humanized monoclonal antibody that can disrupt the link between programmed death 1 (PD-1) and its ligands 1 and 2 (PD-L1; PD-L2). Pembrolizumab has been licenced by the FDA for the treatment of haematological cancers such as melanoma and Hodgkin’s lymphoma [38].
Pembrolizumab therapy was well tolerated in a multicentre, phase II trial in patients with MF, but there were no objective clinical outcomes; hence, the study was stopped after the first stage. Immunological profiling using flow cytometry, T-cell receptor sequencing, and plasma proteomics, on the other hand, revealed alterations in the immune microenvironment of patients, indicating enhanced T-cell responses that may support antitumor immunity. The lack of a therapeutic outcome to these modifications implies that, rather than monotherapy, combined immunotherapeutic treatments may be required to overcome the multiple causes of immune suppression in myeloproliferative neoplasms [32].
Nivolumab is a IgG4 (kappa) isotype and a human monoclonal antibody that binds PD-1. In a phase II study, the effectiveness and safety of single-agent nivolumab were evaluated in 8 adult myelofibrosis patients. Nivolumab was administered at a dose of 3 mg/kg every 2 weeks for 8 doses, and then every 12 weeks for up to 4 years, or until progression of the disease or toxicity, whichever came first. For a median of 3.3 months, five patients exhibited stable symptoms in terms of spleen size, overall symptom score, and blood needs. Two patients were still alive after a median of 57 months of follow-up. The median time to death was 6.1 months. The trial was prematurely ended due to failure to fulfil the specified effectiveness outcome [33].
Sotatercept belongs to a new series of drugs known as activin receptor ligand traps, which can treat anaemia by sequestering TGF-superfamily ligands like growth and differentiation factor 11 (GDF-11), which are naturally produced by bone marrow stromal cells and suppress terminal erythropoiesis via Smad signalling, thus blocking their interaction with activin receptors. TGF− was found to be significantly expressed in the bone marrow of MF patients, and it is thought to have a role in the fibrosis caused by the disease. Sotatercept was proven to be safe and effective in treating anaemia in MF patients in phase II studies [34].
Conclusion
Relying solely on allogeneic HSCT for curing MF has never been the ultimate solution for the treatment of MF despite its effectiveness. JAK inhibitors and non-JAK targeted therapies were a good solution for the reduction of overall symptoms and spleen size, but they had no effect on genetic mutations causing the disease. The clinical trials discussed in this review showed that the improvement in these drugs might achieve our targets especially by combining different drugs. There are still many discoveries and research not yet done in this regard, which we hope will focus on genetic mutations causing MF and how to overcome them. On the other hand, combining ruxolitinib with other medications offers a lot of therapeutic benefits. It can not only compensate for monotherapy’s inadequacies but also improve therapeutic efficacy and lessen side effects. Unfortunately, the trials mentioned here were restricted by their sample sizes, despite the possibility of significant therapeutic values. To validate the clinical effectiveness of ruxolitinib in conjunction with other therapies, large cohorts of patients and multicenter, randomized controlled studies are still needed.
Availability of data and materials
Not applicable
Abbreviations
- AML:
-
Acute myeloid leukaemia
- CML:
-
Chronic myeloid leukaemia
- ET:
-
Essential thrombocytopenia
- FDA:
-
US Food and Drug Administration
- GDF-11:
-
Differentiation factor 11
- HSC:
-
Haematopoietic stem cells
- HSCT:
-
Haematopoietic stem-cell transplantation
- IPSCs:
-
Induced pluripotent stem cell
- JAKs:
-
Janus kinases
- MF:
-
Myelofibrosis
- MPNs:
-
Myeloproliferative neoplasms
- PD-1:
-
Programmed death 1
- PET-MF:
-
Post-ET myelofibrosis
- PMF:
-
Primary myelofibrosis
- PPV-MF:
-
Post-PV myelofibrosis
- PV:
-
Polycythemia vera
- SMF:
-
Secondary marrow fibrosis
- WHO:
-
World health organization
References
Wang F, Qiu T, Wang H, Yang Q (2022) State-of-the-art review on myelofibrosis therapies. Clin Lymphoma Myeloma Leuk 22:e350–e362. https://doi.org/10.1016/j.clml.2021.11.007
Vainchenker W, Kralovics R (2017) Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood, J Am Soc Hematol 129:667–679
Raivola J, Haikarainen T, Abraham BG, Silvennoinen O (2021) Janus kinases in leukemia. Cancers (Basel) 13. https://doi.org/10.3390/cancers13040800
Meyer SC, Levine RL (2014) Molecular pathways: molecular basis for sensitivity and resistance to JAK kinase inhibitors. Clin cancer Res an Off J Am Assoc Cancer Res 20:2051–2059. https://doi.org/10.1158/1078-0432.CCR-13-0279
Zahr AA, Salama ME, Carreau N et al (2016) Bone marrow fibrosis in myelofibrosis: pathogenesis, prognosis and targeted strategies. Haematologica 101:660–671. https://doi.org/10.3324/haematol.2015.141283
Tefferi A, Vaidya R, Caramazza D et al (2011) Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol Off J Am Soc Clin Oncol 29:1356–1363. https://doi.org/10.1200/JCO.2010.32.9490
Zarogoulidis P, Katsikogianni F, Tsiouda T et al (2014) Interleukin-8 and interleukin-17 for cancer. Cancer Invest 32:197–205. https://doi.org/10.3109/07357907.2014.898156
Lu M, Xia L, Liu Y-C et al (2015) Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 126:972–982. https://doi.org/10.1182/blood-2014-12-618595
Schepers K, Pietras EM, Reynaud D et al (2013) Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 13:285–299. https://doi.org/10.1016/j.stem.2013.06.009
Mullally A, Ebert BL (2013) Sinister symbiosis: pathological hematopoietic-stromal interactions in CML. Cell Stem Cell 13:257–258. https://doi.org/10.1016/j.stem.2013.08.009
Tefferi A, Guglielmelli P, Pardanani A, Vannucchi AM (2018) Myelofibrosis Treatment Algorithm 2018. Blood Cancer J 8:72. https://doi.org/10.1038/s41408-018-0109-0
Malato A, Rossi E, Tiribelli M et al (2020) Splenectomy in myelofibrosis: indications, efficacy, and complications. Clin Lymphoma Myeloma Leuk 20:588–595. https://doi.org/10.1016/j.clml.2020.04.015
Giralt S, Bishop MR (2009) Principles and overview of allogeneic hematopoietic stem cell transplantation. Cancer Treat Res 144:1–21. https://doi.org/10.1007/978-0-387-78580-6_1
Tiribelli M, Palandri F, Sant’Antonio E et al (2020) The role of allogeneic stem-cell transplant in myelofibrosis in the era of JAK inhibitors: a case-based review. Bone Marrow Transplant 55:708–716. https://doi.org/10.1038/s41409-019-0683-1
Harrison CN, Vannucchi AM, Kiladjian J-J et al (2016) Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 30:1701–1707
Verstovsek S, Mesa RA, Gotlib J et al (2017) Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 10:1–14
Harrison CN, Schaap N, Vannucchi AM et al (2017) Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol 4:e317–e324
Pardanani A, Harrison C, Cortes JE et al (2015) Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol 1:643–651
Harrison CN, Vannucchi AM, Platzbecker U et al (2018) Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol 5:e73–e81
Mesa RA, Kiladjian J-J, Catalano JV et al (2017) Simplify-1: a phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor–naïve patients with myelofibrosis. J Clin Oncol 35:3844
Mascarenhas J, Hoffman R, Talpaz M et al (2016) Results of the PERSIST-2 phase 3 study of pacritinib (PAC) versus best available therapy (BAT), including ruxolitinib (RUX). Patients with Myelofibrosis Platelet Counts <100.000/ μl. Blood 128(22), LBA-5.
Mesa RA, Vannucchi AM, Mead A et al (2017) Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol 4:e225–e236
Berdeja J, Palandri F, Baer MR et al (2018) Phase 2 study of gandotinib (LY2784544) in patients with myeloproliferative neoplasms. Leuk Res 71:82–88
Verstovsek S, Talpaz M, Ritchie E et al (2017) A phase I, open-label, dose-escalation, multicenter study of the JAK2 inhibitor NS-018 in patients with myelofibrosis. Leukemia 31:393–402
Mascarenhas JO, Talpaz M, Gupta V et al (2017) Primary analysis of a phase II open-label trial of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Haematologica 102:327
Bose P, Verstovsek S (2020) JAK inhibition for the treatment of myelofibrosis: limitations and future perspectives. Hemasphere 4(4).
Deisseroth A, Kaminskas E, Grillo J et al (2012) U.S. Food and Drug Administration approval: ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin cancer Res an Off J Am Assoc Cancer Res 18:3212–3217. https://doi.org/10.1158/1078-0432.CCR-12-0653
Center for Drug Evaluation and Research (2019) FDA approves fedratinib for myelofibrosis. U.S. Food and Drug Administration. Retrieved October 29, 2022, from https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fedratinib-myelofibrosis
Center for Drug Evaluation and Research (2022) FDA approves drug for adults with rare form of bone marrow disorder. U.S. Food and Drug Administration. Retrieved October 29, 2022, from https://www.fda.gov/drugs/news-events-human-drugs/fda-approves-drug-adults-rare-form-bone-marrow-disorder
Gangat N, Marinaccio C, Swords R et al (2019) Aurora kinase a inhibition provides clinical benefit, normalizes megakaryocytes, and reduces bone marrow fibrosis in patients with myelofibrosis: a phase I trial. Clin Cancer Res 25:4898–4906
Mascarenhas J, Komrokji RS, Palandri F et al (2021) Randomized, single-blind, multicenter phase II study of two doses of imetelstat in relapsed or refractory myelofibrosis. J Clin Oncol Off J Am Soc Clin Oncol 39:2881–2892. https://doi.org/10.1200/JCO.20.02864
Hobbs G, Cimen Bozkus C, Moshier E et al (2021) PD-1 inhibition in advanced myeloproliferative neoplasms. Blood Adv 5:5086–5097. https://doi.org/10.1182/bloodadvances.2021005491
Abou Dalle I, Kantarjian H, Daver N et al (2021) Phase II study of single-agent nivolumab in patients with myelofibrosis. Ann Hematol 100:2957–2960
Bose P, Masarova L, Pemmaraju N et al (2021) Final results of a phase 2 study of sotatercept (ACE-011) for anemia of MPN-associated myelofibrosis. Blood 138:144. https://doi.org/10.1182/blood-2021-150908
Woods B, Chen W, Chiu S et al (2019) Activation of JAK/STAT signaling in megakaryocytes sustains myeloproliferation in vivo. Clin Cancer Res 25:5901–5912
Wen Q, Goldenson B, Silver SJ et al (2012) Identification of regulators of polyploidization presents therapeutic targets for treatment of AMKL. Cell 150:575–589
Tefferi A, Lasho TL, Begna KH et al (2015) A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med 373:908–919
Sochacka-Ćwikła A, Mączyński M, Regiec A (2021) FDA-approved drugs for hematological malignancies-the last decade review. Cancers (Basel) 14. https://doi.org/10.3390/cancers14010087
Acknowledgements
Not applicable
Funding
Not applicable
Author information
Authors and Affiliations
Contributions
AG collected the data and drafted the manuscript. The author read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The author declares no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ghit, A. Myelofibrosis treatment history and future prospects. Egypt J Intern Med 34, 81 (2022). https://doi.org/10.1186/s43162-022-00169-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43162-022-00169-x