
Abstract
Background
Early childhood caries is a significant public health concern affecting about 600 million children globally. The etiology of early childhood caries can be explained as an interplay between genetic and environmental factors. Single nucleotide polymorphisms are the most common variations in the human genome. Genetic variations of immune response genes can modify the defense response of the host, and alter the susceptibility to bacterial colonization of the oral cavity and early childhood caries. The aim of this systematic review is to identify genetic variants of immune response genes associated with early childhood caries.
Results
A total of 7124 articles were identified by conducting an elaborate search across various electronic databases and genome-wide association studies databases. Subsequent to exclusion at various stages, fifteen articles qualified to be included into the present review. Risk of bias assessment was done with the Q-genie tool. Quantitative synthesis revealed that the odds ratio for TT and CC genotypes of rs11362 was 1.07 (0.67–1.71) and 1.16 (0.84–1.60), respectively. Gene-based analysis revealed a statistically significant association between variants of tumor necrosis factor-alpha gene and T-cell receptor alpha variable 4 locus with early childhood caries. Gene clustering showed the presence of three functional clusters. To comprehend the protein–protein interaction, the bioinformatic tool of “Search Tools for the Retrieval of Interacting Genes and Proteins” was used. Among the biological processes and the reactome pathways, complement activation through the lectin pathway showed the highest strength of association with early childhood caries. To understand the interaction and functionality of the genes, “gene function prediction using Multiple Association Network Integration Algorithm” was used, which revealed that the genes were linked by physical interaction (39.34%) and through co-expression (34.88%).
Conclusions
Genotype TT of rs7217186 of arachidonate 15-lipoxygenase gene was a risk factor for early childhood caries. Multiple genetic variants of T-cell receptor alpha variable 4 locus and tumor necrosis factor-alpha gene were associated with increased susceptibility to early childhood caries. Polymorphisms of genes regulating the lectin pathway of complement activation can modify the susceptibility to early childhood caries.
Similar content being viewed by others


Background
Early childhood caries (ECC) is a chronic complex disease affecting children characterized by demineralization of calcified tissues and destruction of organic tissues of teeth. Genetic predisposition to dental caries was suggested by Dr G V Black [1]. Variations observed in the susceptibility to caries, following exposure to the same risk factors, can be explained by the innate genetic factors [2]. Genetic contribution to caries development has been reported to range between 40 and 60% [3,4,5] with the heritability of caries in primary dentition being greater than the heritability of caries in permanent dentition [5, 6]. Genes regulating amelogenesis, immune response, taste preferences, glucose metabolism, salivary composition, and flow alter the susceptibility to ECC [7]. Saliva plays a crucial role in oral defense mechanisms and certain proteins secreted in saliva contribute to its anti-microbial properties. Single nucleotide polymorphisms (SNPs) of genes encoding these proteins may modify the antimicrobial property of saliva, thus leading to the establishment of cariogenic microflora.
Polymorphisms of genes regulating Immune response can alter the defense response of the host. Lactotransferrin (LTF), an iron-binding glycoprotein in mammalian secretions exhibits broad-spectrum antimicrobial activity, participates in inflammation, and regulates the immune response [8]. rs1126478 of the LTF gene can influence caries development [8] and genotype AA of rs1126478 displays bioactivities against other acid-producing microbes [9]. Defensin beta 1 (DEFB1) gene regulates microbial colonization and polymorphisms in the promoter region of this gene may alter the caries susceptibility [10,11,12]. Mannose-binding lectin (MBL2) plays an important role in innate immunity [13, 14]. Mutations of codon 54 of MBL2 are associated with recurrent infections and autoimmune diseases [15, 16]. Differences in major histocompatibility complex (MHC) or human leukocyte antigen (HLA) may cause variations in the immune response and influence the susceptibility to ECC [17]. Arachidonate 15-lipoxygenase (ALOX15) regulates inflammation and immune response and TT genotype of rs7217186 is a risk factor for ECC [18].
SNPs of immune response genes may alter the immune response, inflammatory reactions, and cytokine production and may modify the susceptibility to ECC. Studies on the association between polymorphisms of genes regulating immune response and dental caries in children have yielded inconsistent results. This systematic review aims to comprehend the association between genetic variants of immune response genes and ECC.
Methods
Registration of protocol and reporting guidelines
The systematic review was registered with PROSPERO (International Prospective Register of Systematic Reviews) with protocol number CRD42020179922 and is reported as per the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analysis) checklist 2020 [19]. We deviated from the protocol by including only the studies evaluating the polymorphisms of Immune response genes in this review.
Eligibility criteria
The research question of the present review was to ascertain the polymorphisms of immune response genes associated with ECC. The review followed the PECO framework (1) participants/population: children up to 6 years of age; (2) exposure: SNPs and genetic variants of genes regulating immune response (3) comparison: children without the polymorphisms of immune response genes; (4) outcome being ECC.
Observational studies (cross-sectional, case–control, and cohort design) that assessed the association of SNPs and variations in genes regulating immune response with ECC were included. Studies conducted on animals, case reports, case series, and those not in English were excluded.
Search strategy
An extensive search was conducted across various electronic databases such as MEDLINE via PubMed, CINAHL via EBSCO, LILACS, Web of Science, SCOPUS, EMBASE, Cochrane Central, Google Scholar, and Opengrey and GWAS (Genome-Wide Association Study) databases from January 2003 (completion of Human Genome Project) till September 2022. The search strategy has been summarized in Supplementary Table 1. The references of the existing reviews were assessed for relevant studies. A hand search of Journal of Clinical Pediatric Dentistry, Journal of Dentistry for Children, Pediatric Dentistry, International Journal of Paediatric Dentistry, European Archives of Paediatric Dentistry, Caries Research, Pediatric Dental Journal, Journal of Indian Society of Pedodontics and Preventive Dentistry, and Genetic Epidemiology and American Journal of Epidemiology were also conducted.
Selection of studies
The titles and abstracts of the selected studies were screened by two authors (PA and SP) independently and were grouped as included, excluded, and uncertain studies (if the abstract was ambiguous or unavailable). The full texts of the included studies and studies in uncertain categories were evaluated and studies which did not satisfy the eligibility criteria were excluded from the review. Disagreements about the inclusion of the studies were resolved either by consensus or by the third author (MSM). The corresponding author was contacted to elicit any missing, unreported data.
Data extraction
Two authors (PA and SP) recorded the data independently in a customized data extraction form. Data regarding author’s name, institutional affiliation, journal name, year of publication, study design, ethnicity of participants, chromosome, gene, sample size, SNPs analyzed, genotype and allele frequencies, co-variates evaluated, odds ratio (OR) at 95% confidence intervals (CI) and p-value were obtained.
Assessment of risk of bias
The quality of the included studies was assessed by two authors (PA and SP) using the Q-Genie tool [20]. This tool was designed and validated to evaluate the quality of studies analyzing the genetic association. It is a Likert-type scale, consisting of eleven questions. The maximum score for each question is seven and the minimum score is 1. For studies with a control group, a score < 35 indicates poor quality, in the range of 35–45 indicates moderate quality, and > 45 indicates good quality. Similarly, for studies without control groups, a score < 32 indicates low quality, in the range of 32–40 indicates moderate quality, and greater than 40 indicates good quality. Any difference of opinion was resolved by consensus or by another author (VV).
Data synthesis
Review Manager statistical software (RevMan 5.4, The Cochrane Collaboration, London, UK) was used to analyze the results of the included studies. The SNPs analyzed across the included studies were scrutinized and their genotype frequencies were collated to generate the forest plots and the pooled OR at 95% CI was calculated to estimate the effect sizes. The inverse-variance method was used to estimate the weight of the study. Heterogeneity was assessed by evaluating the population and study characteristics. The I2 analysis and chi-square test were conducted to assess heterogeneity between the studies and a random-effects model was used to conduct the meta-analysis.
Plink software and R statistical software were used to perform the gene-based analysis and gene pair-based associations. “BiomaRt” and “BS genome and Homosapiens.UCSC.hg38” packages were used to extract gene coordinates for each corresponding reference SNP cluster ID (RSIDs). The linkage disequilibrium r2 value was computed using Plink. Fisher’s exact approach, Simes approach, ECS (extended chi-square) approach, GATES (Gene-based Association Test using extended Simes procedure), inverse method, weighted truncated product method (TPM), unweighted truncated product method (TPM), and Adaptive Rank Truncated Product (ARTP) were used to perform gene-based analysis, with an error rate of 0.05. The largest test statistic from all the SNP-based tests in a gene was used as a gene-based test statistic.
Significant association within the immune response gene cluster were determined by gene cluster analysis. “GeneGeneInteR 1.22.0”, “BS genome,” “Biobase,” “Biocgenerics,” “Biocmanager,” and “ARTP2” packages were applied to perform gene-based and gene cluster analysis. Significant gene pairs associated with ECC were determined with LD (Linkage Disequilibrium) attenuating rank sum test. The multiple testing for pathway p values was performed using “Benjamini & Hochberg 1995” with a false detection rate (FDR) threshold set at 0.05 [21].
Enrichment analysis with protein–protein interaction (PPI) network construction was done to evaluate the functional impacts of differentially expressed genes. Search Tools for the Retrieval of Interacting Genes and Proteins (STRING) plot was constructed (https://string-db.org) wherein network nodes represent genes and lines of different colors represent different types of evidence used in predicting associations [22]. Possible gene network association and gene interaction were predicted using geneMANIA (gene function prediction using Multiple Association Network Integration Algorithm). The possible interaction network is predicted using many publicly available datasets on gene–gene and protein–protein interaction networks. GeneMANIA (http://www.genemania.org) is used for prioritizing genes for functional assays. Given a list of query genes, geneMANIA extends the list by including other functionally similar genes from the available genomics and proteomics data [23].
Results
Search outcome
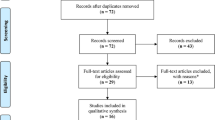
A total of 7124 articles were identified after a comprehensive search of databases. Initial screening resulted in the exclusion of 7011 articles, including duplicates. After a full-text screening of the remaining 113 articles, fifteen articles which satisfied the inclusion criteria were included in the present review. The PRISMA flow diagram depicting this is shown in Fig. 1. The included and excluded studies have been tabulated in Supplementary Table 2 and Supplementary Table 3, respectively.

PRISMA flow diagram
Description of studies
The studies were published from 2006 to 2022. The number of participants varied from 37 [24] to 1005 [25]. The studies were performed on diverse population groups such as Chinese, Turks, Norwegians, Polish, Caucasians, Hispanic Whites and Blacks, Non-Hispanic Whites and Blacks, Brazilians, Iranians, and Saudi Children. Eight studies were designed as case–control studies [25,26,27,28,29,30,31,32], two were cross-sectional [17, 18] and five were cohort studies [24, 33,34,35,36].
Thirteen studies assessed the association between 23 SNPs of seven genes regulating immune response with ECC in 4240 children; one study evaluated the relationship between four SNPs of T-cell receptor alpha variable 4 (TRAV4) locus and ECC in 176 children and one study analyzed the association between various alleles of Human Leucocyte Antigen (HLA) and ECC in seventy-nine participants. Six polymorphisms of the LTF gene were analyzed of which two were coding sequence variants and four were intronic variants. Eight polymorphisms of Lactoperoxidase (LPO) gene were evaluated of which six polymorphisms were intronic and two were Upstream Transcript Variants. Three Upstream transcript variants of DEFB1 and two intronic polymorphisms of ALOX15 were assessed. One coding sequence variant and one Upstream transcript variant of the MBL2 gene, one coding sequence polymorphism of mannose-binding lectin-associated serine protease 2 (MASP2) and a single upstream transcript variant of tumor necrosis factor-alpha (TNF-α) were evaluated for the relationship to ECC.
Quality assessment
The fifteen included studies scored in the range of 36 to 50 with the Q-Genie tool. Eleven studies were of good quality and four studies were of moderate quality [17, 24, 27, 28]. The quality assessment has been tabulated in Table 1.
Quantitative synthesis
Among the fifteen included studies, two studies that analyzed one SNP have been included in the meta-analysis. Studies with various designs were pooled together as the genetic association was analyzed using the inverse variance method with random-effect models. The TT genotype of the polymorphism rs11362 of DEFB1 displayed an OR of 1.03; 95%CI ranging from 0.65 to 1.64 and an insignificant P value of 0.89. The CC genotype revealed an OR of 1.11 and 95% CI ranging from 0.81 to 1.53 and a P value of 0.51 which was not statistically significant. The heterozygous genotype CT returned an OR of 0.83; 95% CI (0.60–1.15) and a P value of 0.26, which lacked statistical significance. The forest plots generated are shown in Fig. 2. Meta-analysis was not performed for rs1126478 as the same data was reported in two studies with a difference in the phenotype [24, 28]. Meta-analysis could not be performed for the other variants as the genotype frequencies were not reported.

a–c Forest plots of CC, CT, and TT genotypes of rs11362
Six polymorphisms of LTF (rs1126478, rs1126477, rs2269436, rs743658, rs4547741, rs17078878) eight polymorphisms of LPO (rs8178350, rs7209537, rs17762644, rs8178281, rs8178290, rs8178307, rs8178329, rs3744093) two variants of DEFB1 (rs1799946, rs1800972) and ALOX15 (rs2619112, rs7217186), three SNPs of MBL2 (rs1800450, rs7096206, rs11003125), four variants of TRAV4 locus (rs1997532, rs8011979, rs7150049, rs1997533), single SNP of MASP2 (rs72550870), and TNF-α (rs1800629) were evaluated in various studies. However, a meta-analysis could not be performed as the genotype frequencies were not reported.
Gene-based and gene cluster analysis
Gene-based analysis of the four variants in the TRAV4 locus revealed a significant association with ECC as reflected by the P values of the various statistical approaches — Fischer’s exact approach (P = 0.0026688), Simes approach (P = 0.002), GATES (P = 0.042), inverse method (P = 0.007375), ECS (P = 0.042), weighted TPM (P = 0.00218), unweighted TPM (P = 0.001962), ARTP (P = 0.001744). The polymorphism rs1800629 of TNF-α also revealed a statistically significant association with ECC under all the above-mentioned statistical approaches except GATES (P = 0.0805) and ECS (P = 0.0805). The results of the gene-based analysis are depicted in Table 2. Gene cluster analysis did not reveal a statistically significant association between polymorphisms of immune response gene cluster and incidence of ECC (Table 3).
Enrichment analysis
The constructed enrichment network consisted of nine nodes (differentially expressed genes) and eight edges with a strength of interaction score set at > 0.8. The PPI enrichment coefficient, average node degree, and average local clustering coefficient were < 2.35e − 06, 1.78, and 0.789, respectively. With the number of pre-defined clusters being three, the network was constructed with the Kmeans hierarchical clustering algorithm. The generated network revealed that ALOX15, DEFB1, HLA-DQB1, HLA-DRB1, and TNF-α constituted one cluster, LTF and LPO defined the second cluster, and MBL2 and MASP2 were in the third cluster. The enrichment analysis with the three clusters is depicted in Fig. 3.

STRING plot showing clustering of genes into 3 clusters and their interaction
Biological pathways under the enrichment analysis revealed that seven genes (LTF, DEFB1, HLA-DRB1, MBL2, HLA-DQB1, MASP2, TNF-α) were associated with humoral immune response. Four genes HLA-DRB1, MBL2, HLA-DQB1, and MASP2 were related to humoral immune response mediated by circulating immunoglobulin. Genes LTF, DEFB1, HLA-DRB1, MBL2, HLA-DQB1, MASP2 were linked to Innate Immune response. The lectin pathway of complement activation was mediated by MBL2 and MASP2. Three genes MBL2, TNF-α, and ALOX15 played a role in the regulation of phagocytosis whereas LTF and DEFB1 were associated with the innate immune response of the mucosa. Two genes TNF- α, ALOX15 were associated with positive regulation of heterotypic cell–cell adhesion. HLA-DRB1 and TNF- α were linked to the regulation of inflammatory response to antigenic stimulus. HLA-DRB1 and HLA-DQB1 were associated with MHC class II receptor activity under the molecular pathways. With respect to the cellular component, two genes HLA-DRB1 and HLA-DQB1 were associated with the MHC class II protein complex, integral component of the lumenal side of endoplasmic reticulum membrane, clathrin-coated endocytic vesicle membrane; HLA-DRB1, HLA-DQB1, TNF-α, and ALOX15 were associated with the side of the membrane and seven genes LTF, LPO, DEFB1, HLA-DRB1, MBL2, MASP2, and TNF-α were related to extracellular space. Reactome pathways revealed that MBL2 and MASP2 were related to the lectin pathway of complement activation; HLA-DRB1 and HLA-DQB1 were associated with translocation of ZAP-70 to immunological synapse, phosphorylation of CD3 and TCR zeta chains, and generation of second messenger molecules and with PD-1 signaling. Wiki pathways showed that HLA-DRB1 and TNF-α were related to Cytokines and inflammatory response and LTF and TNF-α genes were associated with LTF danger signal response pathway. Protein Domains Pfam and SMART (Simple Modular Architecture Research Tools) displayed HLA-DQB1 and HLA-DRB1 were associated with Class II histocompatibility antigen, beta domain. Among the biological processes associated with ECC, the lectin pathway of complement activation had the highest strength of association of 2.69 (false discovery rate of 0.0055), and regulation of inflammatory response to antigenic stimulus had a strength of association of 2.21(false discovery rate of 0.0227). Among the reactome pathways, the lectin pathway of complement activation had a strength of association of 2.79 (false discovery rate of 0.0073). Among the protein domains, class II histocompatibility antigen, beta domain had a strength of association of 2.68 with a false discovery rate of 0.0075. This is depicted in Table 4. GeneMANIA plot revealed that the genes which were prioritized are Mannan binding lectin serine peptidase 1 (MASP1), HLADQA1, HLADQA2, translocase of inner mitochondrial membrane 29 (TIMM29), family with sequence similarity 172 member A (FAM172A), histatin 3 (HTN3), complement (C4A), serpin family G member 1 (SERPING1), complement C2, epididymal peptidase inhibitor (EPPIN), peptidase M20 domain containing 2 (PM20D2), proline-rich acidic protein 1 (PRAP1), ficolin 2 (FCN2), zonadhesin (ZAN), DEAD-box helicase 31 (DDX31), APC down-regulated 1 (APCDD1), phosphatidylethanolamine binding protein 1 (PEBP1), CCAAT enhancer binding protein epsilon (CEBPE), keratin 1 (KRT1), trafficking protein particle complex 2 (TRAPPC2), N-myristoyltransferase 2 (NMT2), and carboxyl ester lipase (CEL). GeneMANIA plot revealed that these genes were linked by physical interaction at 39.34%, co-expression at 34.88%, pathways at 20.08%, co-localization at 2.94%, and shared protein at 2.18%. This is depicted in Fig. 4.

geneMANIA plot showing the interaction between various genes
Discussion
The etiology of ECC can be explained as an interplay of environmental and genetic factors [37]. This systematic review assessed the association between polymorphisms of immune response genes and ECC. Polymorphisms in the coding region as well as in the non-coding regions of the genes regulating the immune response were analyzed to understand their effect on susceptibility to ECC. SNPs in the coding region can change the encoded protein and those in the non-coding region can alter the transcription site and gene expression, thereby altering the predisposition to ECC [38]. Alternate transcription may also result in the synthesis of isoforms of various proteins, thus modifying the susceptibility.
SNPs are the most common variations in the human genome and when analyzing genetic association in complex diseases, a change of a single nucleotide may not always yield results of observable significance. But polymorphisms across the gene can modify the susceptibility significantly. Hence a gene-based analysis identifying the genetic variation across the gene can aid in understanding the alteration in susceptibility as the structure, function, and position of the gene are highly consistent across the individuals with or without the observed phenotype. Differences/inconsistencies arising due to changes in the population sub-structure can also be addressed with this gene-based analysis rather than allele-based association. Clustering helps in grouping of the genes based on similar patterns of gene expression and function. This helps in identifying genes associated with particular biological pathways and evaluation of variation across all such genes can significantly contribute to understanding the genetic underpinnings of the disease.
We deviated from the protocol by including databases Web of Science, SCOPUS, EMBASE, and Google Scholar to increase the specificity of search. Due to the voluminous nature of the included studies, only those studies evaluating the relationship between genetic variants of Immune response genes and ECC were included into the present review. To the best of our knowledge, this is the first systematic review to identify the polymorphisms of immune response genes associated with ECC and to conduct a gene-based and gene cluster analysis to comprehend their effect on ECC.
In this present review, the levels of beta-defensin 1 in the saliva were higher in children without caries [35] and quantitative synthesis revealed that genotype CC of rs11362 could be more in affected children. Genotype TT of rs7217186 of ALOX15 was a risk factor for ECC [32]. The frequency of allele DRB1*04 of HLA was increased in patients with active carious lesions [17]. G variant of rs7096206 of the MBL2 gene is associated with reduced protein levels in the serum, thus increasing the susceptibility to infections [39, 40], and was a risk factor for ECC in Polish children [25]. Mutant genotype GAC of codon 54 was higher in patients with ECC [26]. However, gene-based analysis of the variants of DEFB1 and MBL2 did not reveal statistical significance in contrast to findings of meta-analysis by Chistni et al. where MBL2 was reported to be associated with increased caries experience [41]. MBL-associated serine Protease 2 (MASP2) cleaves C2 and C4 to generate C3 convertase in the lectin pathway of the complement system (https://www.ncbi.nlm.nih.gov/gene/10747) [42]. Enrichment analysis revealed that the biological process of the lectin pathway of complement activation had the highest strength of association with ECC followed by humoral immune response and innate immune response of the mucosa, thus indicating that genes involved in these biological processes may affect ECC susceptibility.
The genotype CT of rs4547741 of LTF was found to be protective against ECC in Turkish children [32]. However, Al-Marshad et al., Wu et al., and Zaorska et al., did not find any association with ECC [29,30,31]. Other polymorphisms of LTF (rs1126477, rs1126478, rs2269436, rs743658, rs17078878) were not reported to be associated with altered susceptibility to caries [24, 28, 30, 31, 43]. However, gene-based analysis of the LTF gene revealed that multiple polymorphisms of the LTF gene were significantly associated with caries [44]. This is in contrast to the findings of the present review and could be accounted by differences in the study populations of the included primary studies. Elevated levels of TNF-α had been detected in saliva samples of patients with caries. rs1800629 of TNF-α is associated with systemic inflammation, and auto-immune diseases and elevated levels were observed in children with caries as a response of the host to pathological stimulus [45]. AG genotype of SNP rs1800629 of TNF-α was protective against ECC [28]. G alleles of rs1997533, rs7150049, and T Alleles of rs8011979 and rs1997532 in TRAV4 locus were associated with low caries experience in Turkish children and mRNA of TRAV4 is expressed to a greater extent in children with lesser caries experience [33]. Gene-based analysis revealed a statistically significant association between variants of TRAV4 locus and TNF-α with ECC.
The main limitations of this review are that of missing data due to which only two studies were included in the meta-analysis. Despite the heterogeneity being minimal (I2 = 0), the authors preferred to use the random-effects model, as it accounts for both within-study and between-study variance and is more conservative as it yields a wider confidence interval. Meta-analysis could not be performed for rs1126478 as the same data set was reported in two different studies with a different criterion for the observed phenotype. Certain studies did not have a control group and divided the study subjects into children with low and moderate caries and those with high caries [25]. If the observed phenotype was evaluated uniformly, the effect of the polymorphisms assessed on the carious phenotype may have been different. Most of the studies analyzed the phenotype using the DMFT/deft index. The evaluation of white spot lesions also is to be considered as they are the initial signs of demineralization and disease.
The relationship between the immune response of a host to an antigen is dynamic and can change as per the age of the patients and the dentition. Genes affecting susceptibility to caries differ between primary and permanent dentitions and the direction of association can also change between primary and permanent dentitions. Hence, longitudinal studies can result in more precise phenotypic characterization by assessing the gene-time interaction and aid in understanding the genetic underpinnings of the observed phenotype.
Conclusions
This review revealed that polymorphisms of TNF-α, ALOX15, TRAV4 locus, and alleles of HLA-DRB1 can modify susceptibility to ECC. Genotype TT of polymorphism rs7217186 of ALOX15 increased the susceptibility to ECC. Polymorphisms of genes regulating the lectin pathway of complement activation can alter the susceptibility to ECC. Quantitative Synthesis of TT and CC genotypes of rs11362 yielded OR greater than one. However, this has to be interpreted with caution as this evidence is not sufficient to state that rs11362 is a risk factor for ECC. The marginally higher OR suggests that the likelihood of these variants being associated with ECC may be higher which can be corroborated with studies being conducted on more number of individuals. Hence, studies with larger sample size, evaluation of the epigenetic mechanisms, transcriptomics, metabolomics, gene–gene interactions, and protein–protein interaction may aid in understanding the effect of genetic variants of immune response genes and ECC. Application of various BioInformatics tools contributes to understanding the genetic interaction and association. More studies evaluating the polymorphisms of functional significance in these immune response genes can aid in understanding their effect on ECC susceptibility and contribute towards “Personalized and Precision Dentistry.”
Availability of data and materials
Supplementary file is available.
Abbreviations
- ALOX15:
-
Arachidonate 15-lipoxygenase
- ARTP:
-
Adaptive Rank Truncated Product
- DEFB1:
-
Defensin beta 1
- DMFT/deft:
-
Decayed missing filled teeth/decayed extracted filled teeth
- ECC:
-
Early childhood caries
- FDR:
-
False detection rate
- GATES:
-
Gene-based Association Test using Extended Simes Procedure
- geneMANIA:
-
Gene function prediction using Multiple Association Network Integration Algorithm
- GWAS:
-
Genome-Wide Association Study
- HLA:
-
Human leukocyte antigen
- HLA-DQB1:
-
Human leukocyte antigen major histocompatibility complex, class II, DQ beta 1
- HLA-DRB1:
-
Human leukocyte antigen major histocompatibility complex, class II, DR beta 1
- LD:
-
Linkage disequilibrium
- LPO:
-
Lactoperoxidase
- LTF:
-
Lactotransferrin
- MASP2:
-
Mannose-binding lectin-associated serine protease 2
- MBL2:
-
Mannose-binding lectin 2
- MHC:
-
Major histocompatibility complex
- PECO:
-
Participants, Exposure, Comparison, Outcome
- PPI:
-
Protein-protein interaction
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analysis
- RSIDs:
-
Reference SNP cluster ID
- SNPs:
-
Single nucleotide polymorphisms
- STRING:
-
Search Tools for Retrieval of Interacting Genes and Proteins
- TPM:
-
Truncated product method
- C2:
-
Complement 2
- C3:
-
Complement 3
- C4:
-
Complement 4
- TNF-α:
-
Tumor necrosis factor-alpha
- TRAV4:
-
T-cell receptor alpha variable 4
- ZAP-70:
-
Zeta chain of T cell receptor-associated protein kinase 70
- CD3:
-
Cluster of differentiation 3
- TCR:
-
T cell receptor
- PD-1:
-
Programmed death 1
- SMART:
-
Simple Modular Architecture Research Tool
- MASP1:
-
Mannan-binding lectin serine peptidase 1
- TIMM29:
-
Translocase of inner mitochondrial membrane 29
- FAM172A:
-
Family with sequence similarity 172 member A
- HTN3:
-
Histatin 3
- C4A:
-
Complement 4A
- SERPING1:
-
Serpin family G member 1
- EPPIN:
-
Epididymal peptidase inhibitor
- PM20D2:
-
Peptidase M20 domain containing 2
- PRAP1:
-
Proline-rich acidic protein 1
- FCN2:
-
Ficolin 2
- ZAN:
-
Zonadhesin
- DDX31:
-
DEAD-box helicase 31
- APCDD1:
-
APC down-regulated 1
- PEBP1:
-
Phosphatidylethanolamine binding protein 1
- CEBPE:
-
CCAAT enhancer binding protein epsilon
- KRT1:
-
Keratin1
- TRAPPC2:
-
Trafficking protein particle complex 2
- NMT2:
-
N-myristoyltransferase 2
- CEL:
-
Carboxyl ester lipase
References
Black GV (1899) Susceptibility and immunity to dental caries. Dental Cosmos 41(9):826–836
Yildiz G, Ermis RB, Calapoglu NS, Celik EU, Türel GY (2016) Gene-environment Interactions in the etiology of dental caries. J Dent Res 95(1):74–79. https://doi.org/10.1177/0022034515605281
Conry JP, Messer LB, Boraas JC, Aeppli DP, Bouchard TJ Jr (1993) Dental caries and treatment characteristics in human twins reared apart. Arch Oral Biol 38(11):937–943. https://doi.org/10.1016/0003-9969(93)90106-v
Bretz WA, Corby PM, Hart TC, Costa S, Coelho MQ, Weyant RJ, Robinson M, Schork NJ (2005) Dental caries and microbial acid production in twins. Caries Res 39(3):168–172. https://doi.org/10.1159/000084793
Wang X, Shaffer JR, Weyant RJ, Cuenco KT, DeSensi RS, Crout R, McNeil DW, Marazita ML (2010) Genes and their effects on dental caries may differ between primary and permanent dentitions. Caries Res 44(3):277–284. https://doi.org/10.1159/000314676
Vieira AR, Modesto A, Marazita ML (2014) Caries: review of human genetics research. Caries Res 48(5):491–506. https://doi.org/10.1159/000358333
Opal S, Garg S, Jain J, Walia I (2015) Genetic factors affecting dental caries risk. Aust Dent J 60(1):2–11. https://doi.org/10.1111/adj.12262
Fine DH, Toruner GA, Velliyagounder K, Sampathkumar V, Godboley D, Furgang D (2013) A lactotransferrin single nucleotide polymorphism demonstrates biological activity that can reduce susceptibility to caries. Infect Immun 81(5):1596–1605. https://doi.org/10.1128/IAI.01063-12
Velliyagounder K, Kaplan JB, Furgang D, Legarda D, Diamond G, Parkin RE, Fine DH (2003) One of two human lactoferrin variants exhibits increased antibacterial and transcriptional activation activities and is associated with localized juvenile periodontitis. Infect Immun 71(11):6141–6147. https://doi.org/10.1128/IAI.71.11.6141-6147.2003
Krisanaprakornkit S, Weinberg A, Perez CN, Dale BA (1998) Expression of the peptide antibiotic human beta-defensin 1 in cultured gingival epithelial cells and gingival tissue. Infect Immun 66(9):4222–4228. https://doi.org/10.1128/IAI.66.9.4222-4228.1998
Mathews M, Jia HP, Guthmiller JM, Losh G, Graham S, Johnson GK, Tack BF, McCray PB Jr (1999) Production of beta-defensin antimicrobial peptides by the oral mucosa and salivary glands. Infect Immun 67(6):2740–2745. https://doi.org/10.1128/IAI.67.6.2740-2745.1999
Dunsche A, Açil Y, Siebert R, Harder J, Schröder JM, Jepsen S (2001) Expression profile of human defensins and antimicrobial proteins in oral tissues. J Oral Pathol 30(3):154–158. https://doi.org/10.1034/j.1600-0714.2001.300305.x
Ezekowitz RA (2003) Role of the mannose-binding lectin in innate immunity. J Infect Dis 187(Suppl 2):S335–S339. https://doi.org/10.1086/374746
Garred P (2008) Mannose-binding lectin genetics: from A to Z. Biochem Soc Trans 36(Pt 6):1461–1466. https://doi.org/10.1042/BST0361461
Hansen S, Holmskov U (1998) Structural aspects of collectins and receptors for collectins. Immunobiology 199(2):165–189. https://doi.org/10.1016/S0171-2985(98)80025-9
Mok MY, Ip WK, Lau CS, Lo Y, Wong WH, Lau YL (2007) Mannose-binding lectin and susceptibility to infection in Chinese patients with systemic lupus erythematosus. J Rheumatol 34(6):1270–1276
Bagherian A, Nematollahi H, Afshari JT, Moheghi N (2008) Comparison of allele frequency for HLA-DR and HLA-DQ between patients with ECC and caries-free children. J Indian Soc Pedod Prev Dent 26(1):18–21. https://doi.org/10.4103/0970-4388.40316
Abbasoğlu Z, Tanboğa İ, Küchler EC, Deeley K, Weber M, Kaspar C, Korachi M, Vieira AR (2015) Early childhood caries is associated with genetic variants in enamel formation and immune response genes. Caries Res 49(1):70–77. https://doi.org/10.1159/000362825
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, Chou R, Glanville J, Grimshaw JM, Hróbjartsson A, Lalu MM, Li T, Loder EW, Mayo-Wilson E, McDonald S, McGuinness LA, … Moher D (2021) The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 372:n71. https://doi.org/10.1136/bmj.n71
Sohani ZN, Meyre D, de Souza RJ, Joseph PG, Gandhi M, Dennis BB, Norman G, Anand SS (2015) Assessing the quality of published genetic association studies in meta-analyses: the quality of genetic studies (Q-Genie) tool. BMC Genet 16:50. https://doi.org/10.1186/s12863-015-0211-2
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B StatMethodol 57(1):289–300
https://www.string-db.org. Search Tools for Retrieval of Interacting Genes and Proteins. Accessed 27 Dec 2022
http://www.genemania.org. gene function prediction using Multiple Association Network Integration Algorithm. Accessed 31 Dec 2022
Krasone K, Lāce B, Akota I, Care R, Deeley K, Küchler EC, Vieira AR (2014) Genetic variation in the promoter region of beta-defensin 1 (DEFB 1) is associated with high caries experience in children born with cleft lip and palate. Acta Odontol Scand 72(3):235–240. https://doi.org/10.3109/00016357.2013.822549
Wang M, Qin M, Xia B (2017) The association of Enamelin, Lactoferrin, and Tumour necrosis factor alpha gene polymorphisms with high caries susceptibility in Chinese children under 4 years old. Arch Oral Biol 80:75–81. https://doi.org/10.1016/j.archoralbio.2017.03.023
Olszowski T, Adler G, Janiszewska-Olszowska J, Safranow K, Kaczmarczyk M (2012) MBL2, MASP2, AMELX, and ENAM gene polymorphisms and dental caries in Polish children. Oral Dis 18(4):389–395. https://doi.org/10.1111/j.1601-0825.2011.01887.x
Yang Y, Wang W, Qin M (2013) Mannose-binding lectin gene polymorphisms are not associated with susceptibility to severe early childhood caries. Hum Immunol 74(1):110–113. https://doi.org/10.1016/j.humimm.2012.08.012
Mubayrik AFB, Deeley K, Patir A, Koruyucu M, Seymen F, Vieira A (2014) Polymorphisms in the antimicrobial peptide DEFB1 are not associated with caries in primary dentition. JPDA 23(02):66
Wang M, Qin M (2018) Lack of association between LTF gene polymorphisms and different caries status in primary dentition. Oral Dis 24(8):1545–1553. https://doi.org/10.1111/odi.12939
AlMarshad LK, AlJobair AM, Al-Anazi MR, Bohol MFF, Wyne AH, Al-Qahtani AA (2021) Association of polymorphisms in genes involved in enamel formation, taste preference and immune response with early childhood caries in Saudi pre-school children. Saudi J Biol Sci 28(4):2388–2395. https://doi.org/10.1016/j.sjbs.2021.01.036
Zaorska K, Szczapa T, Borysewicz-Lewicka M, Nowicki M, Gerreth K (2021) Prediction of early childhood caries based on single nucleotide polymorphisms using neural networks. Genes 12(4):462. https://doi.org/10.3390/genes12040462
Wu L, Li Z, Zhou J, Ma B, Yu F, Zheng X, Hu X, Ma Z, Su X (2022) An association analysis for genetic factors for dental caries susceptibility in a cohort of Chinese children. Oral Dis 28(2):480–494. https://doi.org/10.1111/odi.13758
Briseño-Ruiz J, Shimizu T, Deeley K, Dizak PM, Ruff TD, Faraco IM Jr, Poletta FA, Brancher JA, Pecharki GD, Küchler EC, Tannure PN, Lips A, Vieira TC, Patir A, Koruyucu M, Mereb JC, Resick JM, Brandon CA, Letra A, Silva RM, … Vieira AR (2013) Role of TRAV locus in low caries experience. Hum Genet 132(9):1015–1025. https://doi.org/10.1007/s00439-013-1313-4
Stanley BO, Feingold E, Cooper M, Vanyukov MM, Maher BS, Slayton RL, Willing MC, Reis SE, McNeil DW, Crout RJ, Weyant RJ, Levy SM, Vieira AR, Marazita ML, Shaffer JR (2014) Genetic association of MPPED2 and ACTN2 with dental caries. J Dent Res 93(7):626–632. https://doi.org/10.1177/0022034514534688
Lips A, Antunes LS, Antunes LA, Abreu JGB, Barreiros D, Oliveira DSB, Batista AC, Nelson-Filho P, Silva LABD, Silva RABD, Alves GG, Küchler EC (2017) Genetic polymorphisms in DEFB1 and miRNA202 are involved in salivary human β-Defensin 1 levels and caries experience in children. Caries Res 51(3):209–215. https://doi.org/10.1159/000458537
Weber M, Bogstad Søvik J, Mulic A, Deeley K, Tveit AB, Forella J, Shirey N, Vieira AR (2018) Redefining the phenotype of dental caries. Caries Res 52(4):263–271. https://doi.org/10.1159/000481414
Piekoszewska-Ziętek P, Turska-Szybka A, Olczak-Kowalczyk D (2017) Single nucleotide polymorphism in the aetiology of caries: systematic literature review. Caries Res 51(4):425–435. https://doi.org/10.1159/000476075
Tannure PN, Küchler EC, Lips A, de Costa MC, Luiz RR, Granjeiro JM, Vieira AR (2012) Genetic variation in MMP20 contributes to higher caries experience. J Dent 40(5):381–386. https://doi.org/10.1016/j.jdent.2012.01.015
Eisen DP, Minchinton RM (2003) Impact of mannose-binding lectin on susceptibility to infectious diseases. Clin Infect Dis 37(11):1496–1505. https://doi.org/10.1086/379324
Bouwman LH, Roep BO, Roos A (2006) Mannose-binding lectin: clinical implications for infection, transplantation, and autoimmunity. Hum Immunol 67(4–5):247–256. https://doi.org/10.1016/j.humimm.2006.02.030
Chisini LA, Cademartori MG, Conde MCM, Costa FDS, Tovo-Rodrigues L, Carvalho RV, Demarco FF, Correa MB (2020) Genes and SNPs in the pathway of immune response and caries risk: a systematic review and meta-analysis. Biofouling 36(9):1100–1116. https://doi.org/10.1080/08927014.2020.1856821
https://www.ncbi.nlm.nih.gov/gene/10747. Accessed 11 Jan 2023
Sharifi R, Jahedi S, Mozaffari HR, Imani MM, Sadeghi M, Golshah A, Moradpoor H, Safaei M (2020) Association of LTF, ENAM, and AMELX polymorphisms with dental caries susceptibility: a meta-analysis. BMC Oral Health 20(1):132. https://doi.org/10.1186/s12903-020-01121-7
Li X, Liu D, Sun Y, Yang J, Yu Y (2021) Association of genetic variants in enamel-formation genes with dental caries: a meta- and gene-cluster analysis. Saudi J Biol Sci 28(3):1645–1653. https://doi.org/10.1016/j.sjbs.2020.11.071
Gornowicz A, Bielawska A, Bielawski K, Grabowska SZ, Wójcicka A, Zalewska M, Maciorkowska E (2012) Pro-inflammatory cytokines in saliva of adolescents with dental caries disease. Ann Agric Environ Med 19(4):711–716
Acknowledgements
The authors wish to thank Tannure et al., for sharing the data.
Funding
The authors extend their thanks to the assistance provided under Founder-Chancellor, Shri NPV Ramasamy Udayar Research Fellowship (Ref No. Founder Chancellor Fellowship 2019–20-2) by Sri Ramachandra Institute of Higher Education and Research in conducting this study.
Author information
Authors and Affiliations
Contributions
PA, SP — conceptualisation, database search, data curation, writing the original draft, review and editing of the manuscript. MSM, VV, NS — conceptualisation, methodology, project administration, supervision, review and editing of the manuscript. RK, SA — methodology, software, formal analysis, review and editing of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This work was mainly carried out in the Department of Pediatric and Preventive Dentistry and Department of Human Genetics of Sri Ramachandra Institute of Higher Education and Research, Chennai.
Supplementary Information
Additional file 1: Supplementary Table 1.
Search Strategy. Supplementary Table 2. Table of characteristics of Included studies. Supplementary Table 3. Table of Characteristics of Excluded Studies.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aruna, P., Patil, S.S., Muthu, M.S. et al. Association between polymorphisms of immune response genes and early childhood caries — systematic review, gene-based, gene cluster, and meta-analysis. J Genet Eng Biotechnol 21, 124 (2023). https://doi.org/10.1186/s43141-023-00566-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00566-x