
Abstract
Background
The Himalayas have always been an enigma and, being biodiversity hotspots, are considered extremely important from an ecological point of view. Recent advances in studies regarding high-altitude lakes have garnered relevant importance as these habitats could harbor potential psychrophilic and psychrotrophic microbes with bio-prospective applications. Contemplating the above scenario, the present study has been undertaken to understand the diversity and the functional capacities of the microbes thriving in this lake.
Results
In our present study on Samiti Lake, the abundance of Proteobacteria as the major phylum was seen in both the soil and water samples. Incase of the ABSLW (water) and ABS1 (soil) sample, 148,066 and 239,754 predicted genes, were taken for functional analysis. The KEGG analysis showed that ABSLW and ABS1 had 122,911 and 160,268, genes assigned to KO terms respectively. Whereas in case of COG functional analysis, 104,334 and 130,191 genes were assigned to different COG classes for ABSLW and ABS1 respectively. Further, on studying the glycoside hydrolases, an abundance of GH13, GH2, GH3, GH43, and GH23 in both the soil and water samples were seen.
Conclusion
Our study has provided a comprehensive report about the bacterial diversity and functional capacities of microbes thriving in Samiti Lake. It has also thrown some light on the occurrence of glycoside hydrolases in this region, as they have numerous biotechnological applications in different sectors.
Similar content being viewed by others

Background
Culture-independent approaches to studying microbial diversity in extreme environments have increased significantly because of their bio prospective aspects [1, 2]. These unexplored extreme environments and their functional diversity studies could facilitate various potential bio-prospective solutions to environmental concerns [2]. Recently, ecology and biodiversity relationships with bacterial communities via environmental sampling and the next-generation sequencing technologies have been at the forefront of research [3]. Since most microorganisms are still difficult to cultivate hence, culture-based methods have been found to be insufficient for determining the diversity of microbes [4]. Exploring the microbial diversity via metagenomics has been a boon to science as, it has promoted and enabled a better understanding of the unexplored and extreme realm and diversified our knowledge about microbial adaptations and their interactions in unexplored areas [5, 6]. Numerous metagenomics studies have been carried out in aquatic environments [7, 8]. Diverse metagenomics studies on the psychrophilic Pangong Lake [9], Upper Mississippi River (Minnesota) [10], and Amazon Basin freshwater lakes [11] have recently been reported. The metagenomics methodology has thus paved the way through which novel gene sequences, and metabolic pathways of unculturable microorganisms have been identified as has been elucidated by some recent works [12].
High-altitude lakes are exposed to extreme environments like low nutrient conditions, UV radiation, and low temperatures [13, 14]. Furthermore, these high-altitude lakes are harder to access than low-altitude lakes due to their distant location [15]. Additionally, the mountain ecosystems show altitudinal gradients and adverse environmental factors [16,17,18]. Reports suggest that although the diversity of flora and fauna shows an inverse relationship with increasing altitude, it may not apply to microbes [19, 20]. The drastic seasonal shifts in the physiochemical properties of the soil, climatic changes in altitude gradient and fluctuating subzero temperatures are characteristics of the Himalayan range [1, 21]. The Himalayas are biodiversity hotspots for different flora and fauna, ranging from orchids and rhododendrons to Himalayan tahr, Red panda, Himalayan Musk deer, Black Eagle, Tibetan Partridge [22, 23] etc. Likewise, this region may also hold a great promise of untapped potential for microbial diversity. Recent metagenomics research on Manikaran Hot Springs [24], frozen soil of the northwestern Himalayas of Jammu and Kashmir [25], and high-elevation Himalayan glacial lakes (Parvati kund) imply that these Himalayan regions are undergoing exploration [26]. Further, the Eastern Himalayas provide a plethora of high-altitude lakes [27] and such regions could harbor numerous psychrophilic and psychrotolerant microorganisms [28] that could be the source of numerous cold-adapted enzymes [29]. Psychrophilic enzymes have a wide range of documented uses, including stone washing [30], bioremediation, biotransformation, biomedical [31,32,33], and molecular biology applications [34, 35]. Reports of bio-augmentation with psychrophiles have also been shown to improve the biodegradation of recalcitrant substrates [36]. One such enzyme, glycoside hydrolases (GH), has an increased potential for complex carbohydrate deconstruction [37]. Glycoside hydrolases like cold active cellulases [38], amylases [39, 40], and β-galactosidases [41, 42] have found to be advantageous in a variety of industries like that of detergents, cosmetics, food, textiles, and bakery [43].
Sikkim is a Himalayan state situated in the northeastern part of the Indian territory, close to the Eastern Himalayas [44]. Samiti Lake, is a glacial lake in Sikkim that sits at an elevation of approximately 4200 to 4300 m (13,700 ft.) [45]. The high elevation of the lake and the cold temperatures prevailing in this region, are apt for the detection of diverse microflora having metabolic pathways that they use as a survival tactic in such extreme environments. It is in this regard that the present study was undertaken, to have an insight into the microbial community thriving in this region and to unearth the presence of glycoside hydrolases as they are presently being used extensively in the biotechnological industries.
In the present study, diversity analysis was done using Kaiju, Cognizer was used to obtain functional annotation against the COG, KEGG, Pfam, FIG, and GO databases. Ecological distance matrices were computed and employed to determine the diversity of the species. Further, to study the GH families, the CAZy database was employed.
Materials and method
Collection of environmental sample
Soil samples (ABS1) (100 g) at depths of approximately 10 to 12 cm, beneath the soil surface from Samiti Lake in sterile plastic and glass containers, and water samples (ABSLW) of 2.5 L were collected from three different points in the lake at 1-m depth. After careful homogenization of the soil, they were stored at −20° C. All of the samples were further transported to Xcelris labs for DNA sequencing. The samples were pooled before processing for DNA isolation.
Physicochemical parameters
The physicochemical properties of water like pH and conductivity were analyzed using Eutech’s Cyber Scan PCD650 (a handheld waterproof meter). The soil organic carbon was estimated by chromic acid method proposed by Walkley and Black [46], and the Nitrogen content was measured by the Kjeldah method [47].
Isolation of DNA and library preparation
For metagenomic analysis, the Xcelgen Soil DNA isolation kit was used to isolate the DNA, and 0.8% agarose gel was used to detect the DNA (loaded 3 μl). Covaris was used to shear the DNA. Further, HiFi PCR Master Mix was used to amplify the fragments. The paired-end sequencing library was performed using NEB Next Ultra DNA library Prep Kit [48]. The size of libraries as determined by the Agilent bio analyzer was 470bp and 475bp for ABS1 and ABSLW samples, respectively. Further, the libraries have been submitted in NCBI with accession numbers SAMN13671136 and SAMN13671135. Illumina HiSeq 2500 platform was used to sequence the sample libraries on 2 × 150 bp chemistry to generate ~ 3Gb of data per sample.
Metagenome analysis
Reads obtained from Illumina platform were quality checked using FastQC v0.11.9, and filtered to remove sequencing adapter as well as low-quality bases using Trimmomatic v0.36. Clean reads thus obtained were used for de novo assembly of data. Scaffold generation was done using Metaspades and CLC genomics workbench at default parameters for ABS1 and ABSLW samples. While Metaspades uses de Bruijin graph, CLC genomic workbench uses overlap layout consensus for de novo assembly of reads. The scaffolds thus generated were subject to gene prediction using Prodigal (v2.6.3) followed by diversity analysis using Kaiju [49] which is a very sensitive taxonomy classification tool. It classifies the sequence at protein level using greedy mode. However, functional annotation against COG, KEGG, Pfam, FIG, and GO database was obtained using Cognizer [50] which uses novel-directed search strategy to reduce the computational time.
Ecological distance matrices were calculated and used in this study to find out all the canonical macro-ecological species diversity. To find out the probability that two randomly sampled organisms belong to the same species, Simpson diversity index (D) was calculated [51] according to the following:
where ni is the abundance of ith species and N is the total number of individual present.
Species richness and dominance were calculated in this study via Shannon’s diversity index (H) [52] according to the following:
where ni is the abundance of ith species and N is the total number of individual present.
In an attempt to find out species richness which is independent of sample size, Menhinick index has been calculated [53].
Calculation of Buzas and Gibson’s index [54] helped decipher conservation model and observe the trends of changes in an ecosystem.
Berger-Parker dominance index [55] is simple mathematical expression relating species richness and evenness.
Margalef’s diversity index [56] is a species richness index. Many species richness measures are strongly dependent on sampling effort. The greater the sampling effort, higher the index value. Thus, Margalef’s diversity index considers the number of taxa and total number of individuals.
Annotation against CAZy
The family of glycoside hydrolases (GH) is present in the CAZy database (www.cazy.org) which is a database of Carbohydrate Active Enzymes or CAZymes [57]. Henceforth, the CAZy database was used to analyze the GH families present in samples using dbCAN [58].
Results
Physicochemical parameters
Soil pH is an indicator of the soil’s acidity or alkalinity. The physicochemical parameters of the soil revealed the pH of the soil to be in the range of 6.25±0.14. Organic carbon is the amount of soil organic matter [59] where the carbon content of the soil was found to be around 1.20 ±0.03; this could also be an essential factor to indicate the abundance of Betaproteobacteria in the soil as elucidated from our study. Nitrogen, i.e., significantly important to plant growth [60] was also checked, and it was found to be around 0.10±0.015. This could facilitate the growth of Firmicutes in the soil as they have been reported to play a role in nitrate metabolism [61]. However, the pH of water was found to be neutral around 7.12±0.07, indicating an ideal environment for bacterial growth. Further, conductivity, was found to be around 204.3±0.76 μS cm-1, a factor used to gauge the degree of mineralization [62]. Overall, Physicochemical parameters are significant because its disparity can bring about changes in the microbial community [63].
Taxonomic annotation using standalone Kaiju
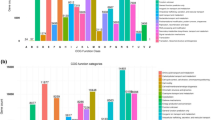
ABS1 taxonomic hits distribution shows Proteobacteria as the most abundant phylum represented by 78,597 genes. Whereas at class level, Betaproteobacteria and Deltaproteobacteria were found to be most abundant represented by 39,405 and 19,928 genes, respectively. At order level, taxonomic hits distribution showed 20,510 genes for Burkholderiales followed by 14,129 for Desulfuromonadales. The taxonomic distribution of genes for the bacterial family revealed 11,413 gene hits on Geobacteraceae followed by 10227 on Commonadaceae. However, at the genus level, there was 5864 gene hits on Geobacter followed by 4775 on Nitrospira. At the species level the taxonomic hit distribution showed 10,279 gene hits of Bacteroidetes bacterium GWB2_41_8 followed by 6135 gene hits on Chloroflexi bacterium (Fig. 1, Supplementary Figure 1)

The figure depicts the abundance of bacterial communities of ABS1 sample at different taxonomic units (Phylum Family and Genus). Lesser than 1% of the distribution were together labeled as "Others"
Taxonomic hit distribution of ABSLW shows Proteobacteria as most abundant phylum represented by 104,745 genes, with 62,779 gene hits of Alphaproteobacteria followed by 26,921 hits of Betaproteobacteria at class level. In the bacterial order distribution, taxonomic hits showed 25,406 gene hits of Burkholderailes followed by 22,912 of Sphingomonadales. The taxonomic distribution of gene hits for the bacterial family revealed 16,940 gene hits on Sphingomonadaceae followed by 16,038 Rhodobacteraceae. In the genus level, there was 10,812 gene hits of Flavobacterium followed by 10,141 on Pseudomonas. At the species level, the taxonomic hit distribution showed 3211 gene hits of Sphingomonadaceae PASS1 followed by 1905 gene hits of Oxalobacteraceae (Fig. 2, Supplementary Figure 2).

The figure depicts the abundance of bacterial communities of ABSLW sample at different taxonomic units (Phylum Family and Genus). Lesser than 1% of the distribution were together labeled as "Others"
COG functional category hit distribution
The COG database is an endeavor to categorize proteins [64]. The COG functional analysis of ABS1 showed 130,191 genes were assigned to COG where maximum genes, i.e., 14,175 genes, were found to belong to general function prediction only (S1). COG functional analysis of ABSLW shows 104,334 genes assigned to COG, where 10,667 genes were belonging to amino acid transport and metabolism and 11,004 genes falling into the category of general function prediction only (S6). The COG classification also depicted the prevalence of the functions associated with amino acid transport and metabolism, energy production and conversion, general function prediction only, carbohydrate transport and metabolism, lipid transport and metabolism, replication, recombination, and repair, signal transduction mechanisms, translation ribosomal structure and biogenesis, post-translational modification, protein turnover, and chaperones in both the samples.
KEGG functional category hit distribution
KEGG is a collection of databases that help to predict the different metabolic pathways of a biological system [65]. In ABS1 (S4), functional analysis showed that from a total of 239,754 genes, 160,268 genes were assigned to KEGG classes. Whereas, in the case of ABSLW (S9), KEGG functional analysis showed that from a total of 148,066 genes 122,911 genes were assigned to KEGG classes. In both the cases, the majority of KOs comprised of metabolism category. ABS1 samples revealed a greater involvement of two component systems, cell cycle response regulator, and cell cycle response regulator. However, methyl-accepting chemotaxis protein, ATP-binding cassette, histidine kinase, and RNA polymerase sigma 70 factor were also some of the abundant terms associated with KO of the ABS1 sample. The most abundant KEGG term in ABSLW was found to be iron complex outer membrane receptor protein. Some of the terms associated with the ABSLW sample were also methyl-accepting chemotaxis protein, acyl CoA dehydrogenase, N-acetylmuramayl-L-alanine amidase, and ATP-binding cassete. The analysis of the KEGG pathway showed diverse pathways and mechanisms that the microbes thriving in this lake use as a survival tactic to endure the extreme conditions in this region.
Pfam functional category hit distribution
Pfam is a useful annotation tool for categorizing protein families [66]. Analysis of ABS1 showed that 140,559 genes were assigned with different PFAM domains (S5). Likewise when ABSLW sample was analysed it resulted in assignment of PFAM domains to 111,508 genes, (S10).
FIGfam functional category hit distribution
Figfams are a set of proteins that share a common function [67]. FIGfams functional analysis of ABS1 (S2) showed that from total of 239,754 genes, 84,544 genes were assigned with different FIG classes. On the other hand 62,074 genes were assigned to different FIG classes for ABSLW (S7).
Gene Ontology (GO) functional category hit distribution
The Gene Ontology project represents gene product properties [68]. Henceforth, GO functional analysis was carried out for ABS1 and ABSLW samples. It resulted in assignment of 181,375 and 143,201 genes with GO classes, for ABS1 and ABSLW respectively (S3,S8).
Alpha (α) diversity index
The Simpson index represents the species diversity in a particular habitat. According to our results, the Simpson index was 0.022 (water) and 0.16 (soil). This index represents higher diversity in the habitat. Simpson’s reciprocal index for water and soil samples was 47 and 6.3, respectively. The Shannon index calculated from water and soil samples was 4.2 and 3, respectively, suggesting an even distribution of species. The Menhinick index for both water (.41) and soil (.054) indicates a richness of species. Here, in our results, Buzas and Gibson’s index were found to be.76 and.8, respectively, for water and soil samples, indicating that the species in almost both the samples were evenly distributed. The Berger Parker dominance of the water sample has an index of 0.077, and the soil sample has an index of 0.3, which suggests the samples have higher richness and more abundance. Margalef’s index, in the water sample (7.8) and in the soil sample (.86), signifies abundant diversity (Table 1).
Glycoside hydrolase
Our study shows an abundance of GH13, GH2, GH3, GH43, and GH23 in both the soil and water samples in high numbers as is evident from the (Fig. 3); however, the presence of GH53 was abundant in ABS1 compared to ABSLW.

The heat map depicts the presence of glycoside hydrolases in ABS1 and ABSLW samples
Discussion
Our study has indicated an abundance of Proteobacteria in this region, which is very similar to the reports by Gangwar et al. [21] who revealed a predominance of Proteobacteria, in Western Himalayas. Reports from Pangong lake, Tsomgo Lake [9, 69], some of the high-altitude Himalayan lakes, Alpine lakes of Hengduan Mountain [70], and Sayram lake [71] have also reported the abundance of Proteobacteria. Many members of Proteobacteria are involved in the metabolism of sulphate and nitrate [61]. Proteobacteria have been reported to play significant roles in carbon sequestration and nitrogen cycling [72]. The abundance of AlphaProteobacteria in water sample is well justified as it requires minimal amount of nutrients [73]. However, the presence of Betaproteobacteria in the soil sample elucidates the fact that it extensively utilizes the nutrient of the soil. The abundance of Firmicutes also sheds light on its role in nitrate, methane, and sulphate metabolism as has been reported by Haldar et al. [61].
Studies conducted widely over polar and non-polar glaciers have also reported the presence of genera like Proteobacteria, Actinobacteria, and Flavobacteria [74]. At the genus level Flavobacterium was the most abundant taxon in water. Flavobacteria have been reported to play an imperative role mineralizing poorly degradable macronutrients and providing their surrounding environments with carbon flux regulators [75]. Genus Flavobacterium are considered as chief mineralizers of organic matter, numerous reports of their isolation from soil [76], water [77], Antarctic regions [78], and glacier samples [79] have been described. Further, it has been reported that many members of Geobacter play a role in carbon and metals cycling in aquatic sediments and also help in the bioremediation of metal-contaminated groundwater and harvesting of electricity [80, 81]. They have been found to play significant roles in both pristine and contaminated subsurface environments [82, 83], also helping in bioremediation [84]. Moreover, some Geobacter species have been reported to help in sulfur reduction, [85] where most of the mechanisms of sulfur reduction relates to elemental sulfur being converted to H2S [86], ultimately, sulfide the ultimate byproduct serves as an electron donor for a wide range of microbial metabolisms [87].
Glycoside hydrolases facilitate carbohydrate degradation and are thus sought after enzyme in different industries [88]. The abundance of GH13 in this lake indicates the predominance of α-amylase (very significant in the hydrolysis of starch and related poly- and oligosaccharides) [89] as its been reported as an essential representative of the GH13 family [89]). Numerous reports of microbial α-amylases in bioremediation and biorefinery [90, 91] also highlights its importance. The prominence of GH43 also indicates the presence of many enzymes under GH43 involved in the breakdown of pectin and hemicellulose polymers [92]. Another major industrially important enzyme β-galactosidases are glycoside hydrolases (GH) that give galactose molecules by hydrolyzing glycosidic bonds [42]. Galactosidases hold great potential in industrial and biotechnological applications [42]. These enzymes are ubiquitous and have been isolated from diverse and extreme environments [42, 93]. The lake also has the presence of high quantity of GH3, and since this lake is located at a very high altitude, it could also possess psychrophilic and psychrotrophic β-glucosidase. Recent, reports on psychrophilic Paenibacillus sp. for production of cold-active β-glucosidase belonging to GH3 family have been elucidated [41]. The abundance of GH2 in this lake predicts that this lake could harbor cold active GH2s, as this lake is located at a very high altitude. The application of numerous cold-active GH2s having potential in alkyl galactopyranosides synthesis and lactose hydrolysis has been reported from Pseudoalteromonas sp. 22b and Pseudoalteromonas haloplanktis [94, 95]. The presence of GH23, which is very prevalent in cell wall degradation [88] has also been reported in this lake. Further, the abundance of GH53 in the soil sample could facilitate the growth of probiotic strains in this lake as GH53 has been reported to break down Galactooligosaccharides [96]. The unknown richness of this lake may also hold a wealth of biotechnological uses, as earlier studies of the Himalayan range have described [97, 98].
Conclusion
The presence of varied microbial diversity in this lake predicts a thriving ecosystem, housing different species living in harmony, and building a repository of varied metabolic functions to keep the lake thriving and alive. It also reports the functional characteristics of the microbial diversity, and the different metabolic approaches employed by these microorganisms for their survival in these extreme conditions. This includes different genes involved in defense mechanisms, signal transduction mechanisms, transcription, co enzyme transport, and metabolism that they use to survive at this high-altitude and extreme changing environmental conditions. The abundance of glycoside hydrolases highlights the fact that this lake could be a repository of numerous psychrophilic and psychrotrophic glycoside hydrolases, thus paving the way towards a novel discovery that could be beneficial in bio refinery and bioremediation sectors. However, further studies based on the intricacies of carbohydrate deconstruction, and the mechanism of the microbial community involved in the breakdown of the polymer could provide a breakthrough in CAZyme research. The diverse metabolic pathways depicted through this study gives us an insight into the larger scope of exploration of microbes with bioprospective potential. However, the recent anthropogenic activity has definitely remained a threat to the prevailing ecosystem, and the effect of climate change in recent times has also threatened the microbial diversity of such ecological hotspots. Moreover, the discovery of novel microbes with psychrophilic properties that are involved in xenobiotic degradation and catabolism of recalcitrant chemicals could also prove to be a promising feat for designing bioremediation approaches if required.
Availability of data and materials
The datasets generated during and analyzed during the current study are available in NCBI with accession numbers SAMN13671136 and SAMN13671135.
Abbreviations
- GH:
-
Glycoside hydrolases
- COG:
-
Cluster of Orthologous Groups of Proteins
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- Pfam:
-
Protein families
- GO:
-
Gene Ontology
References
Sharma N, Kumar J, Abedin M, Sahoo D, Pandey A, Rai AK, Singh SP (2020) Metagenomics revealing molecular profiling of community structure and metabolic pathways in natural hot springs of the Sikkim Himalaya. BMC Microbiol 20(1):1–17.
Liu K, Liu Y, Jiao N, Xu B, Gu Z, Xing T, Xiong J (2017) Bacterial community composition and diversity in Kalakuli, an alpine glacial-fed lake in Muztagh Ata of the westernmost Tibetan plateau. FEMS Microbiol Ecol 93:085. https://doi.org/10.1093/femsec/fix085
Shokralla S, Spall JL, Gibson JF, Hajibabaei M (2012) Next-generation sequencing technologies for environmental DNA research. Mol Ecol 21:1794–1805. https://doi.org/10.1111/j.1365-294X.2012.05538.x
Vaz-Moreira I, Egas C, Nunes OC, Manaia CM (2011). Culture-dependent and culture-independent diversity surveys target different bacteria: a case study in a freshwater sample. Antonie Van Leeuwenhoek 100(2):245–257.
Kirk JL, Beaudette LA, Hart M, Moutoglis P, Klironomos JN, Lee H, Trevors JT (2004) Methods of studying soil microbial diversity. J Microbiol Methods 58:169–188. https://doi.org/10.1016/j.mimet.2004.04.006
Cowan DA, Ramond JB, Makhalanyane TP, De Maayer P (2015) Metagenomics of extreme environments. Curr Opin Microbiol 25:97–102. https://doi.org/10.1016/j.mib.2015.05.005
Baker BJ, Lazar CS, Teske AP, Dick GJ (2015) Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:1–2. https://doi.org/10.1186/s40168-015-0077-6
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Alexander Richter R, Valas R, Novotny M, Yee-Greenbaum J, Selengut JD, Haft DH, Halpern AL (2012) Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6:1186–1199. https://doi.org/10.1038/ismej.2011.189
Rathour R, Gupta J, Kumar M, Hiloidhari M, Mehrotra AK, Thakur IS (2017) Metagenomic sequencing of microbial communities from brackish water of Pangong Lake of the northwest Indian Himalayas. Genome Announc 5:e01029–e01017. https://doi.org/10.1128/genomeA.01029-17
Staley C, Gould TJ, Wang P, Phillips J, Cotner JB, Sadowsky MJ (2014) Core functional traits of bacterial communities in the Upper Mississippi River show limited variation in response to land cover. Front Microbiol 5:414. https://doi.org/10.3389/fmicb.2014.00414
Oyama D, Kishi LT, Santos-Júnior CD, Soares-Costa A, de Oliveira TC, de Miranda FP, Henrique-Silva F (2016) Metagenomics analysis of microorganisms in freshwater lakes of the Amazon Basin. Genome Announc 22:e01440–e01416. https://doi.org/10.1128/genomeA.01440-16 PMID: 28007865
Rathour R, Gupta J, Mishra A, Rajeev AC, Dupont CL, Thakur IS (2020) A comparative metagenomic study reveals microbial diversity and their role in the biogeochemical cycling of Pangong lake. Sci Total Environ 731:139074. https://doi.org/10.1016/j.scitotenv.2020.139074
Llorens-Marès T, Catalan J, Casamayor EO (2020) Taxonomy and functional interactions in upper and bottom waters of an oligotrophic high-mountain deep lake (Redon, Pyrenees) unveiled by microbial metagenomics. Sci Total Environ 707:135929. https://doi.org/10.1016/j.scitotenv.2019.135929
Libkind D, Moliné M, Sampaio JP, Van Broock M (2009) Yeasts from high-altitude lakes: influence of UV radiation. FEMS Microbiol Ecol 69:353–362. https://doi.org/10.1111/j.1574-6941.2009.00728.x
Bhat FA, Yousuf AR, Aftab A, Arshid J, Mahdi MD, Balkhi MH (2011) Ecology and biodiversity in Pangong Tso (lake) and its inlet stream in Ladakh, India. Int J Biodivers Conserv 3:501–511
Bryant JA, Lamanna C, Morlon H (2008) Microbes on mountainsides: contrasting elevational patterns of bacterial and plant diversity. PNAS 105:11505–11511. https://doi.org/10.1073/pnas.0801920105
Körner C (2007) The use of ‘altitude’ in ecological research. Trends Ecol Evol 22:569–574
Wang JT, Cao P, Hu HW, Li J, Han LL, Zhang LM, Zheng YM, He JZ (2015) Altitudinal distribution patterns of soil bacterial and archaeal communities along Mt. Shegyla on the Tibetan Plateau. Microb Ecol 69:135–145. https://doi.org/10.1007/s00248-014-0465-7
Fierer N, McCain CM, Meir P, Zimmermann M, Rapp JM, Silman MR, Knight R (2011) Microbes do not follow the elevational diversity patterns of plants and animals. Ecology 92(4):797–804. https://doi.org/10.1890/10-1170.1
Margesin R, Jud M, Tscherko D, Schinner F (2009) Microbial communities and activities in alpine and subalpine soils. FEMS Microbiol Ecol 67(2):208–218. https://doi.org/10.1111/j.1574-6941.2008.00620.x
Gangwar P, Alam SI, Bansod S, Singh L (2009) Bacterial diversity of soil samples from the western Himalayas, India. Can J Microbiol 55:564–577. https://doi.org/10.1139/W09-011
Chettri N, Sharma E, Deb DC (2005) Bird community structure along a trekking corridor of Sikkim Himalaya: a conservation perspective. Biol Conserv 102(1):1–16
Chettri N, Sharma E (2006) Prospective for developing a transboundary conservation landscape in the Eastern Himalayas. In: McNeely JA, McCarthy TM, Smith A, Whittaker OL (eds) Conservation Biology in Asia. Society for Conservation Biology Asia Section and Resources Himalaya, Kathmandu, Nepal, pp 21–44
Kaur R, Rajesh C, Sharma R, Boparai JK, Sharma PK (2018) Metagenomic investigation of bacterial diversity of hot spring soil from Manikaran, Himachal Pradesh, India. Ecol Genet Genom 6:16–21
Gupta V, Singh I, Rasool S, Verma V (2020) Next generation sequencing and microbiome’s taxonomical characterization of frozen soil of north western Himalayas of Jammu and Kashmir, India. Electron J Biotechnol 45:30–37
Kumar V, Kumar S, Singh D (2022) Metagenomic insights into Himalayan glacial and kettle lake sediments revealed microbial community structure, function, and stress adaptation strategies. Extremophiles 26:1–11. https://doi.org/10.1007/s00792-021-01252-x
Shukla A, Garg PK, Srivastava S (2018) Evolution of glacial and high-altitude lakes in the Sikkim, Eastern Himalaya over the past four decades (1975–2017). Front Environ Sci 6:81
Russell NJ (2003) Psychrophily and resistance to low temperature. In: Gerday C, Glansdorff N (eds) Extremophiles, Vol 2. EOLSS Publishers Co Ltd., pp 03–00
Venkatachalam S, Gowdaman V, Prabagaran SR (2015) Culturable and culture-independent bacterial diversity and the prevalence of cold-adapted enzymes from the Himalayan mountain ranges of India and Nepal. Microb Ecol 69(3):472–491. https://doi.org/10.1007/s00248-014-0476-4
Gerday C, Aittaleb M, Bentahir M, Chessa JP, Claverie P, Collins T, D’Amico S, Dumont J, Garsoux G, Georlette D, Hoyoux A (2000) Cold-adapted enzymes: from fundamentals to biotechnology. Trends Biotechnol 18:103–107. https://doi.org/10.1016/s0167-7799(99)01413-4
Joseph B, Ramteke PW, Thomas G (2008) Cold active microbial lipases: some hot issues and recent developments. Biotechnol Adv 26(5):457-470.
Pulicherla KK, Ghosh M, Kumar PS, Rao KRSS (2011) Psychrozymes- the next generation industrial enzymes. J Marine Sci Res Dev 1:102
Ohgiya S, Hoshino T, Okuyama H, Tanaka S, Ishizaki K (1999) Biotechnology of enzymes from cold-adapted microorganisms. In: Margesin R, Schinner F (eds) Biotechnological Applications of Cold-Adapted Organisms. Springer-Verlag, Heidelberg, pp 17–34
Uma S, Jadhav RS, Seshu Kumar G, Shivaji S, Ray MK (1999) An RNA polymerase with transcriptional activity at 0°C from the Antarctic bacterium Pseudomonas syringae. FEBS Lett 453:313–317. https://doi.org/10.1016/s0014-5793(99)00660-2
Schleper C, Swanson RV, Mathur EJ, DeLong EF (1997) Characterization of a DNA polymerase from the uncultivated psychrophilic archaeon Cenarchaeum symbiosum. J Bacteriol 179:7803–7811. https://doi.org/10.1128/jb.179.24.7803-7811.1997
Margesin R (1999) Biotechnological applications of cold-adapted organisms: with 65 figures and 45 tables. Springer Science & Business Media
Berlemont R, Martiny AC (2015) Genomic potential for polysaccharides deconstruction in bacteria. Appl Environ 81:1513–1519. https://doi.org/10.1128/AEM.03718-14
Souza TV, Araujo JN, da Silva VM, Liberato MV, Pimentel AC, Alvarez TM, Squina FM, Garcia W (2015) Chemical stability of a cold-active cellulase with high tolerance toward surfactants and chaotropic agent. Biotechnol Rep 9:1–8. https://doi.org/10.1016/j.btre.2015.11.001
Kuddus MR, Arif JM, Ramteke PW (2012) Structural adaptation and biocatalytic prospective of microbial cold-active á-amylase. Afr J Microbiol Res 6:206–213
Mihaela C, Teodor N, Gabriela B, Peter S (2009) Cold adapted amylase and protease from new Streptomyces 4 Alga Antarctic strain. Inn Romanian Food Biotechnol 5:23–30
Shipkowski S, Brenchley JE (2005) Characterization of an unusual cold-active beta-glucosidase belonging to family 3 of the glycoside hydrolases from the psychrophilic isolate Paenibacillus sp. strain C7. Appl Environ Microbiol 71:4225–4232. https://doi.org/10.1128/AEM.71.8.4225-4232.2005
Lu L, Guo L, Wang K, Liu Y, Xiao M (2019) β-Galactosidases: a great tool for synthesizing galactose-containing carbohydrates. Biotechnol Adv 39:107465. https://doi.org/10.1016/j.biotechadv.2019.107465
Amin K, Tranchimand S, Benvegnu T, Abdel-Razzak Z, Chamieh H (2021) Glycoside hydrolases and glycosyltransferases from hyperthermophilic archaea: Insights on their characteristics and applications in biotechnology. Biomolecules 11(11):1557
Saha D (2013) Lesser Himalayan sequences in eastern Himalaya and their deformation: implications for Paleoproterozoic tectonic activity along the northern margin of India. J Geoscience Front 4:289–304
Buckley M (2008) Shangri-La: a practical guide to the Himalayan dream. Bradt Travel Guides
Walkley, A., & Black, I. A. (1934). An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci 37(1):29–38
Kjeldahl JGCT (1883) A new method for the estimation of nitrogen in organic compounds. Z Anal Chem 1883:366–382
Rhodes J, Beale MA, Fisher MC (2014) Illuminating choices for library prep: a comparison of library preparation methods for whole genome sequencing of Cryptococcus neoformans using Illumina HiSeq. PLoS One 9(11):e113501. https://doi.org/10.1371/journal.pone.0113501
Menzel P, Ng KL, Krogh A (2016) Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7:1–9. https://doi.org/10.1038/ncomms11257
Bose T, Haque MM, Reddy C, Mande SS (2015) COGNIZER: a framework for functional annotation of metagenomic datasets. PLoS One 10:0142102. https://doi.org/10.1371/journal.pone.0142102
Simpson EH (1949) Measurement of diversity. Nature 163:688–688
Shannon CE (1948) A mathematical theory of communication. Bell Syst Tech J 27:379–423
Menhinick EF (1964) A comparison of some species-individuals diversity indices applied to samples of field insects. Ecol 45:859–861 http://www.jstor.org/stable/1934933
Buzas MA, Gibson TG (1969) Species diversity: benthonic foraminifera in western North Atlantic. Science 163:72–75. https://doi.org/10.1126/science.163.3862.72
Berger WH, Parker FL (1970) Diversity of planktonic foraminifera in deep-sea sediments. Science 168:1345–1347. https://doi.org/10.1126/science.168.3937.1345
Margalef R (1958) Information theory in ecology. General Systems Yearbook 3:36–71
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42D1:D490–D495. https://doi.org/10.1093/nar/gkt1178
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y (2012) dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40:W445–W451. https://doi.org/10.1093/nar/gks479
Abraham J (2013) Organic carbon estimations in soils: analytical protocols and their implications Rubber. Science 26:45–54
Gorde SP, Jadhav MV (2013) Assessment of water quality parameters: a review. Int J Eng Res Appl 3:2029–2035
Haldar S, Nazareth SW (2018) Taxonomic diversity of bacteria from mangrove sediments of Goa: metagenomic and functional analysis. 3 Biotech 8(10):436. https://doi.org/10.1007/s13205-018-1441-6
Singare PU, Trivedi MP, Mishra RM (2011) Assessing the physico-chemical parameters of sediment ecosystem of Vasai Creek at Mumbai, India. Mar Sci 1:22–29
Lew S, Glińska-Lewczuk K, Ziembińska-Buczyńska A (2018) Prokaryotic community composition affected by seasonal changes in physicochemical properties of water in peat bog lakes. Water 10(4):485
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28:33–36. https://doi.org/10.1093/nar/28.1.33
Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ (2004) The Pfam protein families database. Nucleic Acids Res 32:D138–D141. https://doi.org/10.1093/nar/gkh121
Meyer F, Overbeek R, Rodriguez A (2009) FIGfams: yet another set of protein families. Nucleic Acids Res 37:6643–6654. https://doi.org/10.1093/nar/gkp698
The Gene Ontology Consortium (2008) The Gene Ontology project in 2008. Nucleic Acids Res 36:D440–D444. https://doi.org/10.1093/nar/gkm883
Rai A, Bhattacharjee A (2021) Molecular profiling of microbial community structure and their CAZymes via metagenomics, from Tsomgo lake in the Eastern Himalayas. Arch Microbiol 203(6):3135–3146. https://doi.org/10.1007/s00203-021-02278-7
Liao B, Yan X, Zhang J, Chen M, Li Y, Huang J, Lei M, He H, Wang J (2019) Microbial community composition in alpine lake sediments from the Hengduan Mountains. Microbiologyopen J 8:e00832. https://doi.org/10.1002/mbo3.832
Fang L, Chen L, Liu Y, Tao W, Zhang Z, Liu H, Tang Y (2015) Planktonic and sedimentary bacterial diversity of Lake Sayram in summer. Microbiologyopen 4:814–825. https://doi.org/10.1002/mbo3.281
Krishna M, Gupta S, Delgado-Baquerizo M, Morriën E, Garkoti SC, Chaturvedi R, Ahmad S (2020) Successional trajectory of bacterial communities in soil are shaped by plant-driven changes during secondary succession. Sci Rep 10:1–10.
Newton R J, Jones SE, Eiler A, McMahon K. D, Bertilsson S (2011) A guide to the natural history of freshwater lake bacteria. Microbiology and molecular biology reviews : MMBR 75(1):14–49. https://doi.org/10.1128/MMBR.00028-10
Christner BC, Skidmore ML, Priscu JC, Tranter M, Foreman CM (2008) Bacteria in subglacial environments. In: Margesin R, Schinner F, Marx JC, Gerday C (eds) Psychrophiles: from biodiversity to biotechnology. Springer, Heidelberg, pp 51–71. https://doi.org/10.1007/978-3-540-74335-4_4
Kolton M, Sela N, Elad Y, Cytryn E (2013) Comparative genomic analysis indicates that niche adaptation of terrestrial Flavobacteria is strongly linked to plant glycan metabolism. PLoS One 8:e76704. https://doi.org/10.1371/journal.pone.0076704
Weon HY, Song MH, Son JA, Kim BY, Kwon SW, Go SJ, Stackebrandt E (2007) Flavobacterium terrae sp. nov. and Flavobacterium cucumis sp. nov., isolated from green house soil. Int J Syst Evol Microbiol 57:1594–1598. https://doi.org/10.1099/ijs.0.64935-0
Cousin S, Pauker O, Stackebrandt E (2007) Flavobacterium aquidurense sp. nov. and Flavobacterium hercynium sp. nov. from a hard-water creek. Int J Syst Evol Microbiol 57:243–249. https://doi.org/10.1099/ijs.0.64556-0
Yi H, Oh HM, Lee JH, Kim SJ, Chun J (2005) Flavobacterium antarcticum sp. nov., a novel psychrotolerant bacterium isolated from the Antarctic. Int J Syst Evol Microbiol 55:637–641. https://doi.org/10.1099/ijs.0.63423-0
Zhu L, Liu Q, Liu H, Zhang J, Dong X, Zhou Y, Xin Y (2013) Flavobacterium noncentrifugens sp. nov., a psychrotolerant bacterium isolated from Hailuogou Glacier, South west China. Int J Syst Evol Microbiol 63:2032–2037. https://doi.org/10.1099/ijs.0.045534-0
He Y, Gong Y, Su Y, Zhang Y, Zhou X (2019) Bioremediation of Cr (VI) contaminated groundwater by Geobacter sulfurreducens: environmental factors and electron transfer flow studies. Chemosphere 221:793–801.
Lovley DR, Holmes DE, Nevin KP (2004) Dissimilatory Fe(III) and Mn(IV) reduction. Adv Microb Physiol 49:219–286. https://doi.org/10.1016/S0065-2911(04)49005-5
Anderson RT, Rooney-Varga JN, Gaw CV, Lovley DR (1998) Anaerobic benzene oxidation in the Fe(III) reduction zone of petroleum contaminated aquifers. Environ Sci Technol 32:1222–1229
Lovley DR, Anderson RT (2000) Influence of dissimilatory metal reduction on the fate of organic and metal contaminants in the subsurface. Hydogeol J 8:77–88
Ortiz-Bernad I, Anderson RT, Vrionis HA, Lovley DR (2004) Vanadium respiration by Geobacter metallireducens: novel strategy for in situ removal of vanadium from groundwater. Appl Environ Microbiol 70:3091–3095. https://doi.org/10.1128/AEM.70.5.3091-3095.2004
Caccavo F Jr, Lonergan DJ, Lovley DR, Davis M, Stolz JF, McInerney MJ (1994) Geobacter sulfurreducens sp. nov., a hydrogen-and acetate-oxidizing dissimilatory metal-reducing microorganism. Appl Environ Microbiol 60:3752–3759. https://doi.org/10.1128/aem.60.10.3752-3759.1994
Florentino AP, Weijma J, Stams AJ, Sánchez-Andrea I (2016) Ecophysiology and application of acidophilic sulfur-reducing microorganisms. In: Rampelotto PH (ed) Biotechnology of Extremophiles: Advances and Challenges. Grand Challenges in Biology and Biotechnology, Vol. 1. Springer International Publishing, pp 141–175. https://doi.org/10.1007/978-3-319-13521-2_5
Rabus R, Hansen TA, Widdel F (2006) Dissimilatory sulfate- and sulfur-reducing prokaryotes. In: Dworkin M, Falkow S, Rosenberg E, Schleifer KH, Stackebrandt E (eds) The prokaryotes. Springer, New York, pp 659–768. https://doi.org/10.1007/0-387-30742-7_22
Abot A, Arnal G, Auer L, Lazuka A, Labourdette D, Lamarre S, Trouilh L, Laville E, Lombard V, Potocki-Veronese G, Henrissat B (2016) CAZyChip: dynamic assessment of exploration of glycoside hydrolases in microbial ecosystems. BMC Genomics 17:1–12. https://doi.org/10.1186/s12864-016-2988-4
Janeček Š, Svensson B, MacGregor E (2014) α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci 71:1149–1170. https://doi.org/10.1007/s00018-013-1388-z
Pinto ÉSM, Dorn M, Feltes BC (2020) The tale of a versatile enzyme: Alpha-amylase evolution, structure, and potential biotechnological applications for the bioremediation of n-alkanes. Chemosphere 250:126202. https://doi.org/10.1016/j.chemosphere.2020.126202
A Linares-Pasten J, Andersson M, N Karlsson E (2014) Thermostable glycoside hydrolases in biorefinery technologies. Curr Biotechnol 3(1):26–44
Mewis K, Lenfant N, Lombard V (2016) Dividing the large glycoside hydrolase family 43 into subfamilies: a motivation for detailed enzyme characterization. Appl Environ Microbiol 82:1686–1692. https://doi.org/10.1128/AEM.03453-15
Henrissat B, Davies G (1997) Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol 7:637–644. https://doi.org/10.1016/s0959-440x(97)80072-3
Makowski K, Białkowska A, Olczak J, Kur J, Turkiewicz M (2009) Antarctic, cold-adapted β-galactosidase of Pseudoalteromonas sp. 22b as an effective tool for alkyl galactopyranosides synthesis. Enzym Microb Technol 44:59–64.
Van De Voorde I, Goiris K, Syryn E, Van den Bussche C, Aerts G (2014) Evaluation of the cold-active Pseudoalteromonas haloplanktis β-galactosidase enzyme for lactose hydrolysis in whey permeate as primary step of d-tagatose production. Process Biochem 49:2134–2140. https://doi.org/10.1016/j.procbio.2014.09.010
Böger M, Hekelaar J, Van LSS, Dijkhuizen L, Van Bueren AL (2019) Structural and functional characterization of a family GH53 β-1, 4-galactanase from Bacteroides thetaiotaomicron that facilitates degradation of prebiotic galactooligosaccharides. J Struct Biol 205:1–10. https://doi.org/10.1016/j.jsb.2018.12.002
Kumar V, Thakur V, Kumar S, Singh D (2018) Bioplastic reservoir of diverse bacterial communities revealed along altitude gradient of Pangi-Chamba trans-Himalayan region. FEMS Microbiol Lett 365:fny144. https://doi.org/10.1093/femsle/fny144
Thakur V, Kumar V, Kumar S (2018) Diverse culturable bacterial communities with cellulolytic potential revealed from pristine habitat in Indian trans-Himalaya. Can J Microbiol 64:798–808. https://doi.org/10.1139/cjm-2017-0754
Acknowledgements
The authors are thankful to the Forests, Environment & Wildlife Management Department Government of Sikkim, India, for providing the sampling permits, the University of North Bengal, Xcelris Labs, Ahmedabad, Tea Science (NBU) for estimation of some of the physicochemical parameters and the Department of Science and Technology, Government of India (DST/INSPIRE/03/2014/001812), for INSPIRE Fellowship.
Funding
The authors are thankful to the University of North Bengal for infrastructural facilities and AR is thankful to the Department of Science and Technology, Government of India (DST/INSPIRE/03/2014/001812), for INSPIRE Fellowship.
Author information
Authors and Affiliations
Contributions
Material preparation, data collection, and the first draft of the manuscript was written by AR and AB. The analysis was done by AR, TM and SPS. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have given their consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: S1.
The table shows functional annotation against COG database for ABS1 sample from Samiti Lake. S2. The table shows functional annotation against FIG database for ABS1 sample from Samiti Lake. S3. The table shows functional annotation against GO database for ABS1 sample from Samiti Lake. S4. The table shows functional annotation against KEGG database for ABS1 sample from Samiti Lake. S5. The table shows functional annotation against PFAM database for ABS1 sample from Samiti Lake. S6. The table shows functional annotation against COG database for ABSLW sample from Samiti Lake. S7. The table shows functional annotation against FIG database for ABSLW sample from Samiti Lake. S8. The table shows functional annotation against GO database for ABSLW sample from Samiti Lake. S9. The table shows functional annotation against KEGG database for ABSLW sample from Samiti Lake. S10. The table shows functional annotation against PFAM database for ABSLW sample from Samiti Lake
Additional file 2: Figure S1.
The Krona graph shows the phylum Proteobacteria to be highest in ABS1 sample. Figure S2. The Krona graph shows the phylum Proteobacteria to be highest in ABSLW sample.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rai, A., Saha, S.P., Manvar, T. et al. A shotgun approach to explore the bacterial diversity and a brief insight into the glycoside hydrolases of Samiti lake located in the Eastern Himalayas. J Genet Eng Biotechnol 20, 162 (2022). https://doi.org/10.1186/s43141-022-00444-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00444-y