
Abstract
Background
Chlamydia trachomatis is an obligate intracellular gram-negative pathogen, responsible for diverse affections, mainly trachoma and sexually transmitted diseases. Antibiotics are the commonly used drugs to tackle chlamydiae infections. However, when overused or wrongly used this may lead to strains’ resistance to antibiotics, this phenomenon represents a real health problem worldwide. Numerous studies showed the association of Chlamydia trachomatis resistance with mutations in different genes; these mutations could have a deleterious or neutral impacts on the encoded proteins. The aim of this study is to perform an in silico analysis of C. trachomatis rpoB-encoded proteins using numerous bioinformatics tools and to identify the functional and structural-related effects of the mutations and consequently their impact on the bacteria sensitivity to antibiotics.
Results
The analysis revealed that the prediction of the damaging impact related to the mutations in rpoB-encoded proteins showed eight mutations: V136F, Q458K, V466A, A467T, H471N, H471Y, H471L, and I517M with big deleterious effects. Among them, six mutations, V136F, Q458K, V466A, A467T, H471N, and I517M, are located in a highly conserved regions decreasing the protein’s stability. Furthermore, the structures analysis showed that the mutations A467T, H471N, I517M, and V136F models had a high deviation compared to the wild type. Moreover, the prediction of protein-protein network indicated that rpoB wild type interacts strongly with 10 proteins of C. trachomatis, which are playing different roles at different levels.
Conclusion
As conclusion, the present study revealed that the changes observed in the encoded proteins can affect their functions and structures, in addition to their interactions with other proteins which impact the bacteria sensitivity to antibiotics. Consequently, the information revealed through this in silico analysis would be useful for deeper exploration to understand the mechanisms of C. trachomatis resistance and enable managing the infection to avoid its complications. We recommend further investigations and perform deeper experimental analysis with collaboration between bioinformaticians, physicians, biologists, pharmacists, and chemistry and biochemistry scientists.
Similar content being viewed by others
Background
The family Chlamydiaceae comprises a group of obligate intracellular Gram-negative bacteria that are responsible for a broad range of infections in mammals, birds, and humans [1]. Current classification within this family recognizes a single genus within this family, Chlamydia, 14 species [2,3,4] and four Candidatus (Ca.) species (Ca. Chlamydia ibidis, Ca. Chlamydia corallus, Ca. Chlamydia sanzinia, and Ca. Chlamydia testudinis) [5,6,7,8]. Humans are only Chlamydia trachomatis (C. trachomatis) natural hosts [9]; when C. trachomatis bacteria is untreated, it may lead to severe complications. In females, pelvic inflammatory disease leads to tubal infertility [10,11,12,13], ectopic pregnancy [13], and chronic pelvic pain [14]. Furthermore, C. trachomatis can be transferred to the newborn from the infected mothers; they can develop ocular, respiratory, and gastrointestinal infections [15]. In men, it would be urethritis, epididymitis, prostatitis, and proctitis [16, 17]. In addition, lymphogranuloma venereum [18] and reactive arthritis [19] are the less common diseases caused by C. trachomatis in both men and women infected with C. trachomatis. In another hand, C. trachomatis produces chronic ocular infections that can lead to trachoma, which is one of the leading causes of blindness worldwide [20, 21].
The recommended treatment for chlamydia infections is antibiotics. However, the misuse or the overuse of these antibiotics may lead to antibiotic resistance. The acquired resistance occurs when the bacterium that has been sensitive to antibiotics develops resistance via mutation or via acquisition of new DNA [22].
Different studies showed that C. trachomatis may develop resistance to macrolides via mutations in the 23S rRNA gene, to fluoroquinolones via mutations in the gyrA gene, and to rifamycins via mutations in the rpoB gene [20, 21, 23,24,25,26,27]. The resistance to rifamycins was shown to be associated with a nucleotide substitution in rpoB gene, impacting the inhibition of bacterial transcription related to interacting with beta-subunit of bacterial DNA-dependent RNA polymerase [28].
All mutations within the genomic sequence can lead to alterations in the sequence of the encoded protein, which could have a deleterious or neutral impacts on the protein. Furthermore, it may ultimately affect alteration of protein charge, geometry, hydrophobicity dynamics, translation, and inter- or intra-protein interaction set cells in danger [29].
The aim of the present study is to perform an in silico analysis of the retrieved amino acid variations in C. trachomatis rpoB gene-encoded protein and identify the functional and structural-related effects of the protein’s variations, which consequently may impacting the bacteria sensitivity to antibiotics.
Methods
In the present study, we performed an in silico analysis using different machine learning algorithms, following the various steps described below and illustrated in the flowchart (Fig. 1).

Study steps flowchart
Mutations collection
To find the rpoB gene mutations linked to C. trachomatis resistance to antibiotics, we proceeded to extract all the rpoB mutations from various resources, which were gathered in our precedent investigations [22].
Prediction of rpoB mutations’ deleterious effects
The damaging effects of the mutations on the protein were predicted using PredictSNP1.0 (http://loschmidt.chemi.muni.cz/predictsnp1/) [30]; this tool includes nine different Bioinformatics’ tools: SIFT [31], PolyPhen-1 [32], PolyPhen-2 [33], MAPP [34], PhD-SNP [35], SNAP [36], PANTHER [37], PredictSNP [38], and nsSNPAnalyzer [39]. Most of these tools are designed to predict whether a particular substitution is neutral or deleterious, based on various parameters derived from the evolutionary, physicochemical, or structural characteristics.
PredictSNP1.0 displays the confidence scores generated by each tool and a consensus prediction as percentages by using their observed accuracy values to simplify comparisons [38]. We classified the mutations as deleterious if the results of five among the nine tools is identified as damaging.
Prediction of changes on the protein stability
To predict the change on the protein stability, we performed the Sanavia et al. protocol, where the effects of the variants on the protein stability are quantified in terms of the Gibbs free energy of unfolding (ΔG), and the measure of interest is the difference of the unfolding free energy between the mutant and wild type proteins (ΔΔGu); the sign of ΔΔG indicates if the mutation decreases (ΔΔGu < 0) or increases (ΔΔGu > 0) the protein stability [40].
We analyzed the different approaches available and selected I-Mutant 3.0 and MUpro to perform this analysis. I-Mutant 3.0 (http://gpcr2.biocomp.unibo.it/cgi/predictors/I-Mutant3.0/I-Mutant3.0.cgi) is a support vector machine (SVM) and a web-based tool that provides the predicted change in Gibbs free energy (ΔΔG) [41]. MUpro server (http://mupro.proteomics.ics.uci.edu/) is based on SVM [38]. We submitted the input data of the retrieved mutations of rpoB in FASTA format.
Evolutionary conservation analysis
The evolutionary conservation analysis consists in estimating the degree of the amino acid conservation based on multiple sequence alignment using the ConSurf server (https://consurf.tau.ac.il) [42]. The degree to which an amino acid position is evolutionarily conserved is strongly dependent on its structural and functional importance [42, 43]. In ConSurf, the evolutionary rate is estimated based on the evolutionary relatedness between the protein and its homologs and considering the similarity between amino acids as reflected in the substitutions matrix [44, 45]. In the present study, the homologous sequences were collected using BLAST (or PSI-BLAST) search against a selected database.
The grade ranges from 1 to 9 estimates the extent of conservation of the amino acid throughout evolution. Therefore, grade 9 represents the most highly conserved residue, and the numbers descend to 1 representing the least conserved region.
Proteins’ 3D structure modeling and validation
To understand the effect of each mutation on the protein structure, the 3D models of rpoB-encoded protein and its selected mutants are designed with SWISS-MODEL (https://swissmodel.expasy.org/) [46]. According to QMEAN score, and sequence identity, the best quality model was selected. Furthermore, to confirm the models, the Ramachandran plots were generated with PROCHECK [47].
Structure analysis of wild type and mutant models
To compare the native and mutated protein structures, structural similarities were calculated using TM-align tool (https://zhanglab.ccmb.med.umich.edu/TM-align/), based on template modeling score (TM-score) and the root mean square deviation (RMSD) scores [48]. Tm-align produces a result between 0 and 1. TM-score equal to 1 means that there is no difference between wild type and the mutant structure; however, TM-score closer to 0 means higher deviation. Furthermore, the RMSD score is proportional to the deviation between the wild type and the mutated structures. Finally, the visualization of the wild type and mutants’ structures were performed by Chimera tool [49].
Structural effect of point mutation rpoB-encoded protein
The energy minimization is essential to determine the proper molecular arrangement in space; it used to eliminate high energies in the predicted model and achieve local minima that is closer to native structure [50]. For this reason, the energy minimization was performed for the wild type and mutants’ structure; in addition, the structural consequences of mutations were visualized by Chimera tool [49].
Prediction of protein-protein interactions
Protein-protein interaction plays key role in predicting the protein function of target protein and drug ability of molecules. The majority of genes and proteins realize resulting phenotype functions as a set of interactions [51]. To investigate the interaction of rpoB-encoded protein with various proteins, the STRING database was used (https://string-db.org) [52]. This database aims to integrate all known and predicted associations between proteins, including both physical interactions and functional associations [53].
Results
rpoB gene’s reference sequence and mutations’ datasets
In order to identify the rpoB gene mutations associated with C. trachomatis resistance to antibiotics, we performed a literature search pertaining to the topic exhaustively, and a total of nine mutations were retrieved (Table 1). Furthermore, the rpoB gene sequence of C. trachomatis reference strain BU-434/L2 was extracted from NCBI database using the published accession number AY623623.1 [24].
Prediction of rpoB mutations’ deleterious effects
According to the rules and recommendation of predictSNP, the results revealed that all the mutations are deleterious. Indeed, the variations V136F, Q458K, A467T, H471Y, H471L, and I517M were shown to be deleterious with a high confidence score 87%, whereas the mutations V466A and H471N were shown to be deleterious with 61% confidence score (Table 2).
Prediction of changes on the protein stability
The eight mutations predicted as deleterious from the previous step were analyzed with both I-Mutant3.0 and MUpro tools; the results showed that the six mutations, V136F, Q458K, V466A, A467T, H471N, and I517M, were predicted to decrease the rpoB-encoded protein’s stability (Table 3). Furthermore, the most of the mutations that predicted to decrease the stability of the protein were be shown in C. trachomatis strains serovar L2.
Evolutionary conservation analysis
The six mutations, which were shown decreasing the rpoB-encoded protein stability, were analyzed by the ConSurf web server, and the results revealed that these mutations had a high conservation score and located in the highly conserved regions (Table 4); three mutations were predicted as functional and exposed (on protein surface), whereas the rest were predicted to be structural and buried (inside protein core).
Proteins’ 3D structure modeling and validation
The tertiary structures of rpoB wild type and mutants’ proteins were subjected to a modeling process using the Swiss-Model (Table 5). Furthermore, the Ramachandran plot of each model shows that the residues in most favored regions are greater than 80%, which explain the accurate of the modeling results (Fig. 2).

Proteins’ 3D models and Ramachandran plots
Structure analysis of the wild type and mutant models
The comparison between mutants and wild type structural 3D models was performed using TM-align; the results showed that the models, A467T, H471N, I517M, and V136F, had the highest root mean square deviation (RMSD = 3.14); furthermore, all the models had template modeling score (TM-score) near to 1; these results signify that the status of protein folding is identical. In addition, the structural 3D models had high RMSD score which signify a high deviation between mutants and wild type (Table 6). Consequently, the 3D mutants’ models, V136F, A467T, H471N, and I517M, were considered for more explorations and further analysis.
Structural effect of point mutation in rpoB-encoded protein
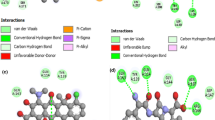
The energy minimization results showed a wide variance between the energy of wild type and the energy of each mutant’s model (Table 7); in addition, the mutants’ structures have more hydrogen bonds (H-Bond) interactions with the adjacent molecules which signify the dispute between wild type and models energies (Fig. 3).

Comparison between native and mutant rpoB-encoded proteins’ tridimensional structures. a Wild type V and mutant F residues at 136th position (V136F). b Wild type Q and mutant K residues at 458th position (Q458K). c Wild type V and mutant A residues at 466th position (V466A). d Wild type A and mutant T residues at 476th position (A476T). e Wild type H and the mutant N residues at 471th position (H471N). f Wild type I and mutant M residues at 517th position (I517M)
Prediction of protein-protein interactions
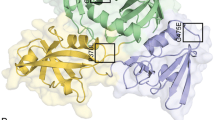
The prediction of protein-protein interactions using STRING indicated that rpoB-encoded protein interacts with 10 proteins from C. trachomatis bacteria, including rpsA, rpoC, rpoA, sigA, greA, nusA, rplL, mfd, fusA, and rpsC (Fig. 4).

rpoB protein-protein interaction network
It is known that any occurred changes in the protein can affect the protein network interaction. Therefore, the results revealed that rpoB network have high confidence interaction scores; moreover, the molecular action of rpoB-encoded protein with other proteins could be modified (Table 8).
Discussion
The treatments adopted commonly against C. trachomatis infections are macrolides, tetracyclines, rifamycins, and quinolones [54]. However, the bacteria can acquire resistant to different antibiotics family via a range of mechanisms. The main causes of resistance to antibiotics are: the abusive usage of antibiotics, the spread of resistant strains, or the spread of genes bearing information able to induce resistance [55].
Different studies revealed that the resistance to rifamycin was associated with mutations in the C. trachomatis RNA polymerase β-subunit gene (rpoB) [20, 21, 24, 25, 56]; these mutations could have a deleterious or neutral impacts on the encoded proteins; moreover, to understand the impact of these mutations on the proteins’ biological functions, stability, and structure, an in silico analysis was performed.
In genomics and proteomics, the in silico analysis plays a significant role to predict the impact of the mutations on the proteins’ function and structure; this analysis can be performed using different bioinformatics tools. However, using these tools could have strengths and weaknesses in the predictions, because every algorithm uses different parameters for prediction [57, 58]. Consequently, to screen and prioritize the candidate functional SNPs requires the implementation of algorithms with different parameters and aspects to combine their advantages, enhance the accuracy and reliability of the predictions, and minimize the errors [59,60,61]. Basically, to perform the SNP prediction, it is recommended to use at least five tools to obtain an agreement on the effect of the variations on the structure and function of the studied proteins [58]. In our approach, nine tools were used to predict the SNP deleterious effects on function and structure of the rpoB-encoded protein (Fig. 1); this protocol was adopted by different investigators [62,63,64]. Our results revealed that the nine used tools showed that all the SNPs (n = 8) have a deleterious effect on the encoded protein (Table 2); indeed, almost the same results of prediction were found by the different used tools; this explains the prediction accuracy and result validity.
It is known that the protein structure governs its stability and determines its function [65]; in our study, the prediction of changes on the proteins’ stability showed differences in the predictions’ results (Table 3). Indeed, the stability prediction using I-Mutant3.0 and MUpro showed discrepant results for the mutations H471Y and H471L, which consist in decreased proteins’ stability by MUpro, and increased proteins’ stability by I-Mutant3.0. In addition, according to the retrieved results, the six mutations, V136F, Q458K, V466A, A467T, H471N, and I517M, were shown to be destabilizing the proteins’ structure by indicating a negative score for the Gibbs free energy. Hence, these mutations can cause misfolding, degradation, or aberrant conglomeration of the rpoB-encoded proteins. The results discrepancies’ can be considered as a negative aspect of the analysis; for that, we suggest performing deeper prediction using new tools.
On the other hand, the ConSurf results revealed that the studied mutations had high conservation scores and located in the highly conserved regions (Table 4). The mutation position can directly affect the proteins’ function and structure; consequently, the mutations position could affect the drug accessibility to the bacteria and so its level of antibiotics sensitivity.
The wild protein 3D structures are essential to more understand the functional and structural effect of mutations; the rpoB protein structure was not available in the Protein Data Bank (PDB). Thus, we predict the 3D structure of wild type and mutants, by changing the six mutations into the native sequence. In addition, Ramachandran plot analysis was performed to validate these protein structures; all the structures had the residues in most favored regions more than 80%, which means that all the structures are valid (Fig. 2). Hence, no negative aspect was notified using the Swiss-Model Server.
Additionally, the structural changes in the encoded proteins were analyzed using TM-Align to compute the RMSD and the TM-score by superimposing models of native and mutant proteins. The results showed that the models with the mutations V136F, A467T, H471N, and I517M had high RMSD values and their TM-score near to 1 (Table 6). Hence, these results indicate a quite large structural dissimilarity between the native and mutant models. Indeed, the structural changes of mutants’ proteins indicate potential alterations in the binding affinity of mutants’ structures of rpoB-encoded proteins with their receptors which may lead to resistance to antibiotics. Regarding the predictions of the mutations Q458K and V466A, the results showed a lowest RMSD score and their TM-score near to 1; these results signify that the status of the protein folding is identical.
In another hand of the analysis, the results of the energy minimization revealed that the total energy of the mutants: V136F, A467T, H471N, and I517M were low compared to that of the wild type protein. Moreover, the mutants present the higher number of H-bonds. In opposition to the high total energy, the wild type structure had the lower number of H-bonds (Table 7). The discrepancy of results found between wild type and mutant structures signifies the dispute between wild type and mutants’ models, which can explain the observed resistance to antibiotics mechanisms.
At the final step of our in silico analysis, the prediction of the protein-protein interaction revealed that rpoB interacts with 10 different bacteria’s proteins (Fig. 4), which are showed playing a role in the bacteria cycle progression and DNA replication events (Table 8). In addition, STRING predicts these protein-protein interactions with high confidence scores; therefore, it can be suggested that any change in the rpoB-encoded protein structure and function might affect the bacteria sensitivity to antibiotics.
Conclusion
This study compiled the mutations in rpoB gene which were revealed to be associated with C. trachomatis resistance to rifamycin and predicts their effects using various bioinformatics tools. The results revealed that the mutations, V136F, A467T, H471N, and I517M, had the most impact on both stability and RMSD, in addition to their location in the highly conserved regions; therefore, they can affect the protein’s function, structure, and their interaction with other proteins. All these changes can explain the observed resistance to antibiotics. Moreover, the study revealed that all mutations are not necessarily translated to strong phenotypic expression. Consequently, the revealed information through this in silico analysis would be useful for deeper exploration to understand the mechanisms of C. trachomatis resistance; this could enable managing the infection and avoid its complications. We recommend further investigations to perform deeper experimental analysis and explore alternative therapies and new drug design by molecular docking; this can be done in collaboration between bioinformaticians, physicians, biologists, pharmacists, and chemistry and biochemistry scientists.
Availability of data and materials
Not applicable.
Abbreviations
- C. trachomatis :
-
Chlamydia trachomatis
- Ca :
-
Candidatus
- SNP:
-
Single nucleotide polymorphism
- ΔG:
-
Gibbs free energy of unfolding
- ΔΔG:
-
Change in Gibbs free energy
- SVM:
-
Support vector machine
- TM-score:
-
Template modeling score
- RMSD:
-
Root means square deviation
- BLAST:
-
Basic Local Alignment Search Tool
- PSI-BLAST:
-
Position-Specific Iterative Basic Local Alignment Search Tool
- QMEAN:
-
Qualitative Model Energy Analysis
- NCBI:
-
National Center for Biotechnology Information
- H-Bond:
-
Hydrogen bond
References
Longbottom D, Coulter LJ (2003) Animal chlamydioses and zoonotic implications. J Comp Pathol 128:217–244. https://doi.org/10.1053/jcpa.2002.0629
Sachse K, Bavoil PM, Kaltenboeck B, Stephens RS, Kuo CC, Rossello-Mora R et al (2015) Emendation of the family Chlamydiaceae: proposal of a single genus, Chlamydia, to include all currently recognized species. Syst Appl Microbiol 38(2):99–103. https://doi.org/10.1016/j.syapm.2014.12.004
Laroucau K, Vorimore F, Aaziz R, Solmonson L, Hsia RC, Bavoil PM et al (2019) Chlamydia buteonis, a new Chlamydia species isolated from a red-shouldered hawk. Syst Appl Microbiol 42(5):125997. https://doi.org/10.1016/j.syapm.2019.06.002
Staub E, Marti H, Biondi R, Levi A, Donati M, Leonard CA et al (2018) Novel Chlamydia species isolated from snakes are temperature-sensitive and exhibit decreased susceptibility to azithromycin. Sci Rep 8(1):5660. https://doi.org/10.1038/s41598-018-23897-z
Vorimore F, Hsia RC, Huot-Creasy H, Bastian S, Deruyter L, Passet A et al (2013) Isolation of a new Chlamydia species from the feral sacred Ibis (Threskiornis aethiopicus): Chlamydia ibidis. PLoS One 8(9):e74823. https://doi.org/10.1371/journal.pone.0074823
Taylor-Brown A, Bachmann NL, Borel N, Polkinghorne A (2016) Culture-independent genomic characterisation of Candidatus chlamydia sanzinia, a novel uncultivated bacterium infecting snakes. BMC Genomics 17:710. https://doi.org/10.1186/s12864-016-3055-x
Taylor-Brown A, Spang L, Borel N, Polkinghorne A (2017) Culture-independent metagenomics supports discovery of uncultivable bacteria within the genus Chlamydia. Sci Rep 7(1):10661. https://doi.org/10.1038/s41598-017-10757-5
Laroucau K, Ortega N, Vorimore F, Aaziz R, Mitura A, Szymanska-Czerwinska M et al (2020) Detection of a novel Chlamydia species in captive spur-thighed tortoises (Testudo graeca) in southeastern Spain and proposal of Candidatus chlamydia testudinis. Syst Appl Microbiol 43(2):126071. https://doi.org/10.1016/j.syapm.2020.126071
Witkin SS, Minis E, Athanasiou A, Leizer J, Linhares IM (2017) Chlamydia trachomatis: the persistent pathogen. Clin Vaccine Immunol 24(10):e00203–e00217. https://doi.org/10.1128/CVI.00203-17
Bakken IJ, Ghaderi S (2009) Incidence of pelvic inflammatory disease in a large cohort of women tested for Chlamydia trachomatis: a historical follow-up study. BMC Infect Dis 9:130. https://doi.org/10.1186/1471-2334-9-130
Bender N, Herrmann B, Andersen B, Hocking JS, van Bergen J, Morgan J et al (2011) Chlamydia infection, pelvic inflammatory disease, ectopic pregnancy and infertility: cross-national study. Sex Transm Infect 87(7):601–608. https://doi.org/10.1136/sextrans-2011-050205
Price MJ, Ades AE, De Angelis D, Welton NJ, Macleod J, Soldan K et al (2013) Risk of pelvic inflammatory disease following Chlamydia trachomatis infection: analysis of prospective studies with a multistate model. Am J Epidemiol 178(3):484–492. https://doi.org/10.1093/aje/kws583
Price MJ, Ades AE, Welton NJ, Simms I, Macleod J, Horner PJ (2016) Proportion of pelvic inflammatory disease cases caused by Chlamydia trachomatis: consistent picture from different methods. J Infect Dis 214(4):617–624. https://doi.org/10.1093/infdis/jiw178
Haggerty CL, Gottlieb SL, Taylor BD, Low N, Xu F, Ness RB (2010) Risk of sequelae after Chlamydia trachomatis genital infection in women. J Infect Dis 201(Suppl 2):S134–S155. https://doi.org/10.1086/652395
Adachi K N, Nielsen-Saines K, Klausner J D (2021) Chlamydia trachomatis Screening and Treatment in Pregnancy to Reduce Adverse Pregnancy and Neonatal Outcomes: A Review. Front Public Health 9:531073. https://doi.org/10.3389/fpubh.2021.531073
Malhotra M, Sood S, Mukherjee A, Muralidhar S, Bala M (2013) Genital Chlamydia trachomatis: an update. Indian J Med Res 138(3):303–316
Mohseni M, Sung S, Takov V. Chlamydia. (2021) In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022.
Mabey D, Peeling RW (2002) Lymphogranuloma venereum. Sex Transm Infect 78(2):90–92
Villareal C, Whittum-Hudson JA, Hudson AP (2002) Persistent Chlamydiae and chronic arthritis. Arthritis Res Ther 4(1):1–5
Rupp J, Solbach W, Gieffers J (2008) Variation in the mutation frequency determining quinolone resistance in Chlamydia trachomatis serovars L2 and D. J Antimicrob Chemother 61(1):91–94
Dreses-Werringloer U, Padubrin I, Köhler L, Hudson AP (2003) Detection of nucleotide variability in rpoB in both rifampin-sensitive and rifampin-resistant strains of Chlamydia trachomatis. Antimicrob Agents Chemother 47(7):2316–2318
Hawkey PM (1998) The origins and molecular basis of antibiotic resistance. BMJ 317(7159):657–660. https://doi.org/10.1136/bmj.317.7159.657
Benamri I, Azzouzi M, Sanak K, Moussa A, Radouani F (2021) An overview of genes and mutations associated with Chlamydiae species’ resistance to antibiotics. Ann Clin Microbiol Antimicrob 20(1):59. https://doi.org/10.1186/s12941-021-00465-4
Kutlin A, Kohlhoff S, Roblin P, Hammerschlag MR, Riska P (2005) Emergence of resistance to rifampin and rifalazil in Chlamydophila pneumoniae and Chlamydia trachomatis. Antimicrob Agents Chemother 49(3):903–907. https://doi.org/10.1128/AAC.49.3.903-907.2005
Suchland RJ, Bourillon A, Denamur E, Stamm WE, Rothstein DM (2005) Rifampin-resistant RNA polymerase mutants of Chlamydia trachomatis remain susceptible to the ansamycin rifalazil. Antimicrob Agents Chemother 49(3):1120–1126
Jiang Y, Zhu H, Yang LN, Liu YJ, Hou SP, Qi ML et al (2015) Differences in 23S ribosomal RNA mutations between wild-type and mutant macrolide-resistant Chlamydia trachomatis isolates. Exp Ther Med 10(3):1189–1193. https://doi.org/10.3892/etm.2015.2595
Deguchi T, Hatazaki K, Ito S, Kondo H, Horie K, Nakane K et al (2018) Macrolide and fluoroquinolone resistance is uncommon in clinical strains of Chlamydia trachomatis. J Infect Chemother 24(8):610–614
Chen LF, Kaye D (2011) Current use for old antibacterial agents: polymyxins, rifamycins, and aminoglycosides. Med Clin North Am 95(4):819–8ix. https://doi.org/10.1016/j.mcna.2011.03.007
Kucukkal TG, Petukh M, Li L, Alexov E (2015) Structural and physico-chemical effects of disease and non-disease nsSNPs on proteins. Curr Opin Struct Biol 32:18–24. https://doi.org/10.1016/j.sbi.2015.01.003
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J et al (2014) PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol 10(1):1003440
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30(17):3894–3900
Adzhubei I, Jordan DM, Sunyaev SR (2013) Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 76(1):7–20
Stone EA, Sidow A (2005) Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15(7):978–986
Capriotti E, Calabrese R, Casadio R (2006) Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 22(22):2729–2734
Bromberg Y, Rost B (2007) SNAP: predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res 35(11):3823–3835
Tang H, Thomas PD (2016) PANTHER-PSEP: predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 32(14):2230–2232
Cheng J, Randall A, Baldi P (2006) Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 62(4):1125–1132
Bao L, Zhou M, Cui Y (2005) nsSNPAnalyzer: identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res 33(Web Server issue):W480–W482. https://doi.org/10.1093/nar/gki372
Sanavia T, Birolo G, Montanucci L, Turina P, Capriotti E, Fariselli P (2020) Limitations and challenges in protein stability prediction upon genome variations: towards future applications in precision medicine. Comput Struct Biotechnol J 18:1968–1979. https://doi.org/10.1016/j.csbj.2020.07.011
AddCapriotti E, Fariselli P, Casadio R (2005) I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res 33(Suppl. 2):306–310
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N (2016) ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res 44(W1):W344–W350
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res 38:W529–W533. https://doi.org/10.1093/nar/gkq399
Pupko T, Bell RE, Mayrose I, Glaser F, Ben-Tal N (2002) Rate4Site: an algorithmic tool for the identification of functional regions in proteins by surface mapping of evolutionary determinants within their homologues. Bioinformatics 18(Suppl. 1):S71–S77
Mayrose I, Graur D, Ben-Tal N, Pupko T (2004) Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol 21:1781–1791
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R et al (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26(2):283–291
Zhang Y, Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33(7):2302–2309. https://doi.org/10.1093/nar/gki524
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Kini RM, Evans HJ (1991) Molecular modeling of proteins: a strategy for energy minimization by molecular mechanics in the AMBER force field. J Biomol Struct Dyn 9(3):475–488. https://doi.org/10.1080/07391102.1991.10507930
Rao VS, Srinivas K, Sujini GN, Kumar GN (2014) Protein-protein interaction detection: methods and analysis. Int J Proteomics 2014:147648. https://doi.org/10.1155/2014/147648
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J et al (2015) STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43(Database issue):D447–D452
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S et al (2021) The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49(D1):D605–D612. https://doi.org/10.1093/nar/gkaa1074
Kohlhoff SA, Hammerschlag MR (2015) Treatment of Chlamydial infections: 2014 update. Expert Opin Pharmacother 16(2):205–212
Laura V M, Vladi M H, Vasile S C (2017) Determining the Antibiotic Resistance of Bacterial Pathogens in Sexually Transmitted Diseases. In: Kumavath R N (ed) Antibacterial Agents. IntechOpen. London. https://doi.org/10.5772/67871
Borel N, Leonard C, Slade J, Schoborg RV (2016) Chlamydial antibiotic resistance and treatment failure in veterinary and human medicine. Curr Clin Microbiol Rep 3:10–18
Chen S, Huo X, Lin Y, Ban H, Lin Y, Li W et al (2010) Association of MDR1 and ERCC1 polymorphisms with response and toxicity to cisplatin-based chemotherapy in non-small-cell lung cancer patients. Int J Hyg Environ Health 213(2):140–145
Brown DK, Bishop ÖT (2017) Role of structural bioinformatics in drug discovery by computational SNP analysis: analyzing variation at the protein level. Glob Heart 12(2):151–161
Doss CGP, Rajith B (2013) A new insight into structural and functional impact of single-nucleotide polymorphisms in PTEN gene. Cell Biochem Biophys 66(2):249–263
Bhatnager R, Dang AS (2018) Comprehensive in-silico prediction of damage associated SNPs in human Prolidase gene. Sci Rep 8(1):9430
Pires AS, Porto WF, Franco OL, Alencar SA (2017) In silico analyses of deleterious missense SNPs of human apolipoprotein E3. Sci Rep 7(1):2509
Pshennikova VG, Barashkov NA, Romanov GP, Teryutin FM, Solov'ev AV, Gotovtsev NN et al (2019) Comparison of predictive in silico tools on missense variants in GJB2, GJB6, and GJB3 genes associated with autosomal recessive deafness 1A (DFNB1A). ScientificWorldJournal:5198931. https://doi.org/10.1155/2019/5198931
Periwal N, Rathod SB, Pal R, Sharma P, Nebhnani L, Barnwal RP et al (2021) In silico characterization of mutations circulating in SARS-CoV-2 structural proteins. J Biomol Struct Dyn:1–16. https://doi.org/10.1080/07391102.2021.1908170
Remali J, Aizat WM, Ng CL, Lim YC, Mohamed-Hussein ZA, Fazry S (2020) In silico analysis on the functional and structural impact of Rad50 mutations involved in DNA strand break repair. PeerJ 8:e9197. https://doi.org/10.7717/peerj.9197
Panja AS, Maiti S, Bandyopadhyay B (2020) Protein stability governed by its structural plasticity is inferred by physicochemical factors and salt bridges. Sci Rep 10(1):1822. https://doi.org/10.1038/s41598-020-58825-7
Acknowledgements
We acknowledge the H3ABionet network (Pan African Bioinformatics Network for the consortium Human Heredity and Health in Africa “H3Africa”) for the provided support.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
IB: data extraction, data curation, data analysis, designed the study, drafting the paper. MA: data curation. AM: reviewing and editing. FR: project conception, following the process, validated the data, reviewing the paper. All the authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Benamri, I., Azzouzi, M., Moussa, A. et al. An in silico analysis of rpoB mutations to affect Chlamydia trachomatis sensitivity to rifamycin. J Genet Eng Biotechnol 20, 146 (2022). https://doi.org/10.1186/s43141-022-00428-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-022-00428-y